一种利用级联放大效应高敏监测水体PAHs的转荧光蛋白基因斑马鱼的研制方法与流程
本发明属于生物技术领域,具体涉及一种利用级联放大效应高敏监测水体PAHs污染物的转荧光蛋白基因斑马鱼的研制方法。
背景技术:
现阶段环境污染尤其是水污染在世界各地普遍存在。水质的污染不仅会影响水生生物的生存与繁殖,而且污染物会通过生物富集浓缩作用到达食物链的终端,严重影响人类健康。近年来,人们在环境中频繁地检测出多环芳烃等有机污染物,研究表明这类污染物已对水环境、土壤及沉积物中生物的生存造成了严重威胁。其中环境水体中的有机污染物以芳香烃类污染物为主。传统检测芳香烃类污染物毒性效应的方法一般分为体外测试(In vitro test)和体内测试(In vivo test)。但是体外测试不能体现对整个生物体的影响,体内测试又具有操作繁琐、实验昂贵且对低浓度暴露的反应不稳定等问题,因此,目前研究中较倾向于利用组合分层式检测方法(Tiered test)测试芳香烃类污染物的毒性效应。但是该方法仍耗时、耗力且需要专业技术人员完成,而且不能用于现场直观监测。
斑马鱼以其独特性可作为理想的环境监测模式生物。现阶段,已有部分国家使用野生型斑马鱼对水体环境进行监测,转基因鱼监测该类污染物的技术也随之产生。2001年Mattingly等首次证实斑马鱼的转录因子可以识别人的AhREs,在TCDD的诱导下可驱动GFP的表达,但是他们仅研究了芳香烃受体及其下游基因的功能,并没有将其应用于活体监测芳香烃类污染物。Payne观察到在石油污染的海水中,可诱导青鲈的CYP1A1活性出现变化,并通过检测苯并芘羟化酶的活性诱导效果监控海上的石油污染。此后,Nebert等利用包含AhREs的启动子驱动LUC报告基因的表达,研制出转基因斑马鱼来检测水体中芳香烃族有机污染物,但他们研制的转基因斑马鱼中LUC报告基因不能传至F2代。
技术实现要素:
为了克服现有技术中所存在的问题,本发明的目的在于提供一种利用级联放大效应高敏监测水体PAHs污染物的转荧光蛋白基因斑马鱼的研制方法。
为了实现上述目的以及其他相关目的,本发明采用如下技术方案:
本发明的第一方面,提供一种监测水体环境污染物的转荧光蛋白基因鱼的培养方法,包括步骤:
(1)提供水体环境污染物响应载体,所述水体环境污染物响应载体带有I-SceI大范围核酸酶转移系统,所述水体环境污染物响应载体包括:水体环境污染物响应元件、转录激活Gal4蛋白的基因序列、UAS序列和荧光蛋白序列;
(2)提供荧光信号放大载体,包括步骤:在步骤(1)所述水体环境污染物响应载体的荧光蛋白序列之后克隆入转录激活因子TALE-VP64编码序列,获得荧光信号放大载体;
(3)将水体环境污染物响应载体和荧光信号放大载体与I-SceI酶共同注射入鱼的受精卵,培养,获得监测水体环境污染物的转荧光蛋白基因鱼。
优选地,所述鱼为斑马鱼,但是在其他鱼类也可适用。
优选地,所述水体环境污染物响应元件为多环芳香烃响应元件,本方法但同样也适用于雌激素反应元件EREs、金属反应原件MREs和亲电性响应元素EpREs等。所述多环芳香烃响应元件含有如SEQ ID NO.16所示的序列。
优选地,所述荧光蛋白为eGFP,但同样也适用其他荧光蛋白如RFP、CFP、YFP和mCherry等。
本发明一优选实施例中,所述水体环境污染物响应载体为多环芳香烃响应载体pI-SceI/3*AhREs-Gal4/UAS-eGFP,所述多环芳香烃响应载体含有如SEQ ID NO.14所示的序列。
优选地,所述转录激活因子TALE-VP64含有如SEQ ID NO.17所示的序列。
在本发明一优选实施例中,所述荧光信号放大载体为pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF,所述荧光信号放大载体含有如SEQ ID NO.15所所示的序列。
优选地,所述培养还包括纯系转荧光蛋白基因鱼的筛选,包括步骤:将注射后的转荧光蛋白基因鱼P0培育至性成熟,然后与野生型鱼杂交,收集鱼卵,TCDD暴露,筛选出可表达荧光蛋白的转荧光蛋白基因鱼F1代;将所述转荧光蛋白基因鱼F1代再与野生型斑马鱼进行杂交,收集鱼卵,TCDD暴露,筛选出可表达荧光蛋白的转荧光蛋白基因鱼杂合体F2代;然后将杂合体F2代雌雄鱼进行自交,筛选出其中转荧光蛋白基因鱼F3代中的纯合体,即为纯系转荧光蛋白基因鱼。
进一步优选地,筛选转荧光蛋白基因鱼F3代中的纯合体,包括步骤:将转荧光蛋白基因鱼F3代继续与野生型鱼杂交,如果后代胚胎发绿色荧光比例为100%,则F4代的亲本F3为纯系 转荧光蛋白基因鱼。
本发明的第二方面,提供了一种由前述方法培养获得的转荧光蛋白基因鱼。
优选地,所述转荧光蛋白基因鱼为转荧光蛋白基因斑马鱼。
所述转荧光蛋白基因鱼能够稳定遗传且具有荧光强度放大功能,能够高敏监测水环境污染物。
本发明的第三方面,提供了前述转荧光蛋白基因鱼用于检测水环境污染物的用途。
本发明的第四方面,提供一种载体组合,包括:水体环境污染物响应载体和荧光信号放大载体,所述水体环境污染物响应载体带有I-SceI大范围核酸酶转移系统,所述水体环境污染物响应载体包括:水体环境污染物响应元件、转录激活Gal4蛋白的基因序列、UAS序列和荧光蛋白序列;所述荧光信号载体为在所述水体环境污染物响应载体的荧光蛋白序列之后克隆入转录激活因子TALE-VP64编码序列获得。
本发明的第五方面,提供了前述载体组合用于培养转荧光蛋白基因鱼的用途。
与现有技术相比,本发明具有如下有益效果:
本发明公开了一种对水体持久性有机污染物多环芳烃(PAHs)高度敏感的转荧光蛋白基因斑马鱼的制备方法。针对目前已有环境监测转荧光蛋白基因斑马鱼存在可监测浓度较低、荧光强度不足的缺陷,本发明提供了一种荧光信号放大系统,研制出对水体中多环芳烃类(PAHs)有机污染物具有更高敏感度的监测斑马鱼。
本发明主要是利用UAS-Gal4基因表达调控系统为基础,用优化重组的多环芳香烃响应元件AhREs替换上游质粒Gal4中的启动子,达到响应水体环境中多环芳香烃,产生的Gal4蛋白结合下游质粒中UAS序列,达到激活荧光蛋白基因eGFP的目的,此为一个2级信号放大系统。同时,我们利用TALE-TF转录激活增强技术,进一步对eGFP基因的转录进行增强,最终达到多重信号放大效应。在利用I-SceI大范围核酸酶转移系统,获得2个品系的转基因斑马鱼后,通过不断筛选最终获得能够稳定遗传且具有荧光强度放大功能的高敏型多环芳香烃类污染物监测荧光斑马鱼。
附图说明
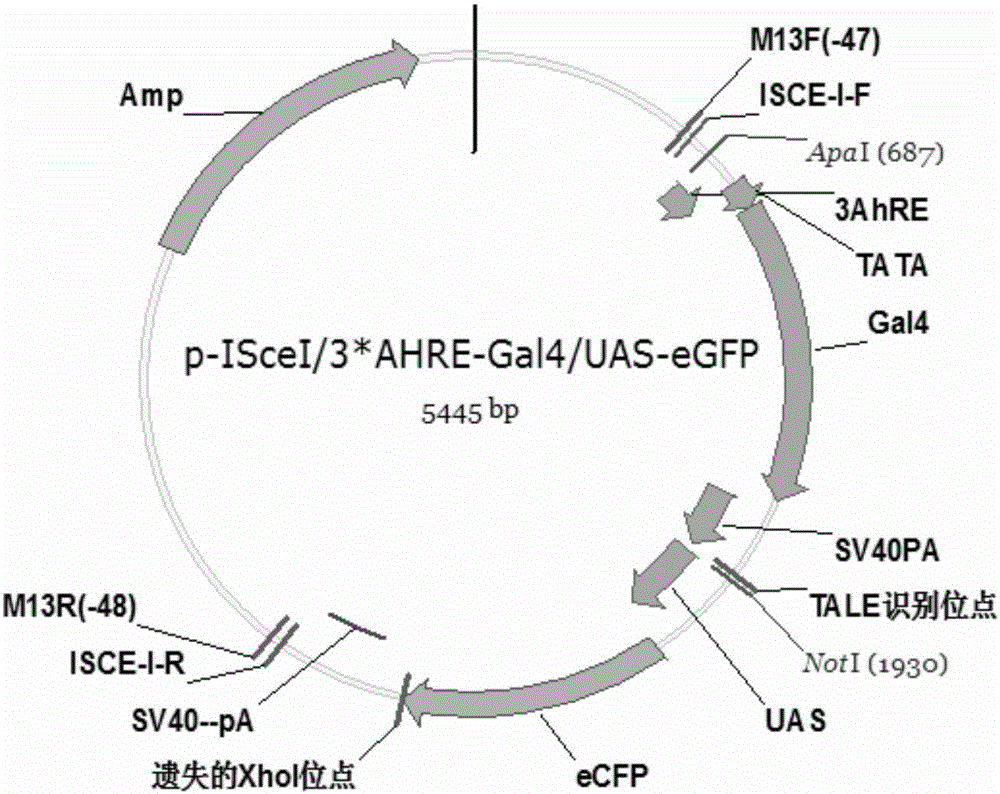
图1:本发明构建的pI-SceI/3*AhREs-Gal4/UAS-eGFP载体示意图。
图2:本发明构建的pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF荧光信号放大载体示意图。
图3:经筛选所得F3代纯合子,通过不同浓度梯度TCDD诱导荧光表达的试验结果。
图4:通过TALE-TF放大的转荧光蛋白基因鱼在TCDD暴露下荧光强度的结果图。
具体实施方式
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
除非另外说明,本发明中所公开的实验方法、检测方法、制备方法均采用本技术领域常规的分子生物学、生物化学、染色质结构和分析、分析化学、细胞培养、重组DNA技术及相关领域的常规技术。这些技术在现有文献中已有完善说明,具体可参见Sambrook等MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989and Third edition,2001;Ausubel等,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987and periodic updates;the series METHODS IN ENZYMOLOGY,Academic Press,San Diego;Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998;METHODS IN ENZYMOLOGY,Vol.304,Chromatin(P.M.Wassarman and A.P.Wolffe,eds.),Academic Press,San Diego,1999;和METHODS IN MOLECULAR BIOLOGY,Vol.119,Chromatin Protocols(P.B.Becker,ed.)Humana Press,Totowa,1999等。
实施例1
一、构建具有响应水体环境中多环芳香烃的GAL4-UAS上游响应载体
1.多环芳香烃响应元件AhREs的合成:
选取AhREs序列的一个突变体:TCCGGTCCTTCTCACGCAACGCCTGGG(SEQ ID NO.1)为基本元件。
人工合成3拷贝数同向串联的,包含相应酶切位点与下游部分TATA-box序列的基因片段NotI-NheI-AhREs-SpeI-M.EcoP15I-NcoI-TATA+-PstI:
5’-GCGGCCGCGCTAGCTCCGGTCCTTCTCACGCAACGCCTGGGTCCGGTCCT TCTCACGCAACGCCTGGGTCCGGTCCTTCTCACGCAACGCCTGGGACTAGTCAGCAGCCATGGTTCGCATATTAAGGTGACGCGTGTGGCCTCGAACACCGAGCGACCCTGCAG-3’(SEQ ID NO.2)(下划线部分为酶切位点,粗体为3倍串联的AhREs突变体序列,斜体为AhREs下游TATA-box的部分序列)。
将人工合成片段NotI-NheI-AhREs-SpeI-EcoP15I-NcoI-TATA+-PstI导入质粒pJWR,构成质粒pJER-A。
11.后续元件GAL4-VP16的设计:
以实验室已有质粒pM3VP16为模板,PCR扩增PstI-TATA--GAL4-VP16-XbaI序列。总序列大小854bp。
具体操作步骤如下:
PCR扩增质粒pM3VP16的GAL4-VP16序列,前引物P1带有PstI酶切位点和AhREs下游TATA-box的剩余部分序列(斜体),P1引物的序列为:PstI-TATA--G+:5’-CTGCAGCGACCCGCTTAAAAGATCCAGCCACCATGAAGCTACTGTCTTCTATCGAACA A-3’(SEQ ID NO.3);后引物P2带有XbaI酶切位点,P2引物的序列为:XbaI-VP16-:5’-TCTAGAACCAAATAATCAGACGACAACCGC-3’(SEQ ID NO.4)。PCR扩增TATA--GAL4-VP16片段:PCR程序为95℃5min;95℃30s,58℃30s,72℃1min共30个循环;72℃10min。PCR产物经1%琼脂糖凝胶电泳检测正确后,用PCR产物纯化试剂盒纯化产物,除去PCR反应体系中的盐离子、酶及引物等杂物。纯化后产物用pMDTM19-T Vector Cloning Kit试剂盒进行TA克隆,反应体系为10μL,其中SolutionI 5μL,PMD19-T vector 0.5μL,TATA--GAL4-VP16片段4.5μL,连接产物16℃连接过夜,后续转化与阳性筛选过程同上。收集阳性菌液并根据质粒提取试剂盒说明书提取质粒,将得到质粒测序验证,测序结果与预期一致,此中间载体命名为pTATA--GAL4-VP16-TA(SEQ ID NO.5)。
以PstI和XbaI双酶切质粒pJER-A和pTATA--GAL4-VP16-TA。反应体系为20μL,其中PstI和XbaI内切酶各1μL,反应缓冲液2μL,DNA模板3μL,无菌水13μL,37℃保温3h。酶切完成后,将所有酶切产物进行琼脂糖凝胶电泳,割取pTATA--GAL4-VP16-TA酶切产物约850bp的片段作为插入片段,割取pJER-A酶切产物中约2900bp片段作为骨架。经凝胶回收试剂盒回收纯化后,用T4 DNA连接酶4℃连接过夜,连接体系为插入片段10μL,骨架0.5μL,缓冲液2μL,T4连接酶1μL,灭菌水6.5μL。后续转化及验证方法同TA克隆,将阳性克隆扩大培养于6mL LB液体培养基,37℃250rpm培养过夜。收集菌液 并根据质粒提取试剂盒说明书提取质粒,将得到质粒测序验证,测序结果与预期一致,此载体即为包含AhREs-TATAbox-GAL4-VP16基因序列载体pAhREs-TATAbox-GAL4-VP16。
以NotI和XbaI双酶切质粒pAhREs-TATAbox-GAL4-VP16和实验室已有质粒pISCE-I-zHSP70-mir30。酶切体系及条件同上。酶切完成后,将所有酶切产物进行琼脂糖凝胶电泳,割取前者产物约1000bp的片段作为插入片段,割取后者约3100bp片段作为骨架。后续连接、转化及验证方法同上,将阳性克隆扩大培养于6mL LB液体培养基,37℃250rpm培养过夜。收集菌液并根据质粒提取试剂盒说明书提取质粒,将得到质粒测序验证,测序结果与预期一致,此载体即为能够响应水体环境中多环芳香烃,并产生GAL4蛋白的质粒pI-SceI/3*AhREs-Gal4(SEQ ID NO.7)。
二、构建具有放大作用的带eGFP荧光蛋白的GAL4-UAS下游显色载体
首先对实验室已有载体pG5-Luc设计引物,上下游引物分别为:
P1-M13-48-NotI:5’-GCGGCCGCAGCGGATAACAATTTCACACAGGA-3’(SEQ ID NO.8);
P2-NcBg-G5UP-A-AgeI:5’-ACCGGTTTTAGCTTCCTTAGCTCCTG-3’(SEQ ID NO.9);
以载体pG5-Luc为模板,PCR扩增UAS+PE1B+TATA片段。PCR程序为95℃5min;95℃30s,56℃30s,72℃30s共30个循环;72℃10min。PCR产物经1%琼脂糖凝胶电泳检测正确后,用PCR产物纯化试剂盒纯化产物,除去PCR反应体系中的盐离子、酶及引物等杂物。纯化后产物用pMDTM19-T Vector Cloning Kit试剂盒进行TA克隆,反应体系为10μL,其中SolutionI 5μL,PMD19-T vector 0.5μL,UAS+PE1B+TATA片段4.5μL,连接产物16℃连接过夜,连接产物10μL转化90μL大肠杆菌感受态细胞DH5α,用移液器轻轻吸打混匀,冰上孵育30min后,42℃热激90s,立即置于冰上2min,加入37℃LB培养基900μL,于37℃180rpm摇床活化1h,活化后菌液5000rpm离心3min富集,吸掉900上清液,100μL菌液混匀后涂于氨苄抗性培养皿上并于37℃温箱中倒置培养过夜。随机挑选10个单菌落利用PCR方法进行检测,将阳性克隆扩大培养于6mL LB液体培养基,37℃250rpm培养过夜。收集菌液并根据质粒提取试剂盒说明书提取质粒,将得到质粒测序验证,测序结果与预期一致,此中间载体命名为pUAS+PE1B+TATA-TA(SEQ ID NO.10)。
以NotI和AgeI双酶切质粒pUAS+PE1B+TATA-TA。反应体系为20μL,其中NotI和AgeI内切酶各1μL,反应缓冲液2μL,DNA模板3μL,无菌水13μL,37℃保温3h。实验室保存质粒pISCE-I-zHSP70-mir30同时进行双酶切,体系如上。酶切完成后,将所有酶切产物进行琼脂糖凝胶电泳,前者割取约280bp的片段作为插入片段,割取质粒pI-SceI-zHSP70-mir30中约4000bp片段作为骨架。经凝胶回收试剂盒回收纯化后,用T4 DNA连接酶4℃连接过夜,连接体系为插入片段10μL,骨架0.5μL,缓冲液2μL,T4连接酶1μL,灭菌水6.5μL。后续转化及验证方法同TA克隆,将阳性克隆扩大培养于6mL LB液体培养基,37℃250rpm培养过夜。收集菌液并根据质粒提取试剂盒说明书提取质粒,将得到质粒测序验证,测序结果与预期一致,此载体即为蓝绿色荧光蛋白载体pI-SceI/UAS-eGFP(SEQ ID NO.11)。
三、构建具有TALE-TF放大eGFP荧光蛋白作用的GAL4-UAS下游显色载体
最后结合TALE-TF技术。在选取pI-SceI/UAS-eGFP质粒上(T)AATACGACTCACTAT(SEQ ID NO.12)作为识别靶序列,人工设计合成NLS-TALE-TF-VP64序列上游,添加2A Peptide序列。最终在此合成T2A-NLS-TALE-TF-VP64序列两端添加BglII和SpeI酶切位点,构建成z质粒pBglII-T2A-NLS-TALE-TF-VP64-SpeI。
以BglII和SpeI双酶切质粒pBglII-T2A-NLS-TALE-TF-VP64-SpeI和pI-SceI-CMV-eGFP。反应体系为20μL,其中BglII和SpeI双内切酶各1μL,反应缓冲液2μL,DNA模板3μL,无菌水13μL,37℃保温3h。酶切完成后,将所有酶切产物进行琼脂糖凝胶电泳,前者割取约2400bp的片段作为插入片段,割取质粒pI-SceI-CMV-eGFP中约4500bp片段作为骨架。经凝胶回收试剂盒回收纯化后,用T4 DNA连接酶4℃连接过夜,连接及后续转化验证方法同上,将阳性克隆扩大培养于6mL LB液体培养基,37℃250rpm培养过夜。收集菌液并根据质粒提取试剂盒说明书提取质粒。将得到质粒继续用AgeI和SpeI双酶切,琼脂糖凝胶电泳后,割取约3200bp的片段作为插入片段。同时AgeI和SpeI双酶切红色荧光蛋白载体pI-SceI/UAS-eGFP,割取其中3500bp片段作为骨架。片段与骨架用T4 DNA连接酶4℃连接过夜,连接体系及后续检测同上。测序结果与预期一致,此载体即为最终具有放大效应的红色荧光显色载体pI-SceI/UAS-eGFP-TALE-TF(SEQ ID NO.13)。
四、构建PAHs诱导型荧光蛋白表达重组转座载体为pI-SceI/3*AhREs-Gal4/UAS-eGFP和pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF
在上述步骤的基础上,利用已经构建好的pI-SceI/3*AhREs-Gal4、pI-SceI/UAS-eGFP和pI-SceI/UAS-eGFP-TALE-TF基础上,通过ApaI和NotI双酶切上述3个质粒,T4 DNA连接酶4℃连接,构建pI-SceI/3*AhREs-Gal4/UAS-eGFP(示意图如图1所示)
和pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF(示意图如图2所示)。
所述pI-SceI/3*AhREs-Gal4/UAS-eGFP含有如SEQ ID NO.14所示的序列。所述pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF含有如SEQ ID NO.15所示的序列。
五、鱼卵显微注射
利用显微注射仪和参照显微注射法,分别将上述实施例中构建好的载体pI-SceI/3*AhREs-Gal4/UAS-eGFP和pI-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF注射斑马鱼单细胞受精卵。注射液体系为10μL,其中质粒30ng/μL,I-SceI缓冲液0.5×,I-SceI酶0.3U/μL,补水至10μL。取2μL注射液注入毛细针管中,将其装入注射仪中,按顺序注射受精卵动物极。整个操作过程在45min内完成,以确保在细胞第一次卵裂之前注射完成。注射后的受精卵培养于28℃,培养至性成熟后。
六、筛选稳定表达的纯合子亲本
筛选稳定遗传纯系转基因斑马鱼的方法,首先将注射后的转基因鱼P0培育至性成熟,与野生型斑马鱼杂交,收集鱼卵,低浓度TCDD暴露,筛选出可表达荧光蛋白的转基因斑马鱼F1代。将F1代转基因斑马鱼再与野生型斑马鱼进行杂交,同样运用低浓度TCDD暴露筛选的方法获得杂合体F2代。将这些发荧光的杂合体F2代雌雄鱼进行自交,其后代F3中,有1/4概率得到Tg(I-SceI/3*AhREs-Gal4/UAS-eGFP)和Tg(I-SceI/3*AhREs-Gal4/UAS-eGFP-TALE-TF)的纯合体F3。为验证纯合子F3,将转荧光蛋白基因鱼F3代继续与野生型鱼杂交,如果后代胚胎发绿色荧光比例为100%,则可以将亲本F3作为纯系来养殖。
如图4所示,通过荧光强度测定,通过TALE-TF放大的转荧光蛋白基因鱼,在相同TCDD暴露浓度下,其荧光强度是没有放大的16~200倍,尤其是在较低浓度下,其放大效应更敏感。
以上所述,仅为本发明的较佳实施例,并非对本发明任何形式上和实质上的限制,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还将可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。凡熟悉本专业的技术人员,在不脱离本发明的精神和范围的情况下,当可利用以上所揭示的技术内容而做出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对上述实施例所作的任何等同变化的更动、修饰与演变,均仍属于本发明的技术方案的范围内。
- 还没有人留言评论。精彩留言会获得点赞!