一种基于自然语言处理的GWAS基因编码方式
本发明涉及一种基于自然语言处理的gwas基因编码方式。
背景技术:
1、全基因组关联分析,英文是genome-wideassociation study,简称gwas。实质是在探究一种因果关系,也就是说x的变化,是否会引起y的变化。在gwas里,x一般指的是snp,即单核苷酸多态性,而y是观察到的表型。snp,一般通过芯片或测序方式得到。换而言之,gwas是确定300万个snp位点中那些部分对某些人类表型,如身高、是否双眼皮、头发颜色、瞳孔颜色的有影响,并从数据角度分析这些snp如何影响人类表型,snp序列原始数据一般是acgt四种不同的碱基组成的位点序列,比如“acgggttaaccaatt”。
2、在现有的技术解决方案中,一般针对该基因序列进行简单编码或独热(one-hot)编码,将其编码为0123或00 01 10 11格式。但是这种技术方案存在一个先验假设,否定了基因序列的时序性,而事实上,相邻区域的snp位点很有可能会互相影响,例如,基因区域casc17通过转录因子与相邻的sox9相互作用,共同影响人类面部的骨骼形成。
技术实现思路
1、本发明的目的是克服现有的全基因组关联分析存在的否定了基因序列的时序性,且相邻区域的snp位点很有可能会互相影响的缺陷。
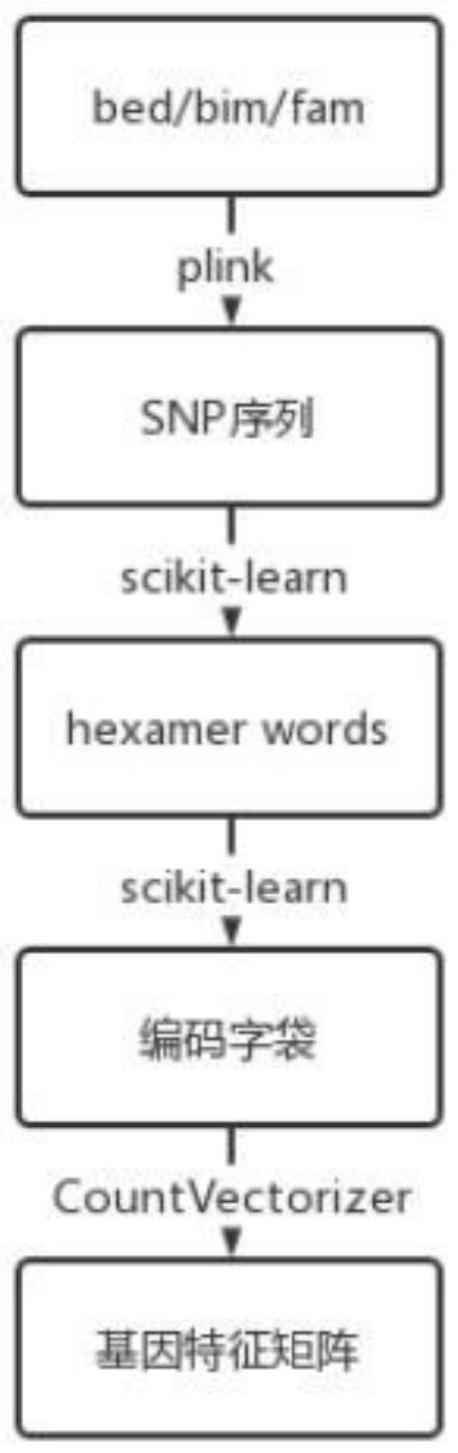
2、为达上述目的,本发明提供了一种基于自然语言处理的gwas基因编码方法,其特殊之处在于,对基因组最原始的.bed、.bid或.fam文件使用plink v1.9软件的raw命令进行处理以得到最原始的snp序列,即aagctaaggtcccaa;然后,将所述snp序列通过python中的scikit-learn库的自然语言处理工具编码为若干个词汇,该词汇同时包含了时序性的特征和snp序列数据本质的特征;之后,使用scikit-learn下的countvectorizert包,对所述词汇建立单词袋模型,countvectorizer包会将输入的文档中的关键词进行对比,得到一个包含权重的编码字袋,终而得到该snp序列对应的特征矩阵,以使用该特征矩阵进行后续分析处理。
3、所述词汇为一个atgcat序列,且选择进制为5的词汇,则所述snp序列被分割为atgca和tgcat两种不同的hexamer words。
4、克服现有的全基因组关联分析存在的否定了基因序列的时序性的缺陷,尤其是与现有的全基因组关联分析相比,所获得的数据具有更高的准确度。
5、以下将结合附图对本发明做进一步详细说明。
技术特征:
1.一种基于自然语言处理的gwas基因编码方法,其特征在于:
2.根据权利要求1所述的基于自然语言处理的gwas基因编码方法,其特征在于:所述词汇为一个atgcat序列,且选择进制为5的词汇,则所述snp序列被分割为atgca和tgcat两种不同的hexamer words。
技术总结
本发明涉及一种基于自然语言处理的GWAS基因编码方法,对基因组最原始的.bed、.bid或.fam文件进行raw命令处理以得到最原始的SNP序列,即AAGCTAAGGTCCCAA;然后,将SNP序列通过Python中的scikit‑learn库的自然语言处理工具编码为若干个词汇,再使用scikit‑learn下的CountVectorizert包,对词汇建立单词袋模型,得到一个包含权重的编码字袋,终而得到该SNP序列对应的特征矩阵,以使用该特征矩阵进行后续分析处理,所获得的数据具有更高的准确度。
技术研发人员:江梓赫,范虹,范晓诺
受保护的技术使用者:陕西师范大学
技术研发日:
技术公布日:2024/1/13
- 还没有人留言评论。精彩留言会获得点赞!