基于马尔科夫模型的噬菌体分类的方法
本发明涉及病毒组分类学,特别涉及基于马尔科夫模型的噬菌体分类的方法。
背景技术:
1、病毒是地球上最大的未开发遗传多样性储存库,随着宏基因组测序的广泛应用,新的病毒基因组序列急剧的累积。噬菌体通过调节自然环境中细菌的丰度,在平衡全球生态系统中发挥着重要作用。噬菌体还与人体的健康密切相关,目前已经发现噬菌体的丰度和组成变化与溃疡性结肠炎、克罗恩病和糖尿病等相关。从宏病毒组数据中鉴定和组装成病毒基因组序列后,对序列进行分类学分类是病毒组研究的基础。但是新鉴定出的病毒中包含大量的新颖病毒序列以及长度从几百碱基到几千碱基的短基因组片段,这些基因组序列给病毒组的分类提出了挑战。
2、目前较为通用的方法是基于blast的搜索方法。先使用prodigal注释基因组编码的所有蛋白,然后使用blast方法从已知序列库中查找基因组包含的每个蛋白匹配的最相似的已知蛋白,最后根据每个蛋白的最佳匹配蛋白所属的家族或属进行投票决定查询基因组的家族或属。该方法优点为假阳性较低,精确度较高;其缺点为对短片断的序列和远同源的基因组预测较为困难。
3、另一类分类方法的代表为vcontact,其基于基因组间的基因共享来构建网络,然后根据网络来聚类成病毒簇从而对噬菌体基因组进行分类,其优点在于能够自动化、可信的给出属级别的分类,可以适用到大的宏基因组数据集;其缺点为较小的基因组或基因组片段难以被分类,存在较多的病毒序列空间未能被其分类。
4、因此,本申请设计了一种基于马尔科夫模型的噬菌体分类的方法。
技术实现思路
1、本发明提供了基于马尔科夫模型的噬菌体分类的方法,其目的是为了解决背景技术存在的上述问题。
2、为了达到上述目的,本发明提供了一种新型的噬菌体自动化分类的方法,适用于从宏基因组或宏病毒组数据中组装形成的基因组片段的分类学分类。
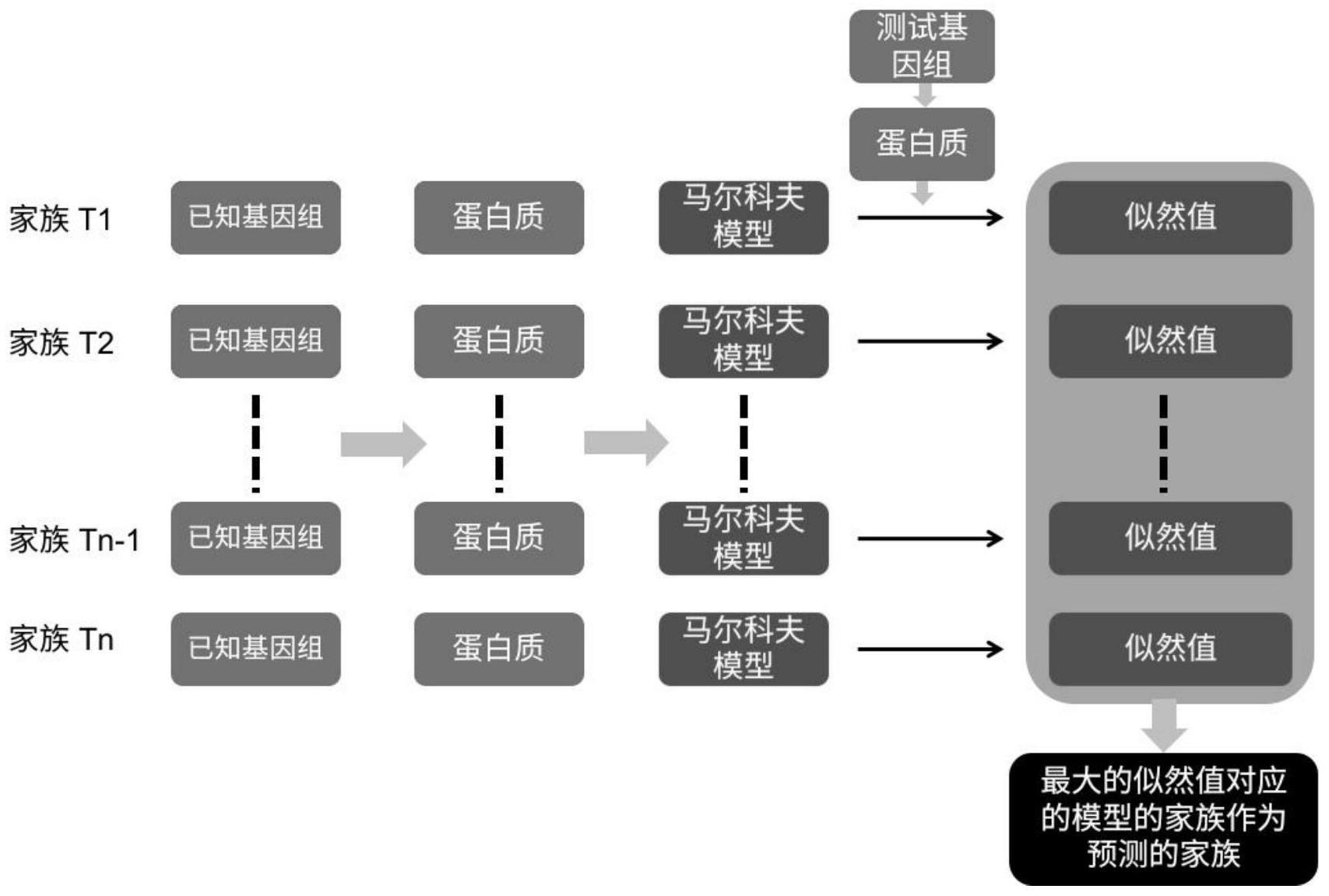
3、本发明的实施例提供了基于马尔科夫模型的噬菌体分类的方法,包括如下步骤:
4、s1.将ncbi噬菌体基因组使用prodigal翻译成蛋白质序列,建立蛋白质库t;对某个分类层级(目、家族或属等)的每个分类单元构建蛋白数据库,以家族层级(family)为例,将已有数据库中的基因组以科为单位整合成单独的基因组数据库,然后使用prodigal进行蛋白质注释转换成每个科的蛋白质库t;
5、s2.取所述蛋白质库t的蛋白质序列,计算长度为k的肽段肽段状态下相邻的下一个氨基酸(nxk+1)的条件概率。nx1...nxk为长度为肽段x1...xk在蛋白库t中的数量,nx1...nx+1k为长度为肽段x1...xk+1在蛋白库t中的数量,得到k阶马尔科夫模型的状态转移概率矩阵,α为可以调整的伪计数,见式(1)
6、
7、将未分类基因组使用prodigal翻译成蛋白质,获得未分类基因组的蛋白质序列v=y1...yn,根据式(1)计算所述未分类基因组的蛋白质序列与蛋白质库t的对数似然值ll,yi+k为起点为i时的k长度的肽段,yi+k-1为起点为i时的k-1长度的肽段,将这两个肽段代入(1)式中得到的状态转移概率矩阵pt,然后对未分类基因组的蛋白质序列v得到的所有概率值取log累加后取均值得到对数似然值ll,见式(2)
8、
9、将所述ll拟合高斯分布的均值和方差,获得所述马尔科夫模型的ll的高斯分布和p值;
10、s3.所述ll得分最高所对应马尔科夫模型的基因组即为预测的噬菌体基因组。预测可信度的评估,以科层级为例,对某个科构建的马尔科夫模型,计算该模型与其他科的病毒基因组的对数似然值ll,使用这些ll值拟合高斯分布的均值和方差从而得到该模型的对数似然值ll的高斯分布;在应用时,针对未分类的基因组得到ll值后,依据模型的高斯分布计算得到p值。后续可以根据p值划定可信度门槛。
11、进一步的,所述p值<0.01时,预测准确率高达98%。
12、本发明的上述方案有如下的有益效果:
13、1、本发明按照分类学将病毒库中的基因组归类后,对每一类中所有的蛋白质序列建模得到每一类的模型,可以更好的评估待分类的病毒基因组的与每一类病毒所有的基因组的整体相似度。
14、2、本发明能够对较短的基因组片段(如几千碱基对长度)进行更准确的分类学分类指定;
15、3、本发明能够对与已知病毒基因组同源性较低的基因组片段预测指定准确的分类学分类。
技术特征:
1.一种基于马尔科夫模型的噬菌体分类的方法,其特征在于,包括如下步骤:
2.根据权利要求1所述的基于马尔科夫模型的噬菌体分类的方法,其特征在于,所述p值<0.01时,预测准确率高达98%。
技术总结
本发明提供了基于马尔科夫模型的噬菌体分类的方法,以科层级为例,对某个科构建的马尔科夫模型,计算该模型与其他科的病毒基因组的对数似然值LL,使用这些LL值拟合高斯分布的均值和方差从而得到该模型的对数似然值LL的高斯分布。在应用时,针对未分类的基因组得到LL值后,依据模型的高斯分布计算得到P值。后续可以根据P值划定可信度门槛。本发明不仅对较短的基因组片段(如几千碱基对长度)进行更准确的分类学分类指定,而且对与已知病毒基因组同源性较低的基因组片段预测指定准确的分类学分类。
技术研发人员:彭友松,卢聪毓
受保护的技术使用者:湖南大学
技术研发日:
技术公布日:2024/1/11
- 还没有人留言评论。精彩留言会获得点赞!