利用Hi-C技术的基因辅助组装方法、染色体水平基因组及应用与流程
本发明涉及一种利用hi-c技术的基因辅助组装方法,组装得到的染色体水平的基因组及应用。
背景技术:
1、染色体水平参考基因组构建是染色体互作、全基因组重测序、染色体进化分化、表观基因组学等研究的基础,可以应用于多个领域,是非常有必要的。
2、hi-c技术(high-throughput/resolution chromosome conformation capture)源于染色体构象捕获(chromosome conformation capture-3c)技术,以整个细胞核为研究对象,利用高通量测序技术,结合生物信息学方法,研究全基因组范围内整个染色体dna的交互关系。hi-c辅助组装即通过捕获染色体dna的交互关系,根据染色体内部互作频率显著高于染色体间互作频率,同时,在同一条染色体上互作频率随着互作距离的增加而减少的原理,将scaffold或者contig聚类到组群,并进一步对组群内的contig/scaffold进行排序及定向,实现趋近于染色体水平的基因组挂载。
3、近年来已发表的高质量动植物基因组,染色体水平的参考基因组占比逐年升高,hi-c在染色体挂载应用中逐渐显现出其优势。
4、目前hi-c辅助组装一般步骤如下:1.数据的统计和过滤;2.hi-c文库评估;3.纠正;4.hi-c辅助组装;5.组装后评估。但是现有hi-c辅助组装的挂载率有待提高,而且需要更成熟的流程进行支撑。
技术实现思路
1、为了解决上述问题,本发明提供了一种利用hi-c技术的基因辅助组装方法,采用本发明的方法,能够将hi-c辅助组装的挂载率提高至95%以上,而且方法更简便、流程更成熟。
2、具体来说,本发明涉及如下方法。
3、1.一种利用hi-c技术的基因辅助组装的方法,包括:
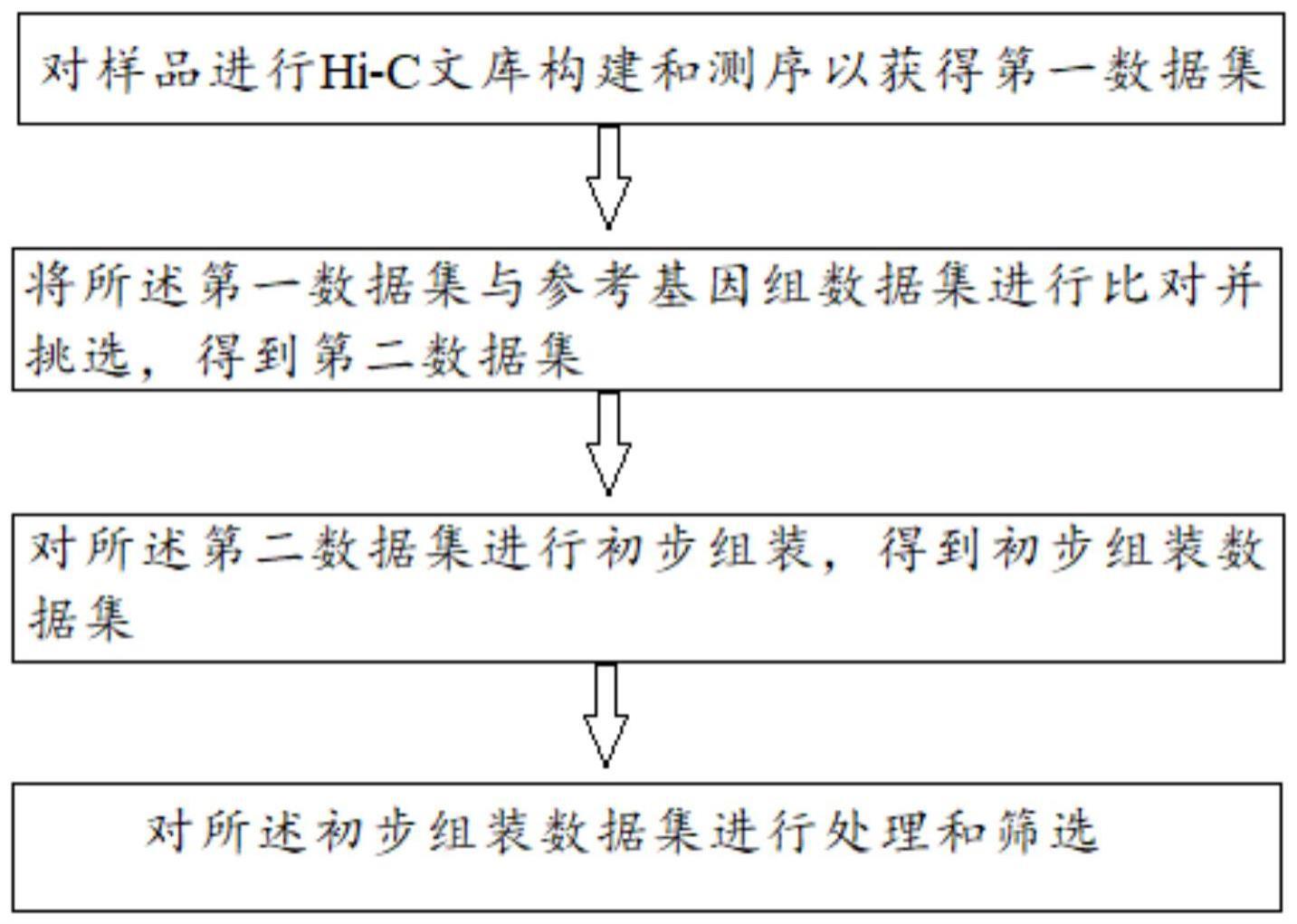
4、对样品进行hi-c文库构建和测序以获得第一数据集;
5、将所述第一数据集与参考基因组数据集进行比对并挑选,得到第二数据集;
6、对所述第二数据集进行初步组装,得到初步组装数据集;
7、对所述初步组装数据集进行处理和筛选;
8、其中,将所述第一数据集与参考基因组数据集进行比对并挑选的方法包括:
9、将第一数据集中的每个数据与参考基因组数据集进行第一比对,得到能比对到参考基因组数据集的第一子集和不能比对到参考基因组数据集的第二子集;
10、将所述第二子集中的每个数据进行酶切之后文库的连接位点搜索,从搜索到的酶切之后文库的连接位点处对第二子集中的数据进行打断,并将打断后的数据再次与参考基因组数据集进行第二比对,得到能够比对到参考基因组数据集上的第三子集;
11、将所述第一子集和第三子集合并、挑选,得到第二数据。
12、2.根据上述的方法,其中,所述数据包括read,和/或,所述数据集包括多个reads。
13、3.根据上述的方法,其中,所述挑选的条件包括:挑选数据的双端均比对到参考基因组数据集唯一位置的数据。
14、4.根据上述的方法,其中,所述初步组装的方法包括:基于细胞核内同一染色体上的互作频率高于不同染色体的互作频率,将第二数据集进行初步组装,得到初步组装的contig/scaffold;
15、优选地,所述初步组装基于lachesis。
16、5.根据上述的方法,其中,对所述初步组装数据集进行处理和筛选的方法包括:
17、将初步组装数据分配到染色体群中;
18、对分配到每一个染色体群中的初步组装数据进行排序、定向和筛选;
19、优选地,对所述初步组装数据集进行处理和筛选基于lachesis;
20、优选地,所述分配的方法包括聚类方法;
21、优选地,所述排序的方法包括:将一个染色体的群组内的contig/scaffold,根据互作关系构建无环的生成树,并从中挑选可信度最高的根部树;
22、优选地,所述定向的方法包括:通过加权的有向无环图遍历contig/scaffold的可能的方向,对于每一个contig/scaffold的顺序判断上,通过对正向的互作关系和反向的互作关系的差异构建一个打分函数并作为可信度判定的依据,并选出可信度最高的定向;
23、优选地,所述筛选的参数选自聚类个数、定向时用到的contig/scaffold的占比、在根部树中的contig/scaffold占比、最短染色体长度占最长染色体长度的比例中的一种或多种。
24、6.根据上述的方法,其中,当筛选出的最优初步组装数据不能作为组装数据输出时,在对第二数据进行初步组装得到初步组装数据中再次改变进行初步组装的条件来重新获得多个初步组装数据,并重新对初步组装数据进行处理和筛选从而筛选出结果最优的初步组装数据作为组装数据。
25、7.根据上述的方法,其中,所述条件选自聚类分析中contig/scaffold序列中的最小酶切位点数、聚类分析中contig/scaffold序列中的最大link深度、聚类回插中contig/scaffold序列与目标cluster互作数同其他cluster互作数比例、排序分析中根部树内contig/scaffold序列中的最小酶切位点数中的一种或多种。
26、8.根据上述的方法,其中,所述参考基因组数据集选自基于二代测序的参考基因草图或基于三代测序的参考基因草图。
27、9.根据上述的利用hi-c技术的基因辅助组装的方法组装得到的染色体水平的基因组。
28、10.上述的利用hi-c技术的基因辅助组装的方法、上述的组装得到的染色体水平的基因组在染色体互作、全基因组重测序、染色体进化分化、表观基因组学中的应用。
技术特征:
1.一种利用hi-c技术的基因辅助组装的方法,包括:
2.根据权利要求1所述的方法,其中,所述数据包括read,和/或,所述数据集包括多个reads。
3.根据权利要求1所述的方法,其中,所述挑选的条件包括:挑选数据的双端均比对到参考基因组数据集唯一位置的数据。
4.根据权利要求1所述的方法,其中,所述初步组装的方法包括:基于细胞核内同一染色体上的互作频率高于不同染色体的互作频率,将第二数据集进行初步组装,得到初步组装的contig/scaffold;
5.根据权利要求1-4中任一项所述的方法,其中,对所述初步组装数据集进行处理和筛选的方法包括:
6.根据权利要求5所述的方法,其中,当筛选出的最优初步组装数据不能作为组装数据输出时,在对第二数据进行初步组装得到初步组装数据中再次改变进行初步组装的条件来重新获得多个初步组装数据,并重新对初步组装数据进行处理和筛选从而筛选出结果最优的初步组装数据作为组装数据。
7.根据权利要求6所述的方法,其中,所述条件选自聚类分析中contig/scaffold序列中的最小酶切位点数、聚类分析中contig/scaffold序列中的最大link深度、聚类回插中contig/scaffold序列与目标cluster互作数同其他cluster互作数比例、排序分析中根部树内contig/scaffold序列中的最小酶切位点数中的一种或多种。
8.根据权利要求1所述的方法,其中,所述参考基因组数据集选自基于二代测序的参考基因草图或基于三代测序的参考基因草图。
9.根据权利要求1-8中任一项所述的利用hi-c技术的基因辅助组装的方法组装得到的染色体水平的基因组。
10.权利要求1-8中任一项所述的利用hi-c技术的基因辅助组装的方法、权利要求9所述的组装得到的染色体水平的基因组在染色体互作、全基因组重测序、染色体进化分化、表观基因组学中的应用。
技术总结
本发明涉及一种利用Hi‑C技术的基因辅助组装方法、染色体水平基因组及应用。组装方法包括:对样品进行Hi‑C文库构建和测序以获得第一数据集;将所述第一数据集与参考基因组数据集进行比对并挑选,得到第二数据集;对所述第二数据集进行初步组装,得到初步组装数据集;对所述初步组装数据集进行处理和筛选。采用本发明的方法,能够将Hi‑C辅助组装的挂载率提高至95%以上。
技术研发人员:任雪,刘涛,李志民,涂成芳,杨伟飞,王娟
受保护的技术使用者:浙江安诺优达生物科技有限公司
技术研发日:
技术公布日:2024/1/14
- 还没有人留言评论。精彩留言会获得点赞!