一种识别增强子与超级增强子的数据处理方法及其系统
1.本发明涉及生物信息学及基因组学技术领域,更具体地,涉及一种识别增强子与超级增强子的数据处理方法及其系统。
背景技术:
2.过去几年,人们对理解基因组区域的调控作用产生了极大的兴趣,调控dna的研究已经取得了新的重要进展。随着基因组高通量测序技术的快速发展,功能性基因组区域数据迅速增加。如何理解这些未知的基因组区域是目前一项紧迫的工作。在人类生物学过程中,功能基因组区域主要通过影响基因表达水平,进而在疾病发生发展中发挥着作用。为了挖掘基因组区域在转录调控中的功能,需要整合现有的、大量的遗传学和表观遗传学信息。
3.研究表明,基因集的功能富集分析是一种非常成功的生物信息学分析方法。它将新发现的基因集逐一与已经明确功能的基因集合进行比较,得到新基因的功能。这种方法有一个关键限制:数据是必须以基因为中心。借鉴基因集合的富集分析方法,研究者们提出了基因组区域富集分析,同样是将现有基因组区域数据与新发现的区域数据相结合,找到两个集合中区域重叠的部分,然后利用统计学方法计算富集显著得分。通过基因组区域富集分析,可以让研究人员更好的探索基因组区域的生物学功能。大量的人类遗传学和表观遗传学研究迅速积累了chip-seq和atac-seq等不同数据集,有效地整理并利用它们,对于研究基因组区域来说显得十分重要。同时,许多已发表的研究表明,增强子、超级增强子等功能基因组区域在人类生物学过程中发挥着难以替代的作用。
4.增强子及其强度的鉴定是生物学研究的热点之一,吸引了大量研究人员。之前研究人员别无选择,只能用实验的方法解决这个问题,如染色质免疫沉淀、深度测序、dnase i超敏反应和组蛋白修饰的全基因组定位等等。但是这些实验方法既昂贵、耗时又效率低。因此急需一些计算方法来识别增强子及其强度。事实上,已有一些研究做了这项工作。例如,2016年,刘等人建立了一个两层的预测器,它不仅可以识别增强子还可以识别它们的强度;贾等人通过组合和选择多种特征建立了一个识别器来发现增强子;两年后,刘等人基于集成学习方法提出了一个模型来识别增强子及其强度;2019年,nguyen等人提出利用卷积神经网络的集成来识别增强子及其强度。但是整体的识别精度并不是很高,并且现有技术也缺乏直接智能化识别普通增强子与超级增强子的方法,因此需发明新的计算方法来识别增强子及其强度,并能够有效识别普通增强子与超级增强子;同时也能够将识别的结果直接作为基因组区域富集分析过程中的数据类型之一,进行基因组区域富集分析,可以让研究人员更好的探索基因组区域的生物学功能。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提供一种识别增强子与超级增强子的数据处理方法,快速、准确地识别增强子及其强度,并能够对普通增强子区域和超级增强子区域做出有效识别;同时,还可将利用此方法得出的处理结果应
用在基因组区域注释富集分析过程中,保证富集分析方法结果的准确性、便携性和系统性,从深层次挖掘隐含在生物数据背后的生命规律,解决相关的生命科学问题。
6.本技术公开一种识别增强子与超级增强子的数据处理方法,包括:
7.获取样本数据;
8.对所述样本数据进行预处理,得到所述样本数据的富集区域,以及所述样本数据的富集区域所对应染色质区域的位置信息和信号强度;
9.对所述位置信息和信号强度分别进行特征提取,得到特征提取后的位置信息特征数据和信号强度特征数据;
10.根据所述染色质区域的位置信息特征数据将相邻所述样本数据的富集区域缝合在一起得到缝合后的所述样本数据的富集区域;
11.基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果;
12.输出所述排序结果。
13.所述缝合后的所述样本数据的富集区域通过以下方式得到:
14.去除距离基因转录起始位点第一阈值内的所述样本数据的富集区域,得到非启动子区域的所述缝合后的所述样本数据的富集区域;
15.基于所述非启动子区域的所述缝合后的所述样本数据的富集区域,将间隔小于第二阈值之内的富集区域缝合在一起得到所述缝合后的所述样本数据的富集区域;所述缝合后的所述样本数据的富集区域为缝合增强子;
16.可选的,所述位置信息特征数据包括:所述富集区域在基因组上的开始位置和结束位置;所述信号强度特征数据包括:所述富集区域的信号峰的高度。
17.所述预处理的过程包括序列比对、识别富集区域;
18.所述序列比对过程包括:获取包含read的fastq文件;采用算法将所述fastq文件比对到参考基因组上,得到read在参考基因组的位置信息;所述read在参考基因组的位置信息被存储在sam文件;
19.所述识别富集区域的过程包括:获取所述sam文件,利用算法对所述sam文件进行分析,得到所述样本数据的富集区域;所述样本数据的富集区域被存储在bed文件;
20.可选的,所述预处理的过程还包括:格式转换;
21.所述格式转换过程包括:获取所述sam文件,利用算法将所述sam格式文件转换为所述bam文件;
22.获取所述bam文件,利用算法对所述bam文件进行分析,得到所述样本数据的富集区域;所述样本数据的富集区域被存储在bed文件。
23.所述基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果的过程包括:
24.对基因组区域内所述样本数据的富集区域信号进行归一化处理,得到所述样本数据的富集区域信号的背景归一化密度水平;根据所述样本数据的富集区域信号的背景归一化密度水平对所述缝合后的所述样本数据的富集区域和未缝合的所述样本数据的富集区域进行排序,得到排序结果。
25.所述数据处理方法还包括:根据所述排序结果进行构图,得到基于所述染色质区
域的信号强度特征数据的分布曲线;
26.计算所述分布曲线的曲线斜率,输出所述曲线斜率;
27.将所述曲线斜率与参考斜率阈值做比较,输出所述曲线斜率大于/等于/小于参考斜率阈值的富集区域;
28.可选的,所述曲线斜率大于/等于参考斜率阈值的富集区域确定为超级增强子,所述曲线斜率小于参考斜率阈值的富集区域确定为普通增强子。
29.所述样本数据包括经筛选处理后的样本数据;所述经筛选处理后的样本数据包括多组样本数据,每组样本数据包括h3k27ac数据和相应对照的input数据;
30.所述经筛选处理后的样本数据的获取方法或步骤包括:
31.获取原始样本数据;所述原始样本数据包括:样本的细胞组织类型、治疗条件、样本号;
32.对所述原始样本数据进行筛选处理,得到经筛选处理后的样本数据;所述经筛选处理后的样本数据为唯一的非冗余的;
33.可选的,所述筛选处理包括:根据唯一的样本号对所述样本数据进行校验;所述校验采用手动校验。
34.一种识别增强子与超级增强子的数据处理系统,包括:
35.获取单元,用于获取样本数据;
36.第一处理单元,用于对所述样本数据进行预处理,得到所述样本数据的富集区域,以及所述样本数据的富集区域所对应染色质区域的位置信息和信号强度;用于对所述位置信息和信号强度分别进行特征提取,得到特征提取后的位置信息特征数据和信号强度特征数据;
37.第二处理单元,用于根据所述染色质区域的位置信息特征数据将相邻所述样本数据的富集区域缝合在一起得到缝合后的所述样本数据的富集区域;
38.第三处理单元,用于基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果;
39.输出单元,用于输出所述排序结果。
40.本技术具有以下有益效果:
41.1、本技术创新性的公开一种识别增强子与超级增强子的数据处理方法,该方法从原始数据入手,通过预处理等步骤得到富集区域,识别增强子及其强度;并采取为富集区域排序、构图的方法,对普通增强子区域和超级增强子区域做出有效识别,大大提高数据分析的精度和深度。
42.2、本技术创新性的利用识别增强子与超级增强子的数据处理方法的处理结果应用在基因组区域注释富集分析的过程中,作为基因组区域注释富集分析的其中一个数据集,保证富集分析方法结果的准确性、便携性和系统性,充分利用了样本数据;还能从深层次挖掘隐含在生物数据背后的生命规律,将其应用在多种疾病的基因组富集分析研究中,具有很大的应用价值。
附图说明
43.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使
用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获取其他的附图。
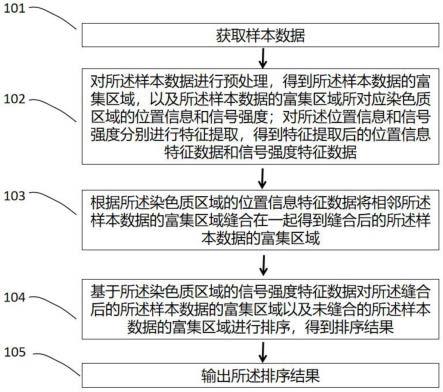
44.图1是本发明实施例提供的一种识别增强子与超级增强子的数据处理方法的分析示意流程图;
45.图2是本发明实施例提供的一种识别增强子与超级增强子的数据处理设备示意图;
46.图3是本发明实施例提供的一种识别增强子与超级增强子的数据处理系统的示意流程图;
47.图4是本发明实施例提供的基于基因组区域注释的富集分析方法的分析示意流程图。
具体实施方式
48.为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。
49.在本发明的说明书和权利要求书及上述附图中的描述的一些流程中,包含了按照特定顺序出现的多个操作,但是应该清楚了解,这些操作可以不按照其在本文中出现的顺序来执行或并行执行,操作的序号如101、102等,仅仅是用于区分开各个不同的操作,序号本身不代表任何的执行顺序。另外,这些流程可以包括更多或更少的操作,并且这些操作可以按顺序执行或并行执行。需要说明的是,本文中的“第一”、“第二”等描述,是用于区分不同的消息、设备、模块等,不代表先后顺序,也不限定“第一”和“第二”是不同的类型。
50.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获取的所有其他实施例,都属于本发明保护的范围。
51.图1是本发明实施例提供的一种识别增强子与超级增强子的数据处理方法示意流程图,具体地,所述方法包括如下步骤:
52.101:获取样本数据;
53.在一个实施例中,所述样本数据包括经筛选处理后的样本数据;所述经筛选处理后的样本数据包括多组样本数据,每组样本数据包括h3k27ac数据和相应对照的input数据;h3k27ac数据通常发挥促进基因表达的作用,被认为是基因激活的标记。chip实验中的input就是输入的总dna,未通过抗体富集处理的,作为分析时的背景参考。在实验过程中,抗体富集以前,需要将input独立出来,然后与富集后的ip一起解交联和纯化建库。input也可以验证ip打断的效果和最后分析的背景资料。
54.在一个实施例中,所述经筛选处理后的样本数据的获取方法或步骤包括:
55.获取原始样本数据;所述原始样本数据为来自ncbi geo/sra、encode、roadmap和ggr的h3k27ac chip-seq数据中处理收集到的542个公开可用的人类样本;在公共数据库中,我们输入关键词“h3k27ac”和“chip-seq”来搜索样本,得到了2,000多个原始样本的信息,包括样本的细胞组织类型、治疗条件和样本号等;
56.对所述原始样本数据进行筛选处理,得到经筛选处理后的样本数据;所述经筛选处理后的样本数据为唯一的非冗余的;所述筛选处理包括:根据唯一的样本号对所述样本数据进行校验;所述校验采用手动校验。最终,从这四个公共数据来源收集了542个公开可用的人类样本。
57.生物信息学将生物与数学、计算机进行了有效结合,主要通过综合运用数学和信息科学等多领域的方法和工具对生物信息进行获取、加工、存储、分析和解释,来阐明大量生物数据所包含的生物学意义,研究重点主要体现在基因组学和蛋白质组学两方面。
58.102:对所述样本数据进行预处理,得到所述样本数据的富集区域,以及所述样本数据的富集区域所对应染色质区域的位置信息和信号强度;对所述位置信息和信号强度分别进行特征提取,得到特征提取后的位置信息特征数据和信号强度特征数据;
59.在一个实施例中,所述预处理的过程包括序列比对、识别富集区域;
60.所述序列比对过程包括:获取包含read的fastq文件;采用算法将所述fastq文件比对到参考基因组上,得到read在参考基因组的位置信息;所述read在参考基因组的位置信息被存储在sam文件;参考基因组为为ucsc下载的hg19参考基因组;序列比对过程采用的软件包括但不限于以下软件:bowtie,具体代码如下:
61.其中-n 2表示在高保真区域允许最大错配碱基数为2;-e 70表示在错配位点phred quality值不能超过70;
62.单末端:bowtie-e 70-k 2-n 2-m 2-s-q genome h3k27ac.fastq h3k27ac.sam
63.单末端:bowtie-e 70-k 2-n 2-m 2-s-q genome input.fastq input.sam
64.双末端:bowtie-e 70-k 2-n 2-m 2-s-q genome-1h3k27ac_1.fastq-2
65.h3k27ac_2.fastq h3k27ac.sam
66.双末端:bowtie-e 70-k 2-n 2-m 2-s-q genome-1input_1.fastq-2input_2.fastq input.sam
67.所述识别富集区域的过程包括:获取所述sam文件,利用算法对所述sam文件进行分析,得到所述样本数据的富集区域;所述样本数据的富集区域被存储在bed文件;识别富集区域的过程采用的软件包括但不限于以下软件:macs;使用macs14找到h3k27ac富集的区域,具体代码如下:其中
‑‑
keep-dup保留重复。默认macs(auto)会使用二项分布估计每个位置上是否存在重复(默认是1,也就是每个位置上出现一个read的概率最大)。输入为bam文件,输出为富集区域的bed文件;
68.macs14-p 1e-9-w-s
‑‑
keep-dup=auto
‑‑
wig
‑‑
single-profile
‑‑
space=50-cinput.sort.bam-t h3k27ac.sort.bam-g hs-n macs
69.可选的,所述预处理的过程还包括:格式转换;
70.所述格式转换过程包括:获取所述sam文件,利用算法将所述sam格式文件转换为所述bam文件;格式转换过程采用的软件包括但不限于以下软件:
71.samtools;使用samtools将sam文件转换为二进制bam文件,具体代码如下:
72.samtools view-b-s h3k27ac.sam》h3k27ac.bam
73.samtools sort h3k27ac.bam》h3k27ac.sort.bam
74.samtools index h3k27ac.sort.bam h3k27ac.sort.bam.bai
75.samtools view-b-s input.sam》input.bam
76.samtools sort input.bam》input.sort.bam
77.samtools index input.sort.bam input.sort.bam.bai
78.获取所述bam文件,利用算法对所述bam文件进行分析,得到所述样本数据的富集区域;所述样本数据的富集区域被存储在bed文件。此步骤采用macs软件。
79.103:根据所述染色质区域的位置信息特征数据将相邻所述样本数据的富集区域缝合在一起得到缝合后的所述样本数据的富集区域;
80.在一个实施例中,采用rose缝合增强子识别超级增强子;所述缝合后的所述样本数据的富集区域通过以下方式得到:
81.去除距离基因转录起始位点第一阈值内的所述样本数据的富集区域,得到非启动子区域的所述缝合后的所述样本数据的富集区域;第一阈值为
±
2kb,保证计算的区域都是非启动子区域;
82.基于所述非启动子区域的所述缝合后的所述样本数据的富集区域,将间隔小于第二阈值之内的富集区域缝合在一起得到所述缝合后的所述样本数据的富集区域;所述缝合后的所述样本数据的富集区域为缝合增强子;第二阈值为12,500bp,定义了一个跨越基因组区域的实体,缝合增强子是由多个增强子元件构成。
83.识别普通增强子和超级增强子的过程采用的软件包括但不限于以下软件:rose;使用rose识别超级增强子与普通增强子,以macs找到的h3k27ac富集区域为基础进行识别。-s 12500为缝合间隔小于12,500bp的富集区域;-t 2000排除tss区域大小,排除与tss前后2,000bp的区域,以排除启动子偏差。输出包含超级增强子与普通增强子位置、样本中排名、构成元件个数等信息的txt格式文件。具体代码如下:
84.python rose_main.py-g hg19-i macs_peaks.gff-c input.sort.bam-r h3k27ac.sort.bam-o name-s 12500-t 2000
85.可选的,所述位置信息特征数据包括:所述富集区域在基因组上的开始位置和结束位置;所述信号强度特征数据包括:所述富集区域的信号峰的高度。
86.104:基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果;
87.在一个实施例中,所述基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果的过程包括:
88.对基因组区域内所述样本数据的富集区域信号进行归一化处理,得到所述样本数据的富集区域信号的背景归一化密度水平;根据所述样本数据的富集区域信号的背景归一化密度水平对所述缝合后的所述样本数据的富集区域和未缝合的所述样本数据的富集区域进行排序,得到排序结果。根据缝合增强子和剩余单个增强子的信号从大到小进行排序。
89.所述数据处理方法还包括:根据所述排序结果进行构图,得到基于所述染色质区域的信号强度特征数据的分布曲线;
90.计算所述分布曲线的曲线斜率,输出所述曲线斜率;
91.将所述曲线斜率与参考斜率阈值做比较,输出所述曲线斜率大于/等于/小于参考斜率阈值的富集区域;
92.可选的,所述曲线斜率大于/等于参考斜率阈值的富集区域确定为超级增强子,所
述曲线斜率小于参考斜率阈值的富集区域确定为普通增强子。
93.105:输出所述排序结果。
94.图2是本发明实施例提供的一种识别增强子与超级增强子的数据处理设备,所述设备包括:存储器和处理器;
95.所述存储器用于存储程序指令;
96.所述处理器用于调用程序指令,当程序指令被执行时,用于执行上述的识别增强子与超级增强子的数据处理方法。
97.图3是本发明实施例提供的一种识别增强子与超级增强子的数据处理系统,包括:
98.获取单元301,用于获取样本数据;
99.第一处理单元302,用于对所述样本数据进行预处理,得到所述样本数据的富集区域,以及所述样本数据的富集区域所对应染色质区域的位置信息和信号强度;用于对所述位置信息和信号强度分别进行特征提取,得到特征提取后的位置信息特征数据和信号强度特征数据;
100.第二处理单元303,用于根据所述染色质区域的位置信息特征数据将相邻所述样本数据的富集区域缝合在一起得到缝合后的所述样本数据的富集区域;
101.第三处理单元304,用于基于所述染色质区域的信号强度特征数据对所述缝合后的所述样本数据的富集区域以及未缝合的所述样本数据的富集区域进行排序,得到排序结果;
102.输出单元305,用于输出所述排序结果。
103.一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现上述的识别增强子与超级增强子的数据处理方法。
104.图4是本发明实施例提供的基于基因组区域注释的富集分析方法的分析示意流程图;
105.一种基于基因组区域注释的富集分析方法,包括:
106.获取基因组区域集合;
107.将所述基因组区域集合与样本数据参考集合进行匹配,计算所述基因组区域集合与样本数据参考集合的富集得分值;所述样本数据参考集合包括基于上述识别到的增强子区域与超级增强子区域;
108.基于所述富集得分值,得到所述基因组区域集合的富集显著排名。
109.一种基于基因组区域注释的富集分析系统,包括:
110.收集单元,用于获取基因组区域集合;
111.分析单元,用于将所述基因组区域集合与样本数据参考集合进行匹配,计算所述基因组区域集合与样本数据参考集合的富集得分值;所述样本数据参考集合包括基于上述识别到的增强子区域与超级增强子区域;
112.排序单元,用于基于所述富集得分值,得到所述基因组区域集合的富集显著排名。
113.在一个实施例中,所述样本数据参考集合还包括以下:基于隐马尔可夫模型算法的染色质状态数据、转录因子与转录辅因子、组蛋白修饰、开放染色质区域、snp数据、甲基化数据、lncrna与mrna;
114.其中,基于隐马尔可夫模型算法的染色质状态数据的获取方法为:收集了来自
roadmap数据库上使用chromhmm计算h3k4me3、h3k4me1、h3k36me3、h3k27me3和h3k9me3五个染色质标记的15个核心染色质状态数据,包括增强子、启动子、绝缘子和异染色质。
115.转录因子与转录辅因子的获取方法为:从cistrome下载了转录因子和转录辅因子chip-seq数据,其中转录因子结合区域信息覆盖超过6,000个样本,包括57种组织类型和2,528个转录因子;转录辅因子结合区域信息涵盖3,000多个样本,包括41种组织类型和973个转录辅因子。
116.组蛋白修饰的获取方法为:从encode中下载了组蛋白修饰chip-seq数据,为研究人员提供一个用户友好的分析。这些chip-seq数据包含了1,400多个样本,涵盖33种组蛋白修饰,如h3k27ac、h3k27me3和h3k4me1等。
117.开放染色质区域的获取方法为:从ncbi geo/sra下载了1493个atac-seq数据,涵盖多种细胞/组织类型。我们使用bowtie2、samtools和macs2的统一流程来识别开放染色质区域,并开发了atacdb数据库。
118.snp数据的获取方法为:从pancanqtl中下载并合并human eqtl数据集。pancanqtl数据包括tcga中不同癌症的eqtl-gene关系对。gwas提供了大量的数据,将遗传变异与常见表型联系起来。本发明从nhgri gwas catalog中收集了风险snp。然后,我们过滤了“variant id”中不属于“rsid”的风险snp。最后,获得了1,515,001个与疾病、性状和表型相关的风险snp。从snp对基因表达的影响出发,本发明将snp位点分别扩大了10kb/1kb、15kb/1kb和20kb/1kb,此扩充位点不做限定。
119.甲基化数据的获取方法为:从encode的450k芯片数据中获取了198,468,712个甲基化位点。本发明根据β值将这些位点分为高甲基化和低甲基化。β值大于0.6的位点被认为是高甲基化,而β值大于0.2且小于0.6的位点被认为是低甲基化。最终,本发明还将甲基化位点分别扩大了10kb/1kb、15kb/1kb和20kb/1kb,此扩充位点不做限定。
120.lncrna与mrna的获取方法为:从lncsea数据库收集了多类lncrna集的数据,包括疾病、药物、亚细胞定位、癌症标志、smorf、外泌体和细胞标记。另外,本发明从cellmarker数据库中收集了mrna的人类细胞标记信息。根据从gencode下载注释文件,将这些mrna的转录起始位点分别扩展为2kb/1kb、5kb/1kb和10kb/1kb的区域并作为mrna参考集合子类(cell_marker_2kb、cell_marker_5kb以及cell_marker_10kb)。除此之外,我们收集包含编码蛋白基因的goterm的组分信息。同样的,将goterm中基因的转录起始位点分别扩展为2kb/1kb、5kb/1kb和10kb/1kb的区域作为mrna参考集合子类(goterm_2kb、goterm_5kb以及goterm_10kb);上述涉及到的扩充位点均不做限定。
121.在一个实施例中,所述计算所述基因组区域集合与样本数据参考集合的富集得分值包括但不限于以下方法:超几何检验方法和位点重叠分析(lola);
122.所述超几何检验方法的分析过程包括:识别所述基因组区域集合与对应所述样本数据参考集合的区域重叠部分,得到所述区域重叠部分的重叠个数;采用超几何检验方法对所述区域重叠部分的重叠个数进行计算,得到所述富集得分值;所述基因组区域集合与对应所述样本数据参考集合的区域重叠部分包括至少一个碱基相交;
123.具体地,超几何检验方法首先是使用bedtools软件找出两个集合的区域重叠部分,然后将重叠区域的个数输入到超几何检验中进行计算富集得分p;富集显著性p值计算为:
[0124][0125]
常规的方法;
[0126]
其中m代表参考集合区域数;n代表由dnasei信号区域组成的背景集合区域数;n代表用户输入区域数;k代表用户输入区域与参考集合重叠的区域数。
[0127]
获取基因组区域集合后,当选择超几何检验方法计算富集得分的时候,系统会将所述基因组区域集合生成一个bed文件存放在服务器中,然后使用bedtools软件找出两个集合的区域重叠部分。在本发明中,认为两个区域之间如果有一个碱基相交就可以被当作重叠区域。还可以通过选择“最小重叠百分比”参数来过滤bedtools找到的重叠区域。最后,系统将重叠区域的个数输入到超几何检验中进行计算富集得分。
[0128]
可选的,所述位点重叠分析的分析过程包括:对所述基因组区域集合进行过滤,得到过滤后的所述基因组区域集合;将所述过滤后的所述基因组区域集合与所述样本数据参考集合做比较,得到重叠区域;采用fisher精确检验方法对所述重叠区域进行计算,得到所述富集得分值。
[0129]
具体地,位点重叠分析(lola)是借助lola的r包,通过引入universe集合对用户输入的区域进行过滤,然后再和参考集合比较才得到重叠区域,使用fisher精确检验计算富集得分。lola通过引入universe文件,灵活的将用户输入的区域进行了过滤,然后根据交叠情况分别获得a、b、c、d四个值,其中a代表用户输入的集合与universe和背景集合都有交集的个数;b代表用户输入的集合只与universe或者只与背景集合有交集的个数;c代表用户输入的集合或者universe只与背景集合有交集的个数;d代表用户输入的集合、universe、背景集合三者直接都没有交集的个数。然后再用fisher精确检验进行p值计算,具体公式如下:
[0130][0131]
其中n代表用户输入集合的区域个数。获取基因组区域集合后,当选择lola方法的时候,系统找到重叠区域的途径就不同于超几何检验方法,而是借助lola的r包。通过引入universe集合对用户输入的区域进行过滤,然后再和参考集合比较才得到重叠区域。计算富集得分的方法则是费希尔精确检验。
[0132]
为了降低假阳性,本系统还用到了多重假设检验对计算好的p值进行校正,例如bonferroni方法和fdr方法。bonferroni方法通过将p值按照检验次数进行降低,以此过滤掉所有的假阳性结果。但是这样可能会造成假阴性,导致富集结果的缺失,所以系统对p值还提供了fdr校正。fdr法则通过控制false discovery rate来校正p值,计算fp/(tp+fp)的期望值,如果期望值小于p值0.05,则认为该检验的结果具有生物学意义。
[0133]
在一个实施例中,所述富集分析系统还包括区域注释单元,区域注释单元包括用于对基因组区域的遗传学注释和基因组区域的表观遗传学注释;基因组区域的遗传学注释包括:common snp,风险snp,eqtl;
[0134]
基因组区域的表观遗传学注释包括:转录因子与转录辅因子,增强子与超级增强子,dna甲基化,组蛋白修饰,染色质开放性。
[0135]
在一个实施例中,所述富集分析系统提供了3种查询方式来搜索基因组区域集合,具体为:
[0136]“通过数据类型搜索”,选择数据类和子类进行查询;
[0137]“通过基因组区域搜索”,选择数据类、子类和输入基因组区域进行查询;
[0138]“通过基因名搜索”,选择数据类、子类和输入和基因名称进行查询。
[0139]
所述富集分析系统提供了多种富集分析策略,可以从不同的角度对获取到的区域集合进行富集分析,增加富集得分的准确性。还收集处理11种不同数据类型,并按每种数据类型的特点进行分类;同时还将对每个区域提供广泛的注释信息,并且还能实现数据可视化。本系统侧重于构建全面的人类基因组区域集,是迄今收集人类基因组区域集合最多的软件,并提供准确的富集分析方法以及广泛的注释信息。不仅能够探索基因组区域的生物学意义,还有利于挖掘基因组区域在转录调控中的功能。
[0140]
此外,利用所述富集分析系统,发明人还对以下疾病做了临床研究:乳腺癌、结肠癌、心脑血管疾病。
[0141]
在乳腺癌案例研究中,本研究将在tcga项目和circlncrnanet数据库中下载的与乳腺癌相关lncrna(log2fc》1,padj《0.05)的转录起始位点周围-2/+1kb的区域作为输入,发现“乳腺癌”集合被显著富集,通过对集合中的lncrna(如hotair)的启动子区域、增强子区域和染色质开放区域进一步研究,找到了重要的调控信息。在hotair的启动子区域,通过chip-seq分析找到了25个相关转录因子的结合。这些转录因子被证实都与乳腺癌相关,证明了本方法的可靠性。
[0142]
在结肠癌案例研究中,本研究将与结肠癌调控相关的转录因子tcf7l2的绑定位点数据作为输入,能够发现tcf7l2是如何通过远端调控来影响结肠癌发生发展的。在本研究中能够清楚的发现在结肠癌细胞系hct116中tcf7l2绑定位点与多个超级增强子区域重叠。本研究还提供了这些超级增强子的相关基因(myc等)、相关转录因子(ctcf等)以及信号通路(tgf_beta_receptor等),这些都能间接证明在结肠癌中,转录因子tcf7l2能够通过调控超级增强子来影响结肠癌致癌基因的转录。当我们把与结肠癌相关的增强子区域作为输入时,也能得到类似的结论。
[0143]
在心脑血管疾病的案例研究中,本研究将一批心衰的差异基因的启动子区域周围-2/+1kb的区域作为输入,显著富集到心肌细胞的gata4转录因子的绑定位点。gata4是心脏基因调节中的关键转录因子,对肥大激动剂有反应。
[0144]
值得注意的是,在本发明的富集分析中,多个与疾病相关的致癌基因和转录因子被注释到,这说明所述富集分析系统对于研究基因组区域在疾病的发生发展中的转录调控具有重要意义。
[0145]
通过对乳腺癌、结肠癌和心脑血管疾病的案例研究,我们发现本研究收集整理的数据背景集合以及富集分析方法对研究基因组区域在疾病发生机制中发挥着重要作用。
[0146]
本验证实施例的验证结果表明,为适应症分配固有权重相对于默认设置来说可以适度改善本方法的性能。
[0147]
所属领域的技术人员可以清楚地了解到,为描述的方便和简洁,上述描述的系统,装置和单元的具体工作过程,可以参考前述方法实施例中的对应过程,在此不再赘述。
[0148]
在本技术所提供的几个实施例中,应该理解到,所揭露的系统,装置和方法,可以
通过其它的方式实现。例如,以上所描述的装置实施例仅仅是示意性的,例如,所述单元的划分,仅仅为一种逻辑功能划分,实际实现时可以有另外的划分方式,例如多个单元或组件可以结合或者可以集成到另一个系统,或一些特征可以忽略,或不执行。另一点,所显示或讨论的相互之间的耦合或直接耦合或通信连接可以是通过一些接口,装置或单元的间接耦合或通信连接,可以是电性,机械或其它的形式。
[0149]
所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个网络单元上。可以根据实际的需要选择其中的部分或者全部单元来实现本实施例方案的目的。
[0150]
另外,在本发明各个实施例中的各功能单元可以集成在一个处理单元中,也可以是各个单元单独物理存在,也可以两个或两个以上单元集成在一个单元中。上述集成的单元既可以采用硬件的形式实现,也可以采用软件功能单元的形式实现。
[0151]
本领域普通技术人员可以理解上述实施例的各种方法中的全部或部分步骤是可以通过程序来指令相关的硬件来完成,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器(rom,read only memory)、随机存取存储器(ram,random access memory)、磁盘或光盘等。
[0152]
本领域普通技术人员可以理解实现上述实施例方法中的全部或部分步骤是可以通过程序来指令相关的硬件完成,所述的程序可以存储于一种计算机可读存储介质中,上述提到的存储介质可以是只读存储器,磁盘或光盘等。
[0153]
以上对本发明所提供的一种计算机设备进行了详细介绍,对于本领域的一般技术人员,依据本发明实施例的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1