提高CRISPR介导的同源重组效率的方法与流程
本发明涉及基因编辑技术领域,尤其涉及一种提高CRISPR介导的同源重组效率的方法。
背景技术:
基因组编辑是一种可以对生物体基因组完成精确修饰的一种技术,利用基因组编辑技术可以将细胞内特定的基因进行突变、敲入和删除等,从而改变生物体的遗传特性。当前的基因组编辑技术是利用某些方法在基因组特定的位置上造成DNA损伤,从而激发细胞内的DNA损伤修复机制,细胞内主要修复DNA双链断裂的机制有两条:一条是非同源末端连接途径(英文全称:Non-homologous End Joining,英文简称:NHEJ),在该途径中细胞对DNA断裂处进行部分酶切,使之产生粘性末端,再将两个末端连接起来,这个过程中往往会导致部分碱基丢失,因此使得该相应基因缺乏几个氨基酸或者发生移码突变而被敲除。另一条重要的修复途径是同源重组途径(英文全称:Homologous Recombination,英文简称:HR),若DNA损伤时的同时恰好遇到与该损伤位点同源性很高的另一条完整的DNA时,损伤位点会以同源的DNA为模板进行损伤修复,若同源基因中被人为引入了其它基因、元件或者点突变则可凭此修改细胞基因组。因此以同源重组的方式对基因组DNA进行修复具有更高的可控性,可以完全产生预期的基因组改变,并且能够在适当的位置引入特定的外源基因。例如可以在较为稳定的基因组位置上插入特定基因进行表达,这对于获得高效表达的重组蛋白细胞株尤其重要。然而,HR只发生在细胞分裂的特定时期,并且效率低下,极大的限制了基于HR的基因编辑技术的应用。
NHEJ和HR修复途径存在一定竞争关系,若削弱了NHEJ途径的修复能力必然会导致HR修复水平适当提升。目前已有大量研究揭示NHEJ和HR途径调控中的各个关键环节及其相关基因在其中所起的重要作用,其中Lig4、DNA-PK和XRCC6等都是NHEJ途径中的关键分子。Lig4是一种DNA连接酶,能将DNA双链断裂中的末端直接连接回来,Lig4和XRCC4形成复合物,并且进一步和DNA-PK形成NHEJ途径所必须的复合物。PNA-PK是一种磷脂酰肌醇-3依赖的丝/苏氨酸蛋白激酶,DNA-PK的自磷酸化使其改变构象允许末端加工酶接近DNA损伤末端。XRCC6又名Ku70,其与Ku80形成异二聚体结合DNA双链损伤末端,也是NHEJ修复途径中所必须的。有研究表明抑制Lig4、DNA-PK和XRCC6能有效促进DNA同源重组修复。所以在基因编辑的同时通过shRNA抑制多个NHEJ途径关键分子可能是提高同源重组效率的有效途径。
将外源基因插入哺乳动物细胞特定的位点能有效防止随机整合导致的突变并且能使细胞在多次分裂后仍然能维持外源蛋白高水平的表达。腺相关病毒位点1(AAVS1)位于蛋白磷酸酶1调控亚基12C(PPP1R12C)基因的第一个内含子中,其所处的位置染色体结构较为开放允许外源基因的插入并持久稳定的表达,并且该位点的基因插入对于其邻近的基因表达干扰较小,因此该位点被认为是转基因插入人类基因组中的一个安全位置,是通过植入外源基因进行基因治疗以及构建稳定高表达重组蛋白细胞株的首选插入位点。目前,在人类多能干细胞的AAVS1位点处插入外源基因产生遗传修饰的干细胞可以很好的用于发育生物学、再生医学和基因治疗等领域的研究。因此通过提高HR修复的效率使得在AAVS1位点特异性的插入外源基因具有重要的理论和实用价值。
技术实现要素:
提供一种提高CRISPR介导的同源重组效率的方法,其基于miRNA转录加工机制同时表达shRNA和sgRNA的方法,在基因编辑的同时抑制NHEJ途径。
一方面,通过sgRNA介导位点特异性DNA断裂,从而激活细胞内的DNA双链损伤修复机制以修复受损DNA;另一方面,通过共表达的shRNA抑制NHEJ途径的关键分子,从而使得DNA损伤只能依靠HR途径修复。本发明具体实施方式提供一种编辑报告基因从而检测HR效率的方法和一种在AAVS1位点上通过同源重组高效整合目的基因的方法。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
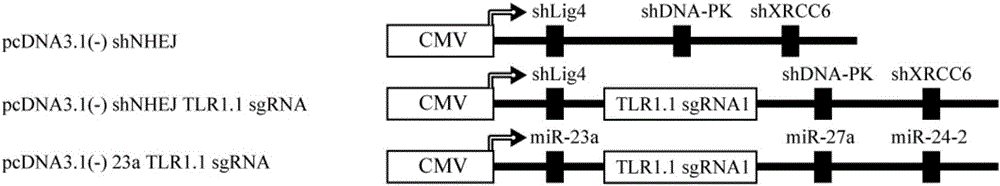
图1为本发明的shNHEJ TLR1.1 sgRNA载体结构图。
图2 shNHRJ TLR1.1 sgRNA载体敲减Lig4、DNA-PK和XRCC6效率的检测。
图3为本发明实施例1中同源重组效率检测结果。其中GFP(左)表示报告载体由HR修复,mCherry(右)表示报告载体由NHEJ修复,同一个样品的左右两个图来自荧光显微镜的同一视野,GFP和mcherry比值可以反应pcDNA3.1(-)-shNHEJ TLR1.1 sgRNA比其它载体更为有效介导同源重组。
图4为pcDNA3.1(-)shNHEJ AAVS1 sgRNA载体结构图;
图5为AAVS1 donor载体(DC-F0215-SH01-10)结构图;
图6为本原理及流程图
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1—shNHEJ TLR1.1 sgRNA载体构建及其介导HR效率检测
shNHEJ TLR1.1 sgRNA载体构建策略如图1所示,具体如下:
shNHEJ sgRNA载体设计;
根据人Lig4、DNA-PK和XRCC6的mRNA序列,分别设计长度在20-24nt之间的shRNA靶位点。shRNA中sense链与靶基因mRNA的序列完全一致,antisense的序列能与sense完全互补配对为宜。Antisense最好进行blast以确保其在全基因组mRNA中没有与其它mRNA有过多的碱基配对,以免有脱靶效应。Sense与antisense之间的loop区域可以自由设计成无配对的区域,但在loop区域含有UGUG模体的加工效率更佳。SgRNA的设计需要考虑靶位点上有相应的PAM位点,我们已SpCas9为例,将含有NGG PAM识别位点前20个碱基作为sgRNA上的引导序列,再这20个碱基后加上SpCas9 sgRNA通用的部分构成完整的sgRNA序列。
本例中ShRNA与sgRNA之间的臂选择miR-23a/27a/24-2clusters上的间隔序列,将sgRNA插入其中,sgRNA靶向pCVL Traffic Light Reporter 1.1(I-Sce target)Ef1a BFP(Plasmid#31481,Addgene)报告载体中的I-SceI酶切位点上的5’-TTATTTGCGTAGGGATAACA GGG-3’序列,其中最后3个GGG为PAM识别位点,仅出现于报告载体中,不存在于sgRNA中。我们构建的sgRNA的DNA序列为:
5’-TTATTTGCGTAGGGATAACATAGAGCTAGAAATAGCAAGTTAAAATAAGG CTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCT-3’。该sgRNA能靶向pCVL Traffic Light Reporter 1.1(I-Sce target)Ef1a BFP报告载体,通过NHEJ途径修复可以使细胞发出红色荧光,通过HR途径修复可以使细胞发出绿色荧光。
shNHRJ sgRNA载体构建外包给公司从头合成,并以XbaI和BamHI克隆至pcDNA3.1(-)中,其序列如下:
AGCCTCTGGCCCCGCCCGGTGCCCCCCTCACCCCTGTGCCACGGCCGGCTAGAGAGAGAATGGCCTATGGAATAGTGAAGCCACAGATGTATTCCATAGGCCATTCTCTCTCTAACCGACCCTGAGCTCTGCCACCGAGGATGCTGCCCGGGGACGGGGTGGCAGAGAGGCCCCGAAGCCTGTGCCTGGCCTGAGGAGCTTGGGTGAAGTTCATCCTAGTTAGTGAAGCCACAGATGTAACTAGGATGAACTTCACCCAACCCCCAGGCCCTCACCTCCTCTGGCCTTGCCGCCTGTCCCCTGCTGCCGCCTGTCTGCCTGCCATCCTGCTGCCTGGCCTCCCTGGGCTCTGCCTCCCGGTGCCAACCTCTTTAGTGATAGTGAAGCCACAGATGTATCACTAAAGAGGTTGGCACGGGTCAAGCCCCCTTGGAGCCTGCAGCCCCTGCCTTCCCTGGGTGGGCTGATGCTTGGA
shNHRJ TLR1.1 sgRNA载体构建外包给公司从头合成,并以XbaI和BamHI克隆至pcDNA3.1(-)中,其序列如下:
AGCCTCTGGCCCCGCCCGGTGCCCCCCTCACCCCTGTGCCACGGCCGGCTAGAGAGAGAATGGCCTATGGAATAGTGAAGCCACAGATGTATTCCATAGGCCATTCTCTCTCTAACCgaccctgagctctgccaccgaggatgctgTTATTTGCGTAGGGATAACAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTcccggggacggggtggcaGAGAGGCCCCGAAGCCTGTGCCTGGCCTGAGGAGCTTGGGTGAAGTTCATCCTAGTTAGTGAAGCCACAGATGTAACTAGGATGAACTTCACCCAACCCCCAGGCCCTCACCTCCTCTGGCCTTGCCGCCTGTCCCCTGCTGCCGCCTGTCTGCCTGCCATCCTGCTGCCTGGCCTCCCTGGGCTCTGCCTCCCGGTGCCAACCTCTTTAGTGATAGTGAAGCCACAGATGTATCACTAAAGAGGTTGGCACGGGTCAAGCCCCCTTGGAGCCTGCAGCCCCTGCCTTCCCTGGGTGGGCTGATGCTTGGA
23a TLR1.1 sgRNA载体构建
23a TLR1.1 sgRNA外包给公司从头合成,并以XbaI和BamHI克隆至pcDNA3.1(-)中,其序列如下:
agcctctggccccgcccggtgcccccctcacccctgtgccacggccggctggggttcctggggatgggatttgcttcctgtcacaaatcacattgccagggatttccaaccgaccctgagctctgccaccgaggatgctgTTATTTGCGTAGGGATAACAGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTcccggggacggggtggcagagaggccccgaagcctgtgcctggcctgaggagcagggcttagctgcttgtgagcagggtccacaccaagtcgtgttcacagtggctaagttccgccccccaggccctcacctcctctggccttgccgcctgtcccctgctgccgcctgtctgcctgccatcctgctgcctggcctccctgggctctgcctcccgtgcctactgagctgaaacacagttggtttgtgtacactggctcagttcagcaggaacaggggtcaagcccccttggagcctgcagcccctgccttccctgggtgggctgatgcttgga
miRNA-based sgRNA载体转染和表达检测
miRNA-based sgRNA载体可以用通用的转染方法导入细胞中表达。本实施例用转染试剂PEI将其与SpCas9表达载体lentiCRISPR(Plasmid#49535,Addgene)共转293T细胞检测其knockdown、HR和NHEJ的效率。具体操作为6孔板细胞,每个孔转染1.5μg shNHEJ TLR1.1 sgRNA或miR-23a TLR1.1 sgRNA以转染pCDH或者pCDH-23a cluster为阴性对照,同时每孔再加入1.5μg lentiCRISPR和1μg pCVL Traffic Light Reporter 1.1(I-Sce target)Ef1a BFP报告载体进行总4μg进行共转实验。每孔加opti-medium 200μl和12μl PEI转染试剂。
本实施例用定量PCR检测Lig4、DNA-PK和XRCC6的knockdown效果。将shNHRJ TLR1.1 sgRNA、SpCas9表达载体、donor载体和TLR1.1报告载体共转293T细胞,转染后2天Trizol裂解细胞并提取RNA,随机引物反转录总RNA,接着用跨内含子的特异性引物进行PCR检测。通过定量PCR检测我们发现Lig4、DNA-PK和XRCC6能有效的被shNHEJ和shNHRJ抑制(图2)。
NHEJ和HR的效率通过报告基因的方法进行检测,将shNHRJ TLR1.1sgRNA、SpCas9表达载体、donor载体和TLR1.1报告载体共转293T细胞,发生位点特异性NHEJ的细胞能发出红色荧光,而发生位点特异性HR的细胞发出绿光。我们发现我们设计的23a TLR1.1 sgRNA对照载体可以使部分细胞发出红色荧光和绿色阳光,因此意味着该载体可以表达有活性的sgRNA并由NHEJ和HR途径共同修复损伤的DNA(图3)。
实施例2—shNHEJ AAVS1 sgRNA载体构建
重组蛋白药物表达需要构建稳定高表达的细胞株,已知整合到人类基因组某些位点的基因表达效率较高且较为稳定,不易在细胞多次传代的过程中发生缺失、沉默等现象,因此将外源基因特异性整合到这些位点有助于提高重组蛋白稳定高表达细胞株的构建效率。其中AAVS1位点我们拟采用我们的策略实现这一高效的位点特异性整合。具体如下:
设计靶向AAVS1位点的sgRNA:GGGGCCACTAGGGACAGGATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCT
将AAVS1 sgRNA替换实施例1中pcDNA3.1(-)shNHEJ TLR1.1 sgRNA中的TLR1.1 sgRNA(图4)
合成如下shNHEJ-AAVS1 sgRNA序列,并以XbaI和BamHI位点克隆至pcDNA3.1(-)载体中。
AGCCTCTGGCCCCGCCCGGTGCCCCCCTCACCCCTGTGCCACGGCCGGCTAGAGAGAGAATGGCCTATGGAATAGTGAAGCCACAGATGTATTCCATAGGCCATTCTCTCTCTAACCgaccctgagctctgccaccgaggatgctgGGGGCCACTAGGGACAGGATGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTcccggggacggggtggcaGAGAGGCCCCGAAGCCTGTGCCTGGCCTGAGGAGCTTGGGTGAAGTTCATCCTAGTTAGTGAAGCCACAGATGTAACTAGGATGAACTTCACCCAACCCCCAGGCCCTCACCTCCTCTGGCCTTGCCGCCTGTCCCCTGCTGCCGCCTGTCTGCCTGCCATCCTGCTGCCTGGCCTCCCTGGGCTCTGCCTCCCGGTGCCAACCTCTTTAGTGATAGTGAAGCCACAGATGTATCACTAAAGAGGTTGGCACGGGTCAAGCCCCCTTGGAGCCTGCAGCCCCTGCCTTCCCTGGGTGGGCTGATGCTTGGA
将上述pcDNA3.1(-)-shNHEJ AAVS1 sgRNA载体1μg与Cas9表达载体1μg以及AAVS1donor载体(DC-F0215-SH01-10,见图5,购买至GeneCopoeia公司)1μg用200μl opti-medium稀释后加9μl PEI转染试剂混匀静止25min后共转染6孔板293T细胞。其中AAVS1donor载体中含有所需要过表达的重组蛋白编码基因。
本领域普通技术人员可知,本发明的技术方案在下述范围内变化时,仍然能够得到与上述实施例相同或相近的技术效果,仍属于本发明的保护范围:
一种提高CRISPR介导的同源重组效率的方法,包括如下步骤:
(1)设计靶向Lig4、DNA-PK和XRCC6的shRNA;
(2)将shRNA的基因序列顺序排列,并sgRNA的基因序列连接于二shRNA之间,形成shRNA-sgRNA多顺反子;
(3)将shRNA sgRNA多顺反子置于RNA聚合酶II或RNA聚合酶III启动子下游,并置于载体上导入人细胞系中表达pri-shRNA-sgRNA;
(4)上述pri-shRNA-sgRNA在细胞中被Dicer和Drosha加工成pre-shRNA-sgRNA,在此过程中,sgRNA和pre-shRNA分别释放出来,其中sgRNA形成sgRNA-Cas9复合物,进而识别相应的靶位点,对所述需要进行编辑的基因进行编辑;pre-shRNA则抑制Lig4、DNA-PK和XRCC6从而抑制NHEJ修复途径。
所述shRNA的正义链和反义链之间具有loop区域。
所述RNA聚合酶II启动子为CMV启动子。
参阅图6,图6为本发明提供的提高CRISPR介导的同源重组效率的方法,该方法如图6所示,包括如下步骤:
步骤S601、设计靶向Lig4、DNA-PK和XRCC6等促进非同源末端连接(NHEJ)途径的基因的shRNA,并将其以miRNA的形式串联成多顺反子表达;
步骤S602、根据目的,在需要进行同源重组(HR)编辑的目的基因上选取合适的sgRNA靶位点;
步骤S603、将上述sgRNA与shRNA融合构成shRNA-sgRNA多顺反子,将多顺反子置于RNA聚合酶II或RNA聚合酶III启动子下游构建表达载体;
步骤S604、将sgRNA两侧约500bp(设定范围)作为重组臂,中间插入目的基因,其中,重组臂序列和细胞基因组上特定位置的序列相同用于引发与细胞基因组之间的同源重组,插入基因可以是插入位置处原本的基因的修饰也可以是需要外源表达的新的基因;
步骤S605、将上述载体与Cas9表达载体以及含重组臂的donor载体共转染动物细胞,以通过转录形成pri-shRNA-sgRNA;
步骤S606、上述pri-shRNA-sgRNA在动物细胞中被Drosha和Dicer加工,在此过程中,sgRNA和pre-shRNA分别释放出来,其中sgRNA的部分与Cas9装配形成位点特异性核酸内切酶,进而识别相应的靶位点,对相应的基因进行编辑;pre-shRNA则被加工进入RISC复合物成为成熟的shRNA,以抑制NHEJ途径的活性。
上述Lig4、DNA-PK和XRCC6的shRNA和特定的sgRNA可以利用miRNA的表达加工机制同时表达抑制Lig4、DNA-PK和XRCC6的shRNA和特点的sgRNA。
在CRISPR基因编辑的同时抑制了NHEJ途径使得更多的断裂的DNA采取HR修复的方式进行修复。HR修复的产物完全是已知的不会像NHEJ途径那样产生难以预计的DNA缺失,因此基因编辑的结果更加可控;HR修复效率的提高有助于增强外源基因的位点特异性整合,从而提高转基因动物以及高表达重组蛋白的细胞株制备的效率。
以上所述,仅为本发明的较佳实施例而已,故不能依此限定本发明实施的范围,即依本发明专利范围及说明书内容所作的等效变化与修饰,皆应仍属本发明涵盖的范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种利用ZFN在哺乳动物细胞中实现多个大片段DNA定点整合的方法与流程
- DNA片段与含有该片段的重组载体、构建重组载体的方法及其应用与流程
- 一种FcγRIIb基因敲除的MDSC及其作为抗肿瘤药物的应用的制造方法与工艺
- 一种药食同源组合物及其应用、植物饮料及其制备方法与流程
- 一种消除PCR反应试剂自带的内源核酸污染的方法与制造工艺
- 一种快速体内验证基因打靶靶点效率的方法与制造工艺
- 用CRISPR/Cas9技术易于筛选获得目的基因敲除细胞系的方法及产品与制造工艺
- 产甘草次酸的重组酿酒酵母、其构建方法以及用途与制造工艺
- 一种用于检测肾癌的引物组及检测方法与制造工艺
- 检测植原体的LAMP引物组及其试剂盒和应用的制造方法与工艺