一种基于矩阵分解算法的微生物-疾病相互作用预测方法
本发明涉及微生物-疾病相互作用预测,具体涉及一种基于矩阵分解算法的微生物-疾病相互作用预测方法。
背景技术:
1、越来越多的研究已经证明,微生物在许多疾病的发展、治疗和预后中发挥着至关重要的作用,包括神经系统疾病和癌症等多种疾病。识别与疾病相关的微生物不仅有助于揭示人类疾病的病理机制,还为疾病的诊断和预后提供了潜在的生物标志物。例如,系统性地识别与疾病病理相关的微生物,可以协助医生和生物学家鉴定在临床或实验中用于诊断和治疗的生物标志物,尤其是对于那些复杂的人类疾病而言。此外,通过计算预测病原微生物,可以帮助药理学家和生物学家更有效地缩小候选化合物的范围,从而进一步指导他们规划实验,降低成本。尽管生物学实验仍然是确认疾病与微生物相关性的黄金标准,但这是一个耗时且成本高昂的过程。因此,迫切需要提出更高效的计算方法,以获得微生物-疾病关联的高概率对,这对于指导生物实验具有至关重要的意义。矩阵分解是一种有效的相互作用预测方法,但目前的模型未能充分提取微生物和疾病的本质特征,并且预测结果也缺乏可解释性。因此,本研究提出了一种基于矩阵分解算法的微生物-疾病相互作用预测方法。
技术实现思路
1、本发明的目的是为了解决上述微生物-疾病相互作用预测领域所存在的困难,提供一种基于矩阵分解算法的微生物-疾病相互作用预测方法,依据目前微生物和疾病的多源数据信息,帮助科研工作者进行高效的预测。本发明的技术方案如下:
2、1、一种基于矩阵分解算法的微生物-疾病相互作用预测方法,包括以下部分:
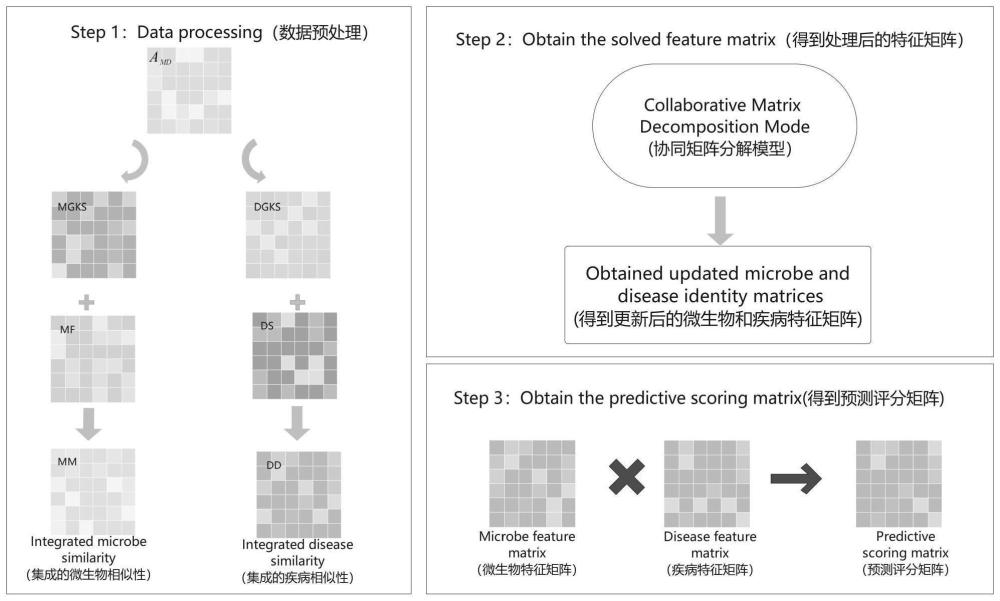
3、a、基于已知的微生物-疾病关联矩阵利用高斯核计算微生物和疾病的高斯相互作用轮廓核相似性。基于微生物的功能相似性和微生物的高斯相互作用轮廓核相似性进行预处理。基于疾病的功能相似性和疾病的高斯相互作用轮廓核相似性进行预处理。
4、b、基于定义的相似性融合策略,对两种微生物相似性进行融合,如果微生物-疾病关联矩阵有关联那么将两种微生物相似性进行求和取平均,否则用微生物的高斯相互作用轮廓核相似性进行替代,对于疾病相似性融合也进行同等操作。
5、c、基于定义的协同矩阵分解模型目标函数,并引入稀疏范数和范围约束项。
6、d、基于交替最小二乘法的迭代算法框架。
7、2、根据权利要求1所述的基于已知的微生物-疾病关联矩阵利用高斯核计算微生物和疾病的高斯相互作用轮廓核相似性。基于微生物的功能相似性和微生物的高斯相互作用轮廓核相似性进行预处理。基于疾病的功能相似性和疾病的高斯相互作用轮廓核相似性进行预处理。本发明基于hmdad,disbiome和combined data数据库进行操作。使用从这三个数据库中下载的已知微生物-疾病关联数据分别计算微生物和疾病的高斯相互作用轮廓核相似性。此外,基于这三个数据库分别收集微生物和疾病的功能相似性。
8、3、根据权利要求1所述的基于定义的相似性融合策略,对两种微生物相似性进行融合,如果微生物-疾病关联矩阵有关联那么将两种微生物相似性进行求和取平均,否则用微生物的高斯相互作用轮廓核进行替代,对于疾病相似性融合也进行同等操作。使用定义的相似性融合策略,减少了使用单一相似度度量相关的潜在偏差,提高了微生物-疾病相互作用预测的精确性。
9、4、根据权利要求1所述的基于定义的协同矩阵分解模型目标函数,并引入稀疏范数和范围约束项。本发明设计了一种改进的协同矩阵分解方法,包含目标矩阵的近似求解项以及正则化项和约束条件。利用目标矩阵的近似求解项得到微生物和疾病的迭代特征矩阵,采用正则化项来避免过拟合。此外,对特征矩阵施加稀疏范数,充分提取了两个特征矩阵的本质特征。此外,将求解的所有矩阵元素值都限制在0-1的范围内,保证数据的有效性和可解释性。
10、5、根据权利要求1所述的基于交替最小二乘法的迭代算法框架。本发明使用基于交替最小二乘法的迭代算法进行优化,交替更新微生物和疾病的特征矩阵,直到收敛。该算法在处理稀疏数据时表现出良好的鲁棒性,并且将矩阵分解成多个小矩阵,每个小矩阵可以独立地进行计算。这使得该算法适用于大规模数据和高性能计算环境。
技术特征:
1.一种基于矩阵分解算法的微生物-疾病相互作用预测方法,包括以下部分:
2.根据权利要求1所述的基于已知的微生物-疾病关联矩阵利用高斯核计算微生物和疾病的高斯相互作用轮廓核相似性。基于微生物的功能相似性和微生物的高斯相互作用轮廓核相似性进行预处理。基于疾病的功能相似性和疾病的高斯相互作用轮廓核相似性进行预处理。本发明基于hmdad,disbiome和combined data数据库进行操作。使用从这三个数据库中下载的已知微生物-疾病关联数据分别计算微生物和疾病的高斯相互作用轮廓核相似性。此外,基于这三个数据库分别收集微生物和疾病的功能相似性。
3.根据权利要求1所述的基于定义的相似性融合策略,对两种微生物相似性进行融合,如果微生物-疾病关联矩阵有关联那么将两种微生物相似性进行求和取平均,否则用微生物的高斯相互作用轮廓核进行替代,对于疾病相似性融合也进行同等操作。使用定义的相似性融合策略,减少了使用单一相似度度量相关的潜在偏差,提高了微生物-疾病相互作用预测的精确性。
4.根据权利要求1所述的基于定义的协同矩阵分解模型目标函数,并引入稀疏范数和范围约束项。本发明设计了一种改进的协同矩阵分解方法,包含目标矩阵的近似求解项以及正则化项和约束条件。利用目标矩阵的近似求解项得到微生物和疾病的迭代特征矩阵,采用正则化项来避免过拟合。此外,对特征矩阵施加稀疏范数,充分提取了两个特征矩阵的本质特征。此外,将求解的所有矩阵元素值都限制在0-1的范围内,保证数据的有效性和可解释性。
5.根据权利要求1所述的基于交替最小二乘法的迭代算法框架。本发明使用基于交替最小二乘法的迭代算法进行优化,交替更新微生物和疾病的特征矩阵,直到收敛。该算法在处理稀疏数据时表现出良好的鲁棒性,并且将矩阵分解成多个小矩阵,每个小矩阵可以独立地进行计算。这使得该算法适用于大规模数据和高性能计算环境。
技术总结
本发明提供了一种基于矩阵分解算法的微生物‑疾病相互作用预测系统,涉及生物信息、深度学习领域。首先,我们基于已知的微生物‑疾病关联矩阵利用高斯核计算出微生物和疾病的高斯相互作用轮廓核相似性并且收集微生物和疾病的功能相似性数据。然后,通过自定义的相似性融合策略,将不同计算方法得到的相似性集成到微生物和疾病的综合相似性中。然后我们构建定义的协同矩阵分解模型目标函数并引入稀疏范数和范围约束项,使用基于交替最小二乘法的迭代算法进行优化,交替更新微生物和疾病特征矩阵,直到收敛。最后,将得到的微生物和疾病特征矩阵相乘得到预测评分矩阵。
技术研发人员:王淑栋,刘体耀,谭效东,范耘滔,范子璇,朱晓冉
受保护的技术使用者:中国石油大学(华东)
技术研发日:
技术公布日:2024/5/6
- 还没有人留言评论。精彩留言会获得点赞!