用于检测人类HLA‑B*5701等位基因的核酸、试剂盒及方法与流程
本发明属于生物技术领域,具体涉及一种用于检测人类HLA-B*5701等位基因的核酸、试剂盒及方法。
背景技术:
人类免疫缺陷病毒(Human Immunodeficiency Virus;abbr:HIV),即艾滋病(AIDS,获得性免疫缺陷综合征)病毒,是造成人类免疫系统缺陷的一种病毒。1983年,人类免疫缺陷病毒在美国首次发现。它是一种感染人类免疫系统细胞的慢病毒(Lentivirus),属逆转录病毒的一种。
阿巴卡韦(abacavir,ABC)是一种抗艾滋病新药,为新的碳环2'-脱氧鸟苷核苷类药物,其口服生物利用度高,易渗入中枢神经系统。与其他核苷类逆转录酶抑制剂一样,它是一个无活性的前药,在体内经4个步骤代谢成为具活性的三磷酸酯,并通过以下2条途径发挥抑制人免疫缺陷病毒(HIV)逆转录酶的作用:①竞争性地抑制2'-脱氧鸟苷三磷酸酯(dGTP)(DNA合成片段之一)结合进入核酸链。②通过阻止新碱基的加入而有效地终止DNA链的合成。
阿巴卡韦(Abacavir,ABC)是一种核苷类反转录酶抑制剂,从1999年即作为经典的联合用药中的一种药物,被欧美一些国家采用,目前,仍是美国食品与药品管理局推荐的初次服用的首选药物。但是,据现有的资料报道,服用阿巴卡韦药物后,大约有5%-8%的人会出现ABC HSR。ABC HSR是一种多器官综合征,会出现两种或两种以上的体征或症状,甚至可以导致偶发的、严重的致死性超敏反应。
ABC从一开始应用即被发现对部分服用者,有较严重的超敏反应(Hyper sensitivity reactio n,HSR),为了避免严重超敏反应的发生,众多研究者致力于发展一种安全有效的筛查方法。2002年,Mallal等首次研究发现,阿巴卡韦斑贴实验阳性者人群遗传的MHC位点的57.1单体型基因频率更高。由此推测发病机制,可能是MHC基因的这个位点与阿巴卡韦代谢生成一种具有免疫性的内源蛋白有关,HLA位点上的HLA-B*5701基因被推测为重要的危险因素。在细胞以及体外实验中,α-肿瘤坏死因子(TNF-α)及γ-干扰素(IFN-γ)的强烈应答,以及CD8+细胞在体外暴露于阿巴卡韦后,产生的增殖均证明了,ABC HSR是由HLA-B*5701限制性的CD8+T淋巴细胞介导的I类主要组织相容性复合体疾病,其阳性预测阿巴卡韦过敏反应准确率超过70%,阴性预测准确率达95%-98%。
2008年7月,FDA对有关研究进行回顾分析后提出建议:在治疗前对患者进行H LA-B*5701等位基因筛选,并对检测为阳性的患者选择替代疗法,除非用药的潜在效益大于风险,否则不建议HLA-B*5701阳性患者使用阿巴卡韦治疗。目前,欧美一些国家已经将HLA-B*5701等位基因筛查应用于临床常规检测,在治疗前对患者进行HLA-B*5701等位基因筛选,并对检测为阳性的患者选择替代疗法,除非用药的潜在效益大于风险,否则不建议HLA-B*5701阳性患者使用阿巴卡韦治疗。
HLA-B*5701等位基因筛查的应用,对于艾滋病的治疗是有极其重要的意义的:第一,它降低了ABC HSR的发生率,使HIV的治疗风险降低;第二,它的特异度高于临床诊断、皮肤斑贴实验,可以使更多的患者接受ABC的有效治疗,避免了临床过度诊断而错过阿巴卡韦药物治疗机会;第三,HLA-B*5701等位基因筛查大大节约了医疗费用。
目前检测该等位基因的方法主要有PCR-SSP、PCR-SSO和PCR-SBT法。PCR-SSP和PCR-SSO检测范围广,操作繁琐,检测失败率高,易出现假阴性,产品内容需每年更新,成本昂贵;PCR-SBT检测范围广,检测周期长,产品内容需每年更新,成本昂贵。上述三种方法都不适用于只检测某一种等位基因型。
技术实现要素:
为克服以上现有检测方法的不足,本发明的目的是提供一组用于检测人类HLA-B*5701等位基因的核酸。本发明的另一个目的是提供一种用于检测人类HLA-B*5701等位基因的试剂盒及其检测方法。
为实现上述目的,本发明采用以下技术方案:
一组用于检测人类HLA-B*5701等位基因的核酸,该核酸包括用于2孔反应体系检测的引物对1和检测探针1,引物对2和检测探针2;其中,所述引物对1的核苷酸序列如SEQ ID No.1和SEQ ID No.2所示,所述检测探针1的核苷酸序列如SEQ ID No.3所示,所述引物对2的核苷酸序列如SEQ ID No.4和SEQ ID No.5所示,所述检测探针2的核苷酸序列如SEQ ID No.6所示。
如上所述的核酸,优选地,所述核酸还包括内部质控,所述内部质控包括质控引物对和质控探针,所述质控引物对的核苷酸序列如SEQ ID No.7和SEQ ID No.8所示,所述质控探针的核苷酸序列如SEQ ID No.9所示。
如上所述的核酸,优选地,所述核酸还包括用作引物对1扩增的阳性对照物1、与用作引物对2扩增的阳性对照物2和用作内部质控的质控阳性对照物,其中,所述阳性对照物1含有如SEQ ID No.10所示的核苷酸序列,所述阳性对照物2含有如SEQ ID No.11所示的核苷酸序列,和/或所述质控阳性对照物含有如SEQ ID No.12所示的核苷酸序列。
一种用于检测人类HLA-B*5701等位基因的试剂盒,所述试剂盒包括如上所述的核酸,所述检测探针1和检测探针2的5'端连接荧光基团,3'端连接淬灭基团,和/或质控探针的5'端连接有不同于所述检测探针1和检测探针2的荧光基团,3'端连接有不同于所述检测探针1和检测探针2的淬灭基团。
如上所述的试剂盒,优选地,所述试剂盒还包括:10×PCR缓冲液、DNA聚合酶、dNTPs、矿物油、阴性对照物:水。
如上所述的试剂盒,优选地,所述荧光基团为FAM、JOE、CY3、HEX中任意一种,所述淬灭基团为MGB、BHQ1、TAMRA、BHQ2中任意一种。
一种检测人类HLA-B*5701等位基因的方法,该方法包括以下步骤:
(1)从样品中提取DNA;
(2)对提取的所述DNA,同时进行反应体系1和反应体系2的实时荧光PCR扩增;其中,所述反应体系1中引物对1的核苷酸序列如SEQ ID No.1和SEQ ID No.2所示,检测探针1的核苷酸序列如SEQ ID No.3所示;所述反应体系2中引物对2的核苷酸序列如SEQ ID No.4和SEQ ID No.5所示,检测探针2的核苷酸序列如SEQ ID No.6所示,所述检测探针1和检测探针2的5'端连接荧光基团,3'端连接淬灭基团;
(3)收集荧光信号,选择对应荧光基团的荧光检测模式,基线调整取3~15个循环的荧光信号,以阈值线刚好超过正常阴性对照的最高点设定阈值线;
(4)结果判定:待测样品的所述反应体系1和所述反应体系2的荧光增长曲线均超过阈值线,有明显的扩增曲线,则判断为阳性,如所述反应体系1和所述反应体系2中任一反应体系无明显的扩增曲线或均无明显的扩增曲线,判断为阴性。
如上所述的方法,优选地,在步骤(2)中,所述反应体系1和所述反应体系2还包括有作为内部质控的质控引物对和质控探针,所述质控引物对的核苷酸序列如SEQ ID No.7和SEQ ID No.8所示,所述质控探针的核苷酸序列如SEQ ID No.9所示,该质控探针的5'端连接有不同于所述检测探针1和检测探针2的荧光基团,3'端连接有淬灭基团。
如上所述的方法,优选地,在步骤(2)中,在所述反应体系1和所述反应体系2中,各条检测引物的终浓度为150-250nmol/L;检测探针的终浓度为150-200nmol/L,所述质控引物对和质控探针的终浓度为100-150nmol/L;所述荧光基团为FAM、JOE、CY3、HEX中任意一种,所述淬灭基团为MGB、BHQ1、TAMRA、BHQ2中任意一种。
如上所述的方法,优选地,步骤(2)中,所述实时荧光PCR扩增的反应程序为:
第一阶段:95℃3min;
第二阶段:95℃20s,60℃25s,72℃30s,15个循环;
第三阶段:95℃20s,60℃25s,72℃30s,30个循环;
信号收集:第三阶段72℃时收集荧光信号。
如上所述的方法,优选地,在步骤(2)中,在各自所述反应体系1和所述反应体系2中,所述检测引物对和检测探针的终浓度均为150nmol/L,所述内部质控的引物对和质控探针的终浓度均为100nmol/L,所述检测探针的5'端连接FAM,3'端连接MGB,所述质控探针的5'端连接JOE,3'端连接BHQ1;
如上所述的方法,优选地,在步骤(4)中,所述待测样本的所述反应体系1和反应体系2的FAM、JOE均有明显的扩增曲线,FAM信号的Ct值≤25,JOE信号的Ct值≤25为阳性;所述反应体系1和反应体系2的JOE均有明显的扩增曲线,且JOE信号的Ct值≤25,两体系中任一体系FAM没有明显的扩增曲线或均无扩增曲线,或FAM信号的Ct值>25为阴性。
如上所述的方法,优选地,在步骤(2)中,还设有阳性对照和阴性对照,所述阳性对照为以含有如SEQ ID No.10和SEQ ID No.11所示的核苷酸序列和含有如SEQ ID No.12所示的核苷酸序列为模板,进行所述反应体系1和反应体系2的实时荧光PCR扩增;所述阴性对照为以水为模板,进行所述反应体系1和反应体系2的实时荧光PCR扩增;
在步骤(4)中,所述阳性对照的反应体系1和2的FAM均有明显的扩增曲线且FAM信号的Ct值≤20,JOE有明显的扩增曲线且JOE信号的Ct值≤25,符合要求;所述阴性对照的反应体系1和2的FAM、JOE均没有明显的扩增曲线,符合要求;否则,应重新进行检测。
本发明提供的检测方法采用完全闭管操作,操作简单、方便快捷、通过直接探测PCR过程中荧光信号值的获得检测的结果,不需要PCR后处理或电泳检测,克服了常规PCR技术的易污染、出现假阳性,能有效避免非特异性扩增难题,并且适合大批量样本的检测。
本发明提供的检测试剂盒及方法,用于检测人类HLA-B*5701等位基因时,为了避免漏检、及初步判断样本DNA量是否在允许范围内,设有内部质控,为了判断检测体系是否正常,还设有阴性对照和检测引物的阳性对照;为了避免假阴性及漏检,设有阳性对照,其阳性对照检测呈阳性结果,说明检测体系没有问题,不会出现假阴性结果及漏检;为了避免假阳性及漏检,设有阴性对照,其阴性对照检测呈阴性结果,说明检测体系没有问题,不会出现假阳性结果及漏检;检测试剂盒及方法设计严谨,有效避免漏检、错检的可能。
本发明建立了实时荧光PCR扩增反应体系实现对HLA-B*5701等位基因的快速检测;且操作简单,结果易读。该方法的灵敏度高,可实现30copies/μL的稳定检出;特异性好,对非目的等位基因组DNA均无非特异性扩增,检测能力高达100%,还具有实验周期短,操作简单,安全无毒,成本低等显著优点。
附图说明
图1为本发明优选的试剂盒检测检测内部质控的扩增曲线。
图2为本发明优选的试剂盒检测检测阴性对照的扩增曲线。
图3为本发明优选的试剂盒检测野生型阳性对照的扩增曲线。
图4为本发明优选的试剂盒反应体系1检测灵敏度的扩增曲线。
图5为本发明优选的试剂盒反应体系2检测灵敏度的扩增曲线。
图6为本发明优选的试剂盒反应体系1检测精密度的扩增曲线。
图7为本发明优选的试剂盒反应体系2检测精密度的扩增曲线。
图8为本发明优选的试剂盒检测HLA-B*5703质粒稀释液的扩增曲线。
图9为本发明优选的试剂盒检测HLA-B*5801质粒稀释液的扩增曲线。
图10为本发明优选的试剂盒检测阳性对照物1的扩增曲线。
图11为本发明优选的试剂盒检测阳性对照物2的扩增曲线。
图12为本发明优选的试剂盒检测最低检测线样本内控检测的扩增曲线。
图13为本发明优选的试剂盒检测上海华山医院临床样本扩增曲线,两条起线的为两个体系的阳性对照。
具体实施方式
以下结合具体实例对本发明做进一步详细说明,并非对本发明的限定,本发明的实施方式并不限于此,本发明提供的核苷酸序列的互补序列也可实现本发明,如无特殊说明,所用试剂为常规试剂,因此凡依照本公开内容所作出的本领域的等同替换,均属于本发明的保护范围。
实施例1引物、探针、验证模板设计
本发明是针对人类HLA-B*5701基因(NCBI GenBank:AJ458991.3)1-7号外显子序列查找相应的等位基因特异性位点通过对检测引物、检测探针的筛选和体系的优化,建立了实时荧光PCR等位基因检测体系,实现对HLA-B*5701等位基因检测的高灵敏度和高特异性检测。在本发明实时荧光PCR等位基因检测体系中,对目的基因特异性区域进行PCR扩增。其中,特异性检测引物与目的基因序列匹配度高于其他等位基因序列;在此基础上,特异性探针与目的序列特异性结合,有效抑制了其他等位基因型的非特异性扩增,进一步提高体系的特异性水平,从而实现了在众多等位基因型背景下对目的等位基因序列的检出。应用2孔反应体系检测HLA-B*5701等位基因,其中一对用于检测的引物对1和检测探针1,另一对为引物对2和检测探针2;其中,所述引物对1的核苷酸序列如SEQ ID No.1和SEQ ID No.2所示,所述检测探针1的核苷酸序列如SEQ ID No.3所示,所述引物对2的核苷酸序列如SEQ ID No.4和SEQ ID No.5所示,所述检测探针2的核苷酸序列如SEQ ID No.6所示,其核苷酸序列如下:
5701-F1(SEQ ID No.1):5’-CTCGGTAAGTCTGCGCGG-3’;
5701-R1(SEQ ID NO.2):5’-CGAGTCCGAGGATGGCG-3’;
5701-P1(SEQ ID NO.3):5’-CCTTCATGTTCCGTGTCTCCCCG-3’;
5701-F2(SEQ ID NO.4):5’-CGTCGTAGGCGGACTGGTC-3’
5701-R2(SEQ ID NO.5):5’-TTGAGGCCAAAATCCCCG-3
5701-P2(SEQ ID NO.6):5’-TCACCTGGATGATGTGAG-3
其中,引物对1扩增产物长度为105bp,扩增产物可作为阳性对照,即阳性对照物1含SEQ ID NO.10所示的核苷酸序列;引物对2扩增片段长度为166bp,阳性对照物2含SEQ ID NO.11所示的核苷酸序列,具体如下:
SEQ ID NO.10:
5’-CGAGTCCGAGGATGGCGCCCCGGGCGCCATGGATAGAGCAGGAGGGGCCGGAGTATTGGGACGGGGAGACACGGAACATGAAGGCCTCCGCGCAGACTTACCGAG-3’;
SEQ ID NO.11:
5’-TTGAGGCCAAAATCCCCGCGGGTTGGTCAGGGCGGGGCGGGGCTCGGGGGGACGGGGCTGACCGCGGGGCCGGGGCCAGGGTCTCACATCATCCAGGTGATGTATGGCTGCGACGTGGGGCCGGACGGGCGCCTCCTCCGCGGGCATGACCAGTCCGCCTACGACG-3’。
进一步,为了对实时荧光PCR反应体系和实验操作过程进行质控,同时检测样本的质量,避免漏检,本发明选用根据NCBI数据库公布的人类ACTB基因(NCBI Reference Sequence:NG_007992.1)4号外显子序列针对这段序列设计的内部控制引物和内控探针,并对内控的上下游引物进行筛选,使非模版体系(NTC)不起线,样本起线正常,并且要求设计的质控引物、质控探针、及质控扩增产物均不会有交叉反应。通过内部质控的质控引物对、质控探针的筛选和体系优化,建立了实时荧光PCR内部质控检测体系,其中,质控引物对(ACTB-F、ACTB-R)、质控探针(ACTB-P)最后确定的核苷酸序列如下:
ACTB-F(SEQ ID No.7):5’-CCCTGGAGAAGAGCTACGA-3’;
ACTB-R(SEQ ID No.8):5’-GAAGGAAGGCTGGAAGAGTG-3’;
ACTB-P(SEQ ID No.9):5’-CTGCCTGACGGCCAGGTCAT-3’。
作为内部质控的质控引物对进行PCR扩增时,其扩增序列的大小为95bp,将扩增序列作为内部质控阳性对照物,即包含如SEQ ID No.12所示序列,作为验证是否漏检的阳性对照。
SEQ ID No.12:
5’-CCCTGGAGAAGAGCTACGAGCTGCCTGACGGCCAGGTCATCACCATTGGCAATGAGCGGTTCCGCTGCCCTGAGGCACTCTTCCAGCCTTCCTTC-3’。
实施例2采用实时荧光PCR检测人类HLA-B*5701等位基因的方法
将实施例1筛选设计的引物对1和检测探针1,引物对2和检测探针2进行合成,并在检测探针1和2的5'端连接荧光基团,3'端连接淬灭基团,其中,荧光基团可为FAM、JOE、CY3、HEX中任意一种,淬灭基团可为MGB、BHQ1、TAMRA、BHQ2中任意一种。
实时荧光PCR检测人类HLA-B*5701等位基因的方法如下:
(1)从样品中提取DNA或获取样品DNA;
(2)对提取的样品DNA,进行人类HLA-B*5701等位基因的实时荧光PCR扩增检测,采用反应体系1和反应体系2,其中,所述反应体系1中含有引物对1的核苷酸序列如SEQ ID No.1和SEQ ID No.2所示,检测探针1的核苷酸序列如SEQ ID No.3所示;所述反应体系2中含有引物对2的核苷酸序列如SEQ ID No.4和SEQ ID No.5所示,检测探针2的核苷酸序列如SEQ ID No.6所示,所述检测探针1和检测探针2的5'端连接荧光基团,3'端连接淬灭基团;
实时荧光PCR扩增反应条件优选为:
第1阶段:95℃3min;
第2阶段:95℃20s,60℃25s,72℃30s,15个循环;
第3阶段:95℃20s,60℃25s,72℃30s,30个循环;
第3阶段:30个循环中,72℃时收集荧光信号;
第2阶段15个循环,60℃的退火温度,72℃的延伸温度,有利于目的基因模板的富集。在第3阶段(30个循环),60℃的退火温度,72℃的延伸温度可以保证更好的扩增特异性。
(3)收集荧光信号,选择对应荧光基团的荧光检测模式,基线调整取3~15个循环的荧光信号,以阈值线刚好超过正常阴性对照的最高点设定阈值线;
(4)结果判定:待测样品的所述反应体系1和所述反应体系2的荧光增长曲线均超过阈值线,有明显的扩增曲线,则判断为阳性,如所述反应体系1和所述反应体系2中任一反应体系无明显的扩增曲线或均无明显的扩增曲线,判断为阴性。
将HLA-B*5701基因的阳性对照物1和阳性对照物2进行上述实时荧光PCR扩增反应,结果显示,所设计的检测引物1和2及反应体系1和2可有效扩增出HLA-B*5701基因的等位基因,对EDTA抗凝血和口腔拭子提取的基因组DNA以及全血细胞去红细胞后的白细胞都具有检测能力。
对样品进行检测时,为避免漏检,如避免有的反应孔中未加检测样本的情况出现,在每个反应孔中设置有内部质控,其质控引物对和质控探针的核苷酸序列如实施例1中所述。应当注意的是检测探针1和2与质控探针荧光基团标记为不同检测模式的荧光基团,荧光基团不同即可,淬灭基团相同或不同不会对结果产生影响。具体的实时荧光PCR扩增反应体系组成按如下表1中PCR反应体系配置,PCR反应总体积40μL,含30μL的反应体系,最后加入10μL的检测模板(阳性对照、非模板对照、质粒模板或待测样本基因组DNA)。对于人血液样品或咽拭子样品的DNA检测时,质控探针的信号应有明显的扩增曲线。
表1.实时荧光PCR扩增反应体系组成
在探针合成时,探针与荧光和淬灭基团相连,检测探针1和2的荧光-淬灭基团为优选为FAM-MGB,质控探针的荧光-淬灭基团优选为JOE-BHQ1,当然,其它荧光-淬灭基团组合也同样适用,比如,HEX-BHQ1、HEX-TAMRA、FAM-TAMRA、FAM-BHQ1、CY3-BHQ1、CY3-BHQ2等,经实验验证,均可检测人类HLA-B*5701基因的基因型。
为了避免假阴性及漏检,检测方法中还设有反应体系1、反应体系2和内部质控的阳性对照(STD),以含有如SEQ ID No.10所示的核苷酸序列、含有如SEQ ID No.11所示的核苷酸序列、含有如SEQ ID No.12所示的核苷酸序列为模板,进行所述反应体系1和反应体系2的实时荧光PCR扩增;其阳性对照检测呈阳性结果,检测荧光信号均有明显的扩增曲线(均起线),说明检测体系没有问题,不会出现假阴性结果及漏检。
为了避免假阳性及漏检,设有检测引物和内部质控的阴性对照(NTC),以水为模板,进行所述反应体系1和反应体系2的实时荧光PCR扩增;其阴性对照检测呈阴性结果,检测荧光信号均无明显的扩增曲线(均不起线),说明检测体系没有问题,不会出现假阳性结果及漏检;否则,应重新进行反应体系的配置及检测。阳性对照(STD)FAM信号Ct值作为实验数据是否有效的判定标准。
上述实时荧光PCR反应可使用的仪器包括ABI实时PCR系统(例如7000,7300,7500,7900等);BioRad实时PCR检测系统、Stratagene定量多聚酶链反应仪(例如MX4000,MX3000,MX3005)。
实施例3用于检测人类HLA-B*5701等位基因的试剂盒
用于检测人类HLA-B*5701等位基因的实时荧光PCR试剂盒包括以下成分:
引物1上游引物:核苷酸序列如SEQ ID No.1所示;
引物1下游引物:核苷酸序列如SEQ ID No.2所示;
检测探针1:核苷酸序列如SEQ ID No.3所示;
引物2上游引物:核苷酸序列如SEQ ID No.4所示,
引物2下游引物:核苷酸序列如SEQ ID No.5所示;
检测探针2:核苷酸序列如SEQ ID No.6所示,所述检测探针1和2的5'端连接荧光基团,3'端连接淬灭基团;
阳性对照物1:含有如SEQ ID No.10所示的核苷酸序列;
阳性对照物2:含有如SEQ ID No.11所示的核苷酸序列。
为了避免漏检,错检,还包括:作为内部质控的质控引物对、质控探针和质控阳性对照物,
其中,质控引物对的核苷酸序列如SEQ ID No.7和SEQ ID No.8所示,质控探针的核苷酸序列如SEQ ID No.9所示,该质控探针的5'端连接有不同于所述检测探针1和2的荧光基团,3'端连接有淬灭基团,质控阳性对照物含有如SEQ ID No.12所示的核苷酸序列。
为了防止反应体系在PCR扩增过程中挥发,影响检测效果,试剂盒还包括有矿物油。
为了便于反应体系的配置,试剂盒还包括10×PCR缓冲液;DNA聚合酶,dNTPs,阴性对照物:水。其中DNA聚合酶可采用Fast Advanced Master Mix,可采用ABI(美国应用生物系统公司);10×PCRbuffer(Mg2+Plus)采用TaKaRa宝生物工程(大连)有限公司,也可应用市场上其他公司的PCR缓冲液与DNA聚合酶。
实施例4快速检测试剂盒的制备
本实施例的试剂盒是在实施例3的基础上,制备成可以直接加入检测样品后,进行检测的试剂盒。该试剂盒有检测HLA-B*5701的8联PCR反应条以及验证试剂盒性能分析时所需的HLA-B*5701质粒1、HLA-B*5701质粒2、内控质粒,也就是HLA-B*5701阳性对照物1和2及质控阳性对照物组成,各成分如表2所示,具体反应体系的配置见表3。每条HLA-B*5701-8联PCR反应条的奇数管(1/3/5/7或A/C/E/G)对应检测体系为反应体系1的A1反应液,;偶数管(2/4/6/8或B/D/F/H)对应的检测体系为反应体系1的A2反应液。每管主要成分为特异性引物、荧光探针和PCR反应液等,其中检测探针1和2的5'端连接FAM,3'端连接MGB,则检测样品信号类型为FAM信号;内部质控的质控探针5'端连接JOE,3'端连接BHQ1,内部质控信号类型为JOE信号(内控作为对试剂盒、DNA质量以及操作本身的质控);检测引物、探针和阳性对照物委托上海英俊生物技术有限公司合成。
表2试剂盒组成
表3反应体系的配置
其中,AB premix的全称Fast Advanced Master Mix,含有DNA聚合酶和PCR buffer(Mg2+Plus),购自ABI(美国应用生物系统公司);dNTPs购自TaKaRa宝生物工程(大连)有限公司,经过大量试验、验证该试剂盒为一种高灵敏、高特异性、高效的检测HLA-B*5701基因多态性的检测试剂盒。
实施例5:对实施例4中制备的试剂盒进行性能分析实验
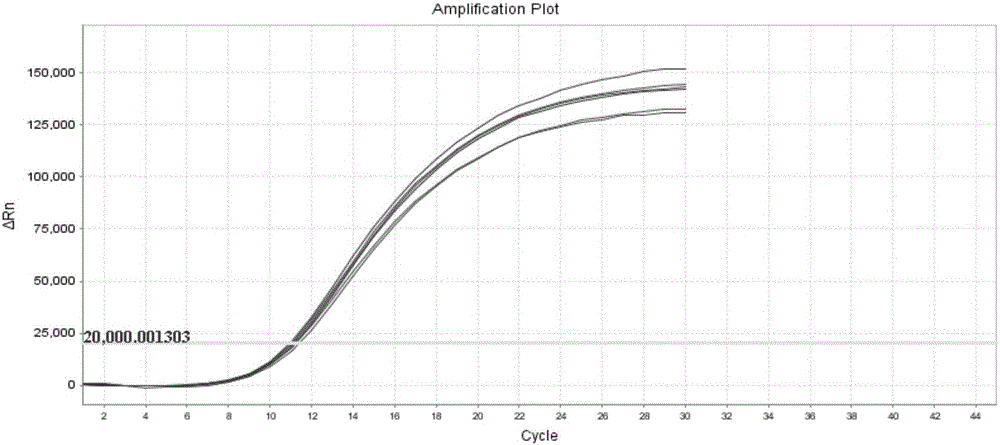
在每次检测具体样品时,为了保证数据真实、可靠、避免漏检、错检,需同时进行阴性对照(非模板对照,即NTC)实验和阳性对照(STD)实验。采用实施例4制备的试剂盒,运用实施例2所用的检测方法,经多次试验验证,图1为内控检测结果,Ct内控≤25,符合要求;图2为NTC检测结果,反应体系1~2的FAM均不起线,符合要求;图3为STD检测结果,反应体系1~2的FAM信号均起线且Ct≤20,符合要求。
并对实施例4制备的试剂盒进行如下的性能分析实验:
(1)灵敏度实验
将以现有基因工程技术合成的含有HLA-B*5701基因特异性片段序列的5701质粒1和5701质粒2的干粉用TE(10mmol/L,PH8.0)复溶,定量后稀释。分别取3×104copies/μL的5701质粒1和5701质粒2进行10倍稀释,稀释3个梯度,然后分别以3种浓度梯度(3×103copies/μL和、3×102copies/μL、30copies/μL)的5701质粒1和5701质粒2为模板,进行实施例2中所述实时荧光PCR扩增反应方法,检测结果见图4和图5所示,由图4和图5可见本发明的荧光PCR方法灵敏度高,30copies/μL的基因即可检出。
(2)精密度实验
以3×102copies/μL的质粒5701质粒1和5701质粒2为模板,进行反应体系1和2的如上实时荧光PCR扩增反应,检测结果见图6和图7。结果表明本发明的实时荧光PCR扩增反应重复性好(反应体系1~2各10次重复实验结果稳定,CV<5%)。
(3)特异性
将以现有基因工程技术合成的与HLA-B*5701目的基因特异性片段相似度较高的等位基因HLA-B*5703和HLA-B*5801特异性片段序列的质粒DNA干粉用用TE(10mmol/L,PH8.0)复溶,定量后稀释。以上述3×104copies/μL的HLA-B*5703和HLA-B*5801的质粒DNA为模板,运用实施例4的试剂盒进行实时荧光PCR扩增反应,扩增结果见图8和图9所示。结果为阴性,说明本发明设计的引物对1和2及检测探针1和2的特异性强,与非目的基因无交叉反应。
实施例6:临床样本DNA的检测
1、获取待测样本基因组DNA
具体的,血液样本的提取使用武汉海吉力血液基因提取试剂盒(Cat.No.HGN-tq1050)。按照说明书提取来源于上海华山医院的临床46例患者血液基因组DNA,操作步骤简述如下:
取含抗凝剂的血液200μL加入到离心管中,加入50μL Proteinase K,吹吸混匀或涡旋振荡10s混匀;在离心管中加入250μL裂解液BL,涡旋振荡10s混匀;室温静置10min充分裂解细胞;在离心管中加入200μL无水乙醇,涡旋振荡10s混匀;将DNA吸附柱置于2mL收集管中,将上述溶液(700μL)全部转移到DNA吸附柱中,13,000rpm离心1min,弃滤液和收集管;将DNA吸附柱置于新的2mL收集管中,向DNA吸附柱中加入500μL洗涤液BWB1,13,000rpm离心1min,弃滤液;将DNA吸附柱重新放回2mL收集管中,加入500μL洗涤液BWB2,13,000rpm离心1min,弃滤液;将DNA吸附柱置于新的1.5mL无核酸酶污染的离心管中,于室温静置或超净工作台风干3-5min,以彻底挥发残余的乙醇;向吸附柱柱膜中央上方小心加入50-100μL洗脱液TE,室温静置1-3min,13,000rpm离心1min。洗脱液即为DNA溶液。
将抽提好的DNA样本,用紫外分光光度计检测浓度和DNA质量,DNA的浓度要求≥10ng/μL,DNA的质量OD260/280在1.8~2.0之间,定量后,母液-20℃保存。
如果DNA的浓度小于10ng/μL,则说明样本浓度提取过低,为防止后续PCR扩增失败,应重新提取;如果样本质量纯度OD 260/280值不在1.8~2.0之间,则说明提取的样本DNA母液含有杂质,可能会导致后续PCR扩增失败,应重新提取。
2、检测待测样本基因组DNA
取本发明实施例4制备的人类HLA-B*5701等位基因检测试剂盒,将上述符合要求的基因组DNA用TE(10mmol/L,PH8.0)稀释到1~10ng/μL作为检测模板,进行实时荧光PCR扩增反应。具体步骤如下:
1)将5701阳性对照取出解冻后震荡混匀,短时离心备用。
2)将8联PCR反应条取出解冻后离心30s,确保管壁、管盖处无液滴残留。
3)将ddH2O作为NTC模板;将待测样本基因组DNA作为待测样本模板。
4)将NTC模板、5701阳性对照和待测的46例样本的DNA模板分别加入到反应体系1~2中,每孔10μL。
5)将以上PCR反应条盖好管盖于离心机上离心30s,确保管壁上无液滴残留。
6)将8联PCR反应条放于实时荧光定量PCR仪器中,按照实施例2中进行反应条件设置并运行程序。
3、检测结果分析
根据荧光定量PCR扩增仪Ct值分析检测结果:以JOE信号Ct值作为PCR反应是否成功的判定标准(JOE信号Ct值≤25,扩增正常),以非模板对照(NTC)和阳性对照(STD)FAM信号Ct值作为实验数据是否有效的判定标准,非模板对照FAM信号不起线,阳性对照FAM信号Ct值≤20,其中,该值是用两种体系同时三复孔检测阳性对照,同时根据阈值线的上下浮动,得出其波动的Ct值范围,保守计算得出,具体见图10和图11,体系性能良好,结果有效;
以内控检测孔JOE信号Ct值作为上样量质控标准,Ct内控≤25,该值是做样本浓度的最低检测线,通过上样10μL样本DNA(浓度为0.1ng/μL),检测其内控Ct值,保守计算得出,具体见图11;Ct内控>25或无扩增曲线,说明加入的基因组DNA浓度过低或存在降解,建议重新提取该样本基因组DNA后进行检测,以待测样本等位基因检测孔FAM信号的Ct值作为阴阳性判定标准,反应体系1~2:Ct≤25为阳性,该值是做阳性样本浓度的最低检测线,通过上样10μL样本DNA(浓度为0.1ng/μL),检测FAM信号Ct值,保守计算得出,具体见图12,Ct>25或无扩增曲线则为阴性。
对上述上海华山医院的临床46例样本DNA、阳性对照和NTC为模板,进行实时荧光PCR扩增反应结果见图13所示。结果说明运用本发明的试剂盒及检测方法检测这46例样本DNA为阴性。
同时,用美国One Lambda,Inc公司生产的PCR-SSO法的HLA-DNA分型试剂盒(SSO法)做参比试剂,确定这46例样本DNA,其全部为非目的等位基因型,具体基因型为HLA-B*07:02:01、HLA-B*08:01:01、HLA-B*13:01:01、HLA-B*13:02:01、HLA-B*15:01:01:01、HLA-B*15:02:01、HLA-B*15:08:01、HLA-B*15:18:01、HLA-B*15:25:01、HLA-B*15:32:01、HLA-B*27:04:01、HLA-B*27:05:02、HLA-B*35:01:01:01、HLA-B*35:02:01、HLA-B*35:03:01、HLA-B*37:01:01、HLA-B*38:02:01、HLA-B*39:01:01:01、HLA-B*40:01:01、HLA-B*40:02:01、HLA-B*40:06:01:01、HLA-B*44:03:01、HLA-B*44:03:02、HLA-B*46:01:01、HLA-B*48:01:01、HLA-B*51:01:01:01、HLA-B*51:02:01、HLA-B*52:01:01:01、HLA-B*54:01:01、HLA-B*55:02:02、HLA-B*56:01:02、HLA-B*58:01:01、HLA-B*67:01:02。可见,本发明设计的引物对和探针对上述非目的等位基因型无非特异性扩增,特异性强。
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。
SEQUENCE LISTING
<110> 武汉海吉力生物科技有限公司
<120> 用于检测人类HLA-B*5701等位基因的核酸、试剂盒及方法
<130>
<160> 12
<170> PatentIn version 3.5
<210> 1
<211> 18
<212> DNA
<213> 人工合成
<400> 1
ctcggtaagt ctgcgcgg 18
<210> 2
<211> 17
<212> DNA
<213> 人工合成
<400> 2
cgagtccgag gatggcg 17
<210> 3
<211> 23
<212> DNA
<213> 人工合成
<400> 3
ccttcatgtt ccgtgtctcc ccg 23
<210> 4
<211> 19
<212> DNA
<213> 人工合成
<400> 4
cgtcgtaggc ggactggtc 19
<210> 5
<211> 18
<212> DNA
<213> 人工合成
<400> 5
ttgaggccaa aatccccg 18
<210> 6
<211> 18
<212> DNA
<213> 人工合成
<400> 6
tcacctggat gatgtgag 18
<210> 7
<211> 19
<212> DNA
<213> 人工合成
<400> 7
ccctggagaa gagctacga 19
<210> 8
<211> 20
<212> DNA
<213> 人工合成
<400> 8
gaaggaaggc tggaagagtg 20
<210> 9
<211> 20
<212> DNA
<213> 人工合成
<400> 9
ctgcctgacg gccaggtcat 20
<210> 10
<211> 105
<212> DNA
<213> 人工合成
<400> 10
cgagtccgag gatggcgccc cgggcgccat ggatagagca ggaggggccg gagtattggg 60
acggggagac acggaacatg aaggcctccg cgcagactta ccgag 105
<210> 11
<211> 166
<212> DNA
<213> 人工合成
<400> 11
ttgaggccaa aatccccgcg ggttggtcag ggcggggcgg ggctcggggg gacggggctg 60
accgcggggc cggggccagg gtctcacatc atccaggtga tgtatggctg cgacgtgggg 120
ccggacgggc gcctcctccg cgggcatgac cagtccgcct acgacg 166
<210> 12
<211> 95
<212> DNA
<213> 人工合成
<400> 12
ccctggagaa gagctacgag ctgcctgacg gccaggtcat caccattggc aatgagcggt 60
tccgctgccc tgaggcactc ttccagcctt ccttc 95
- 还没有人留言评论。精彩留言会获得点赞!
- 油菜BnaSPT1基因在促进双子叶植物角果生长中的应用的制造方法与工艺
- 油菜BnaSPT3基因在促进双子叶植物角果生长中的应用的制造方法与工艺
- 一种木质素合成途径中上位性效应检测方法与制造工艺
- 用于检测人类HLA‑B*5701等位基因的核酸、试剂盒及方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- 与癌症相关的基因融合体和基因变异体的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 一种高通量测序检测无创亲子鉴定的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 通过分析预示性基因集诊断亚临床和临床的急性排异的方法与制造工艺