一种基于分子动力学模拟的多孔炭构建方法
本发明涉及一种基于分子动力学模拟的多孔炭构建方法,尤其涉及一种利用materials studio、gromacs和vmd软件搭建的多孔炭材料,其可用于捕捉或吸附多种物质来研究其吸附特性。
背景技术:
1、多孔炭材料不仅具有碳材料化学稳定性高、导电性好等优点,由于多孔结构的引入,还具有比表面积高、孔结构丰富、孔隙可调节以及易于操作等优点,被广泛应用于吸附领域。而孔道结构是影响多孔炭吸附性能的重要因素,结合不同几何孔隙的优点,通过测量比表面积、孔隙率、孔容,从微观结构出发预测新型多孔炭材料的特性,通过改变几何形状等参数,研究其吸附性能。
2、相比于实验方法,模拟计算能够从微观角度观测到分子之间相互作用的动态变化,可以从分子水平出发阐释作用机制。其中在构建和研究某些微观结构上,分子动力学模拟拥有很大的优势。分子动力学模拟是从分子水平探索物质微观作用本质的有效手段,弥补了现有实验技术中不能够从分子水平上描述多孔炭材料结构特征的不足。而且分子动力学模拟能够通过微观角度分析分子轨迹来考察运动过程,也更好地理解多孔炭材料中的传质和吸附现象,从而制备吸附效果优异的多孔炭材料,为多孔炭材料的设计研发和结构优化提供了理论参考。
技术实现思路
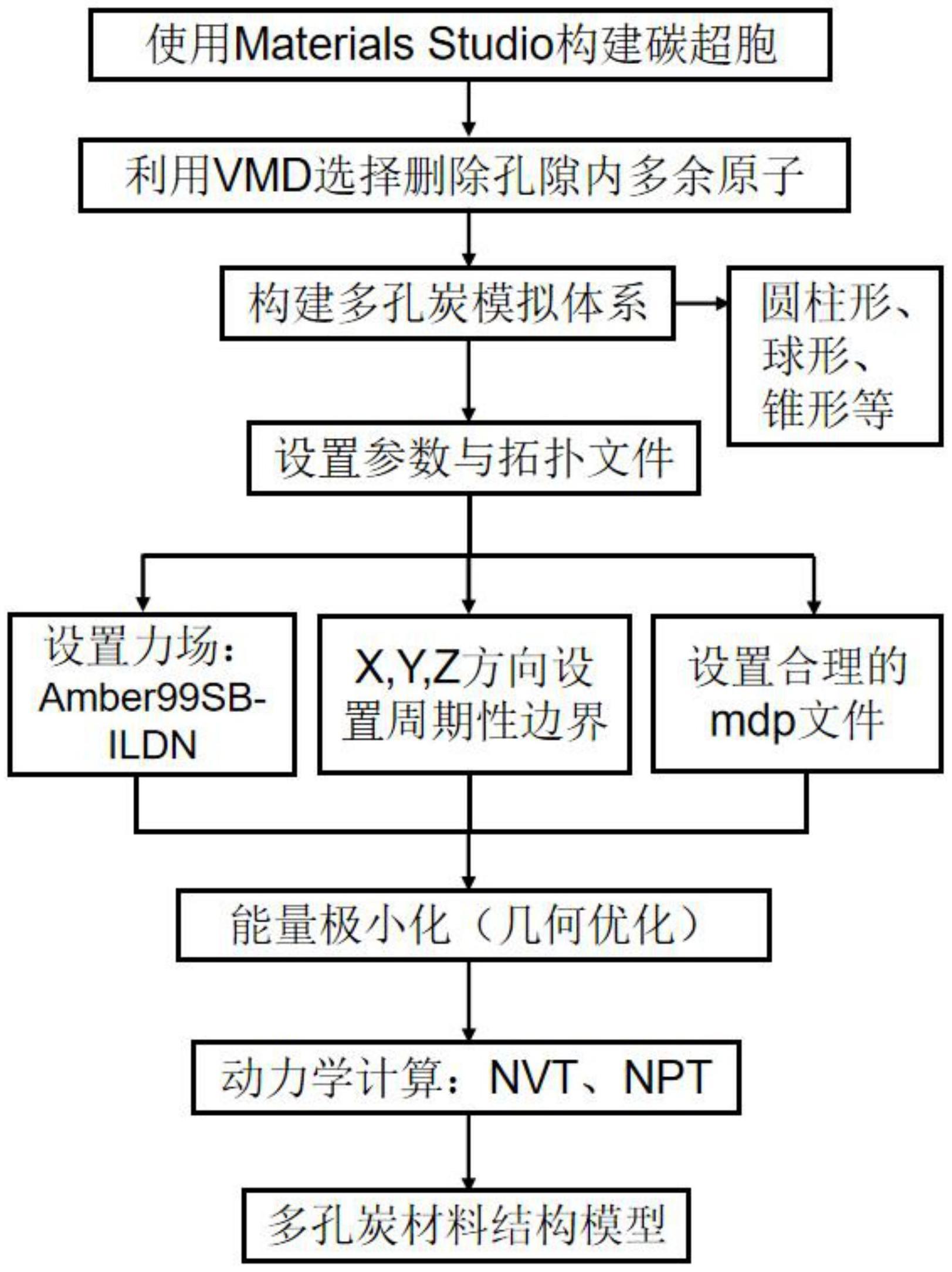
1、本发明的目的在于,提供一种基于分子动力学模拟的多孔炭构建方法。本发明基于分子动力学模拟方法,通过materials studio软件构建一个表面多孔的长方形碳超胞,利用gromacs软件设置适当的力场参数,通过vmd软件包,编写合理的拓扑文件,并进行几何优化,从而可以为构建多孔炭材料的几何结构为其设计与开发提供理论参考。
2、本发明的技术方案:一种基于分子动力学模拟的多孔炭构建方法,包括以下步骤:
3、步骤一:首先采用materials studio软件的visualizer模块平台搭建炭吸附剂骨架模型,导入石墨单胞模型,构建石墨超胞,超胞模型尺寸为6.8768×6.8064×5.3568nm3,导出pdb格式文件;
4、步骤二:将vmd安装包压缩复制到gromacs软件中解压,安装结束后,打开vmd软件中的graphics representations模块,选择selected atoms并删除孔内原子可以得到多孔炭模型。再利用materials studio软件中的“atom volume&surfaces”模块进行计算四种孔隙的比表面积、孔隙率以及孔容。
5、步骤三:将步骤二构建的多孔炭结构模型导出,再利用gromacs软件把得到的炭吸附剂骨架模型使用sobtop脚本对炭材料的结构进行几何优化,设定多孔炭的周期性,在结构文件中设置模拟盒子大小,最终得到不同几何结构的炭吸附剂材料体系模型盒子。
6、所述的分子动力学模拟步骤一包括以下几个方面:
7、步骤1(a)利用materials studio软件,导入石墨晶胞的结构文件。单胞结构单元的尺寸参数为α=β=γ=90°(a、b、c为为晶胞边长,α、β、γ为三维坐标角度)。
8、步骤1(b):选择build→symmetry→supercell,然后在supercell对话框里设置超胞尺度,所选超胞参数为a:28,b:16,c:8。超胞模型尺寸为6.8768×6.8064×5.3568nm3。
9、进一步的,所述步骤二包括以下步骤:
10、步骤2(a):利用gromacs软件,打开vmd软件中graphics representations模块选择selected atoms一栏,通过vmd范围选择语句基本语法选取指定范围,半径取selected atoms一栏输入“resname mol and((x-34.384)^2+(y-34.032)^2>25^2andz<53.568)”,选择删除并保存剩余碳原子,得到半径为高为的圆柱形孔道,以此类推得到圆柱形孔、球形孔、锥形孔等。
11、步骤2(b):再利用materials studio软件中的“atom volume&surfaces”模块,将探针分子的半径设定为等同于氮气的半径,然后选择“solvent surfaces”和“connolly surfaces”分别进行计算四种孔隙的比表面积和孔隙率。孔容计算:选择“connolly surfaces”进行计算得到的超胞自由体积(cell free volume),利用下列方程进行计算得到最终的孔容。
12、
13、进一步的,所述步骤三包括以下步骤:
14、步骤3(a):设定模拟初始参数,使用sobtop脚本对炭材料的结构进行几何优化,同时采用amber99sb-ildn力场对多孔炭进行参数化,在结构文件中设置模拟盒子大小尺寸为6.8768×6.8064×5.3568nm3,设置模拟空间三个方向边界均为周期性边界,最终得到不同几何结构的炭吸附剂材料体系模型盒子。
15、步骤3(b):对由步骤3(a)得到的含有不同几何结构孔隙的炭材料模型体系进行动力学计算,在gromacs软件中使用v-rescale恒温器和berendsen恒压器分别控制系统在298k的温度和1bar的恒定压力下预平衡200ps。随后,使用v-rescale恒温器和parrinello-rahman恒压器分别控制系统在298k的温度和1bar的恒定压力下模拟10ns,得到体系产生相。截断半径为1.2nm,时间步长为2fs,每20ps收集一次轨迹。
16、根据本发明的一种基于分子动力学模拟的多孔炭构建方法,其中,提供了一种构建如上所述的不同几何结构多孔炭模型的方法。
17、根据本发明的一种基于分子动力学模拟的多孔炭构建方法,其中,所述的构建的炭吸附剂模型应用于捕捉或吸附多种物质来研究其吸附特性。
18、与现有技术相比,本发明主要具有下述优点:实现了对不同几何结构多孔炭材料的制备,可以很好的解释在微观运动过程中的贡献。同时,本发明方法便于实现多种物质与具有不同几何结构的多孔炭材料吸附过程的模拟,应用灵活,适用范围广。
19、本发明所提出的利用分子动力学模拟构建多孔炭孔结构模型可以更好地从微观角度观测到分子之间相互作用的动态变化,同时缩短了实验研发周期,大大降低了实验成本,也为多孔炭材料的设计研发和结构优化提供了理论参考。
技术特征:
1.本发明公开了一种基于分子动力学模拟的多孔炭构建方法,主要包括构建模型、几何优化并进行分子动力学模拟三个步骤,其特征在于利用materials studio、vmd构建多孔炭模型,并利用gromacs软件包进行几何优化,最后完成分子动力学模拟,为多孔炭材料的设计研发和结构优化提供了理论参考。
2.根据权利要求1所述的一种基于分子动力学模拟的多孔炭构建方法,其特征在于:利用materials studio、vmd软件建模的版块构建不同几何形状的多孔炭材料,其中,所述不同几何结构的多孔炭包含圆柱形、球形和锥形等。最后利用gromacs软件包进行几何优化,完成分子动力学模拟,得到的多孔炭材料应用于捕捉或吸附多种物质来研究其吸附特性。建模步骤如下:
3.根据权利要求2所述的一种基于分子动力学模拟的多孔炭构建方法,其特征在于:所述的多孔炭材料通过materials studio、vmd构建多孔炭模型,得到实验所需的不同结构的多孔炭材料。
4.根据权利要求2所述的一种基于分子动力学模拟的多孔炭构建方法,其特征在于:所述的多孔炭材料可以通过materials studio软件计算多孔炭的比表面积、孔隙率以及孔容。
5.根据权利要求2所述的一种基于分子动力学模拟的多孔炭构建方法,其特征在于:所述的多孔炭材料是利用gromacs软件编写合理的拓扑文件,使用sobtop脚本对其结构进行几何优化,最终得到拥有吸附能力的多孔炭。
6.根据权利要求2所述的一种基于分子动力学模拟的多孔炭构建方法,其特征在于:制备的含有不同几何结构的多孔炭材料用于实现多种物质与具有不同几何结构的多孔炭材料吸附过程的模拟。
技术总结
本发明公开了一种基于分子动力学模拟的多孔炭构建方法,涉及纳米炭材料技术领域以及分子动力学模拟领域,更具体地说,涉及一种构建不同几结构多孔炭的分子动力学模拟方法。该方法利用Materials Studio构建多孔炭模型,使用VMD软件中Graphics Representations模块,选择SelectedAtoms删除孔内原子得到不同几结构多孔炭,例如圆柱形、球形、锥形等,紧接着利用GROMACS软件包编写合理的拓扑文件,利用Sobtop脚本对其结构进行几何优化,最终得到不同几何结构的炭吸附剂材料体系模型盒子,应用于捕捉或吸附多种物质,并研究其吸附特性。本发明方法便于实现多种物质与具有不同几何结构的多孔炭材料吸附过程的模拟,应用灵活,适用范围广。本发明还涉及利用分子动力学模拟构建具有不同几何结构多孔炭模型的方法,可以快速、大致判断出吸附剂的吸附效果,缩短了实验研发周期,大大降低了实验成本,也为多孔炭材料的设计研发和结构优化提供了理论参考。
技术研发人员:王子璇,徐立恒,魏芳,丁涛,张明
受保护的技术使用者:中国计量大学
技术研发日:
技术公布日:2024/1/15
- 还没有人留言评论。精彩留言会获得点赞!