一种具有抗菌作用的化合物、制备方法与流程
1.本发明属于中药化学及医药技术领域,具体涉及一种具有抗菌作用的吡啶类新化合物、 制备方法。
背景技术:
2.无叶假木贼(anabasis aphylla l.)又称毒藜、无叶毒藜等,系藜科假木贼属植物,主要分 布于甘肃西部、新疆等地,欧洲、中亚也有分布,喜生长于荒漠、戈壁、沙丘及干旱山坡。 无叶假木贼始载于《新华本草纲要》,性寒,味辛、苦,归心经,全株有毒,具有杀虫止痒 之功效,可用于治疗疥癣、疥疮、湿疹痒痛,民间主要用于防治菜青虫、蚜虫等多种害虫。 现代药理学研究表明,无叶假木贼含有生物碱、萜类、皂苷类和生物大分子等化学成分, 表现出神经活性、抗胆碱酯酶、抗微生物、杀虫等活性。
3.关于无叶假木贼化学成分及活性的研究、报道较少。1998年,tilyabaev z等发表文章 报道称,假木贼属植物有很高的抑制胆碱酯酶的活性;2004年,宋素琴等研究发现,新疆 无叶假木贼中多种化学成分表现出抗菌活性;2007年,杜华等发表文章《无叶假木贼和盐 爪爪提取物的抗菌活性》,报道无叶假木贼生物碱对细菌表现出较强的抗菌活性;2010年, 杨燕等人从无叶假木贼中提取分离出一种新化合物p
‑
acetyl
‑
phenol 1
‑
o
‑
β
‑
d
‑
xylopyranosyl
ꢀ‑
(1
→
2)
‑
β
‑
d
‑
glucopyranoside以及5种已知化合物;2011年,李卫林等人无叶假木贼中提取 分离得到一种新化合物顺式咖啡酰二十烷醇酯以及12种已知化合物。然而,对于无叶假木 贼药材还需要积极开发其有效成分,以获得更多的应用价值。
技术实现要素:
4.基于上述现有技术,本发明的目的在于提供具有抗菌活性的从无叶假木贼中分离得到 的两种新的吡啶类化合物和制备方法及其在制备杀菌、抗菌、抑菌药物中的应用。
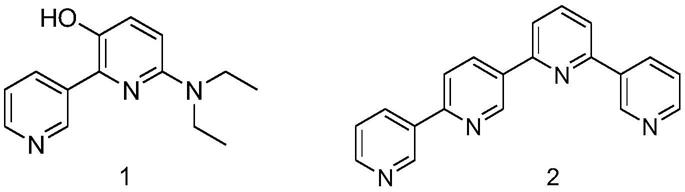
5.本发明的第一个目的,是提供两种吡啶类化合物,分子式分别为c
14
h
17
n3o和c
20
h
14
n4, 分别命名为6
‑
(diethylamino)
‑
[2,3'
‑
bipyridin]
‑3‑
ol和3”,2':5',2:6,3”'
‑
quaterpyridine,结构式分别 如下所示:
[0006][0007]
本发明的另一个目的,是提供所述式1和式2化合物的制备方法。
[0008]
所述式1和式2吡啶类新化合物是以无叶假木贼为原料进行提取、分离、纯化得到的。 以无叶假木贼药材为原料,经醇提得粗提物,经乙酸乙酯萃取,再经氯仿萃取,浓缩,得 氯仿萃取物,然后经柱色谱法初步分离,薄层色谱法鉴别收集显色相似的洗脱流出液,洗 脱流出液再经柱色谱法进一步精制、纯化、干燥分别得式1和式2所示化合物。
[0009]
本发明所述式1化合物的制备方法,包括如下步骤:
[0010]
1)醇提:以干燥的无叶假木贼地上部分为原料,用乙醇加热提取,过滤,收集滤液, 减压浓缩至无醇味,得乙醇粗提物,备用;
[0011]
2)萃取:将步骤1)所得乙醇粗提物加蒸馏水加热溶解,制成水混悬液,调节ph, 依次用乙酸乙酯、氯仿进行萃取,浓缩,得氯仿萃取物;
[0012]
3)柱色谱层析:将步骤2)所得氯仿萃取物依次经凝胶层析法及硅胶层析法分离后, 采用石油醚—二乙胺溶剂体系进行梯度洗脱,得目标洗脱液,经薄层色谱进行检测,显色, 合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、fr.4、fr.5、fr.6;
[0013]
4)精制:将步骤3)所得洗脱组分fr.2经色谱法纯化干燥,得式1化合物。
[0014]
优选地,所述步骤1)加入5~15倍量75~95%乙醇加热回流提取1~3次。
[0015]
优选地,步骤2)中水混悬液先调节ph3.0~4.0,乙酸乙酯萃取,过滤,浓缩回收乙酸 乙酯组分;再调节ph8.5~9.5,采用氯仿萃取,过滤收集萃取液,减压浓缩得氯仿萃取物。
[0016]
优选地,乙酸乙酯萃取过程加入乙酸乙酯的量为1~3倍,萃取1~6次。
[0017]
进一步优选地,氯仿萃取过程加入氯仿的量为1~3倍,萃取1~6次。
[0018]
优选地,步骤3)所述的柱色谱层析包括凝胶柱层析和硅胶柱层析。
[0019]
进一步优选地,凝胶柱层析是将步骤2)所得氯仿萃取物经sephadex lh
‑
20凝胶柱层 析,以氯仿和甲醇的混合溶剂为洗脱剂洗脱层析柱,收集流出液,减压浓缩得浓缩液。
[0020]
优选地,步骤3)凝胶柱层析后将浓缩液进一步经硅胶柱层析。
[0021]
优选地,硅胶柱层析采用干法上样,将浓缩液加甲醇溶解后,用40~80目硅胶拌样, 将拌好的样品采用200~300目硅胶柱层析分离。
[0022]
进一步优选地,步骤3)硅胶柱层析以石油醚
‑
二乙胺为洗脱剂进行梯度洗脱。
[0023]
进一步优选地,所述梯度洗脱过程中,洗脱剂为石油醚与二乙胺的体积比为6:1~1:1, 洗脱过程中薄层色谱进行检测,显色相似的洗脱部位合并收集,浓缩,分别得到6段洗脱 组分,分别为fr.1、fr.2、fr.3、fr.4、fr.5、fr.6。
[0024]
需要说明的是,梯度洗脱过程即变换洗脱剂石油醚与二乙胺的体积比,当石油醚:二 乙胺体积比为6:1洗脱硅胶层析柱时,收集流出液得洗脱组分fr.1;当石油醚:二乙胺体积 比为5:1洗脱硅胶层析柱时,收集流出液得洗脱组分fr.2;当石油醚:二乙胺体积比为4:1 洗脱硅胶层析柱时,收集流出液得洗脱组分fr.3;当石油醚:二乙胺体积比为3:1洗脱硅胶 层析柱时,收集流出液得洗脱组分fr.4;当石油醚:二乙胺体积比为2:1洗脱硅胶层析柱时, 收集流出液得洗脱组分fr.5;当石油醚:二乙胺体积比为1:1洗脱硅胶层析柱时,收集流出 液得洗脱组分fr.6。
[0025]
优选地,步骤4)所述的色谱法为sephadex lh
‑
20凝胶柱层析,以甲醇为洗脱剂洗脱, 收集流出液,浓缩后重结晶得式1化合物。
[0026]
进一步优选地,所述的重结晶方法为降温至0~5℃析晶。
[0027]
本发明所得到的式1化合物为黄色结晶,经改良碘化铋钾溶液喷雾显色呈红色斑点, 推断可能为生物碱类化合物。
[0028]
式1化合物经过电喷雾质谱法(esi
‑
ms(positive))、红外光谱(ir)、核磁共振1h
‑
nmr 及
13
c
‑
nmr图谱分析;式1化合物的主要hmbc相关如图1所示,hmbc图谱如图2所 示,1h
‑
nmr图谱和
13
c
‑
nmr图谱如图3
‑
4所示,hmqc图谱如图5所示。
[0029]
结果如下:
[0030]
ei
‑
ms m/z:243(m
+
,80),228(90),214(100),200(93);positive esi
‑
ms m/z:244(m
+ +h),266(m
+
+na)。通过esi
‑
ms图谱综合分析,可以确定该化合物的分子量为243;因 分子量为奇数,又为生物碱类化合物,推测可能含奇数氮;根据1h
‑
nmr、
13
c
‑
nmr、dept 谱分析结果确定其分子式为c
14
h
17
n3o。
[0031]1h
‑
nmr(400mhz,dmso)低场区δ:9.24(1h,s),8.46(1h,d,j=4.8hz),8.40(1h,d, j=8.0hz),7.42(1h,dd,j=4.8,8.0hz),提示有3
‑
取代吡啶结构单元;δ:7.20(1h,d,j=8.4 hz),6.55(1h,d,j=8.4hz),显示有相邻芳香质子;在高场区δ:3.45(4h,q,j=7.2hz), 1.10(6h,t,j=7.2hz),显示有对称乙基结构片段以及羟基氢的信号。
[0032]
13
c
‑
nmr(100mhz,dmso)低场区除了取代吡啶结构信息δ:149.5(d,c
‑
2'),134.4(s, c
‑
3'),135.5(d,c
‑
4'),123.0(d,c
‑
5'),147.8(d,c
‑
6')外,还有5个芳香碳原子信息δ:151.0(s), 142.8(s),136.9(s),127.9(d),106.8(d),提示含有另外一个取代吡啶结构单元。在高场区有 二乙胺基结构信息δ:42.1(t),13.1(q)。
[0033]
从hmbc谱可以看出,9.24(h
‑
2')分别与142.8(c
‑
3)及36.9(c
‑
2)相关,8.40(h
‑
4')与136.9(c
‑
2)相关,由此确定两个吡啶结构单元的连接方式。另外从hmbc图谱可以观察到 7.20(h
‑
4)分别与136.9(c
‑
2)及151.0(c
‑
6)相关,6.55(h
‑
5)与142.8(c
‑
3)相关, 3.45(h
‑1″
)与151.0(c
‑
6)相关,从而确定另外一个吡啶结构单元的取代位置,二乙胺连接 在吡啶的c
‑
6上,羟基连接在c
‑
3上。
[0034]
由此,化合物1的结构最终确定为式1所示,系统命名为6
‑
(diethylamino)
‑
[2,3'
‑
bipyridin]
ꢀ‑3‑
ol。
[0035]
氢谱(400mhz,dmso
‑
d6)和碳谱(100mhz,dmso)数据见表1。
[0036]
表1式1化合物的氢谱(400mhz,dmso)和碳谱(100mhz)数据
[0037][0038]
本发明还提供一种所述式2化合物的制备方法。
[0039]
一种式2所述化合物的制备方法包括如下步骤:
[0040]
1)醇提:以干燥的无叶假木贼地上部分为原料,用乙醇加热提取,过滤,收集滤液, 减压浓缩至无醇味,得乙醇粗提物,备用;
[0041]
2)萃取:将步骤1)所得乙醇粗提物加蒸馏水加热溶解,制成水悬液,调节ph,依 次用乙酸乙酯、氯仿进行萃取,浓缩,得氯仿萃取物;
[0042]
3)柱色谱层析:将步骤2)所得氯仿萃取物依次经凝胶层析法及硅胶层析法分离后, 采用石油醚—二乙胺溶剂体系进行梯度洗脱,得目标洗脱液,经薄层色谱进行检测,显
核磁共振1h
‑
nmr及
13
c
‑
nmr图谱的分析,式2化合物的主要hmbc相关如图6所示, hmbc图谱如图7所示,1h
‑
nmr图谱和
13
c
‑
nmr图谱如图8
‑
9所示,hmqc图谱如图 10所示。
[0063]
结果如下:
[0064]
ei
‑
ms m/z:310(m
+
,100);positive esi
‑
ms m/z:311(m
+
+h);推断其分子式为 c
20
h
14
n4。
[0065]
13
c
‑
nmr谱给出20个碳信号峰,结合dept谱将它们归属为:14个叔碳和6个季碳, 由ei
‑
ms中的碎片峰以及质荷比接近吡啶的裂解方式m
‑
chn和m
‑
chn
‑
h(分析化学手册 编辑委员会,2000),推测化合物的结构中有四个吡啶环。
[0066]
通过1h
‑
nmr(400mhz,cdcl3)以及1h
‑1hcosy谱分析,低场区显示有两个3
‑
取代 吡啶结构单元,分别是:δ9.35(1h,d,j=2.0hz,h
‑
2”),8.69(1h,m,h
‑
6”),8.42(1h,m, h
‑
4”),7.46(1h,m,h
‑
5”)和δ9.29(1h,d,j=1.6hz,h
‑
2”'),8.69(1h,m,h
‑
6”'),8.49(1h, m,h
‑
4”'),7.46(1h,m,h
‑
5”');一个2,5
‑
二取代吡啶结构单元:δ9.44(1h,d,j=2.0hz,h
‑6’
), 8.59(1h,dd,j=2.0,8.4hz,h
‑
4'),7.92(1h,d,j=8.4hz,h
‑
3');一个2,6
‑
二取代吡啶结构单 元:δ7.94(1h,t,j=8.0hz,h
‑
4),7.84(1h,d,j=8.0hz,h
‑
3),7.80(1h,d,j=8.0hz,h
‑
5)。
[0067]
从hmbc图谱可以观察到,9.29(h
‑
2”'),8.49(h
‑
4”')分别与154.6(c
‑
6)相关,9.44 (h
‑
6'),8.59(h
‑
4'),8.42(h
‑
4”)分别与154.8(c
‑
2')相关,8.59(h
‑
3')与134.3(c
‑
3”)相 关,7.84(h
‑
3)分别与133.4(c
‑
5')和154.1(c
‑
2)相关,从而确定四个吡啶环连接顺序。 通过hmqc谱将c和h的关系进行了归属。
[0068]
由此,化合物2的结构最终确定为式2所示,系统命名为3”,2':5',2:6,3”'
‑
quaterpyridine。
[0069]1h nmr(400mhz,cdcl3)和
13
c nmr(100mhz,cdcl3)数据见表2。
[0070]
表2式2化合物的氢谱(400mhz,cdcl3)和碳谱(100mhz)数据
[0071][0072][0073]
本发明的第三个目的,是提供所述式1和式2化合物在杀菌、抗菌、抑菌药物中的应 用。
[0074]
优选地,所述的药物为抑制植物细菌、真菌的药物或制剂。
水乙醇)、硫酸链霉素阳性对照组(简称“阳性对照组”)、式1化合物组、式2化合物组, 每个处理组设定4个平行孔;测定ic
50
时,式1和式2化合物分别检测,分别设置空白对 照组(h2o)、溶剂对照组(无水乙醇)、阳性对照组(硫酸链霉素)、200.0μg/ml组、100.0 μg/ml组、50.0μg/ml组、20.0μg/ml组、10.0μg/ml组共8个分组,每组设定4个平行 孔。
[0092]
2.3.2加样:取清洁无菌的96孔培养板,每孔分别加入90μl 106cfu/ml的菌液,然 后每孔分别加入10μl的测试样品液;将96孔培养板四周用封口膜封好以减少水分挥发, 缓慢振荡(12rpm),28℃暗培养24h;每孔中加入5mg/ml 10μl mtt溶液,28℃下继 续振荡培养4h;
[0093]
2.3.3结果观察:
[0094]
测定mic时,离心(1500g,20min)后,弃去上清液,每孔中加入150μl二甲基亚 砜(dmso),振荡30min,以终止反应并溶解蓝色沉淀物质。
[0095]
测定ic
50
时,离心(1500g,20min)后,将上清液转移到另一个新的96孔板中,置 酶标仪590nm处测定吸光值(od
590nm
),按下式计算单体化合物的抑制百分率。
[0096][0097]
3检测指标及方法
[0098]
3.1最低抑制浓度(mic) 采用mtt比色法测定2个单体化合物对4种靶标细菌的 最低抑制浓度(mic)。肉眼观测,没有生成蓝紫色沉淀的化合物最低浓度为最低抑制浓度 (mic)。
[0099]
3.2抑制中浓度(ic
50
) 根据mic的测试结果,将化合物配制成一系列的浓度(200.0 μg/ml、100.0μg/ml、50.0μg/ml、20.0μg/ml、10.0μg/ml),测定单体化合物对靶标细菌 的抑制中浓度(ic
50
),对抗细菌活性作进一步筛选。根据单体化合物在590nm处测定的吸 光值(od
590nm
),计算各个浓度单体化合物对靶标细菌的抑制率。
[0100]
实验结果采用microsoft excel软件进行数据整理。在分析中,供试样品浓度取对数(x), 抑菌率换算成生物统计几率值(y)(华南农业大学,1987),求得相关毒力回归方程 (y=ax+b);由“反应率
‑
几率值转换值表”可以查得,当抑制率几率值y=5时,所对应的 抑制率为50%,此时可以通过回归方程求出x的值,再求x值的反对数,得出对应的浓度 值,此浓度值即为抑制中浓度(ic
50
)。
[0101]
4实验结果及结论
[0102]
单体化合物对细菌的最低抑制浓度(mic)测定结果见表3,单体化合物对细菌的抑制 中浓度(ic
50
)测定结果见表4。
[0103]
根据抑制中浓度(ic
50
)的测定结果可知,式1化合物对枯草芽孢杆菌、番茄疮痂病菌、 根癌土壤杆菌、黄瓜角斑病菌的ic
50
为132.10μg/ml、69.57μg/ml、65.60μg/ml、54.50 μg/ml;式2化合物对枯草芽孢杆菌、番茄疮痂病菌、根癌土壤杆菌、黄瓜角斑病菌的ic
50
为65.84μg/ml、68.03μg/ml、42.95μg/ml、75.65μg/ml。
[0104]
以上实验结果表明,式1化合物和式2化合物对多种细菌有抑制作用,有确切的抗菌 效果;其中式1化合物对黄瓜角斑病菌抑制效果最显著,式2化合物对根癌土壤杆菌抑制 效果最显著。
[0105]
表3式1化合物和式2化合物对植物致病菌的最低抑制浓度
[0106][0107][0108]
表4式1化合物和式2化合物对致病菌的抑制中浓度
[0109][0110]
实验例2式1化合物和式2化合物的抗真菌活性测定
[0111]
1对白色念珠菌的抑制作用
[0112]
实验方法同实验例1。以多菌灵为阳性对照药,先采用mtt比色法测定式1化合物、 式2化合物对白色念珠菌(c.albicans)的最低抑制浓度(mic);根据mic的测定结果, 将化合物配制成一系列浓度梯度的溶液,进一步测定两种新化合物对白色念珠菌的抑制中 浓度(ic
50
)。
[0113]
结果如表5所示,式1和式2化合物对白色念珠菌均有抑制作用,ic
50
分别为91.52 μg/ml、82.66μg/ml,其中式2化合物对白色念珠菌的抑制作用较强。
[0114]
表5单体化合物对白色念珠菌的抑制作用
[0115][0116]
2对稻瘟菌孢子萌发的抑制作用
[0117]
2.1化合物抑制稻瘟菌孢子萌发的活性初步筛选
[0118]
以多菌灵为阳性对照药,采用孢子萌发法初步筛选式1化合物和式2化合物对稻瘟菌 (m.oryzae)孢子萌发的抑制作用,化合物的检测浓度为500μg/ml、250μg/ml、125μg/ml、 62.5μg/ml,通过此实验初步了解两种化合物对稻瘟菌孢子萌发抑制活性的强弱。结果表 明,在250μg/ml浓度下,式1化合物和式2化合物对稻瘟菌孢子萌发的抑制率分别为 58.5%、36.5%,具有一定的抑菌活性。
[0119]
2.2化合物对稻瘟菌孢子萌发的抑制中浓度
[0120]
确定式1化合物和式2化合物的浓度范围,进一步采用悬浮培养法测定式1化合物 和式2化合物对稻瘟菌孢子萌发的抑制中浓度(ic
50
),结果见表6。式1化合物和式2 化合物对稻瘟菌孢子萌发的抑制中浓度(ic
50
)分别为229.19μg/ml、278.48μg/ml,表 明两种新化合物对稻瘟菌孢子萌发有明显的抑制作用,其中式1化合物的抑制效果更好。
[0121]
表6化合物对稻瘟菌孢子萌发的抑制中浓度
[0122][0123]
与现有技术相比,本发明具有以下有益效果:
[0124]
本发明首次发现并提取分离出两种无叶假木贼吡啶类新化合物,其分离及药理研究未 被现有论文期刊所报道;本发明提供该两种新化合物的制备方法,该制备方法依次进行提 取、萃取、柱色谱层析及精制等步骤,简便快速,成本低廉,所得新化合物纯度高;经研 究表明,本发明所得两种无叶假木贼吡啶类新化合物具有抗菌活性,可用于制备抗菌药物。
[0125]
说明书附图
[0126]
图1:式1化合物的主要hmbc相关;
[0127]
图2:式1化合物的hmbc图谱;
[0128]
图3:式1化合物的1h
‑
nmr图谱;
[0129]
图4:式1化合物的13c
‑
nmr图谱;
[0130]
图5:式1化合物的hmqc图谱;
[0131]
图6:式2化合物的主要hmbc相关;
[0132]
图7:式2化合物的hmbc图谱;
[0133]
图8:式2化合物的1h
‑
nmr图谱;
[0134]
图9:式2化合物的13c
‑
nmr图谱;
[0135]
图10:式2化合物的hmqc图谱。
具体实施方式
[0136]
下通过具体实施例进一步说明本发明,但本领域相关技术人员理应知晓,所述实施例 并不以任何方式限制本发明。
[0137]
实施例1式1、式2化合物的制备
[0138]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用8倍量80%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0139]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.5,乙酸乙酯萃取4次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至9.0,氯仿萃取4次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物118.5g。
[0140]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,体积比为1:1的甲醇和氯仿洗脱收集得到生物总碱;将所得生物总碱加 适量甲醇溶解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采 用石油醚
‑
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄 层色谱进行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、 fr.3、fr.4、fr.5、fr.6;
[0141]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析纯化,甲醇洗脱, 洗脱流出液浓缩后降温0~5℃重结晶,得黄色结晶43.2mg即为式1化合物;步骤3)所得 洗脱组分fr.3经sephadex lh
‑
20凝胶柱层析,以体积比为1:1的氯仿:甲醇为洗脱剂洗脱, 再经反相硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30, ods柱,检测波长为254nm)分离纯化,得到淡黄色油状物27.6mg即为式2化合物。
[0142]
实施例2式1、式2化合物的制备
[0143]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用10倍量85%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0144]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.0,乙酸乙酯萃取3次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至8.5,氯仿萃取3次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物143.5g。
[0145]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,体积比氯仿:甲醇=1:1洗脱收集得到生物总碱组分;将所得生物总碱加适 量甲醇溶解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分
离。采用 石油醚
‑
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄层 色谱进行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、 fr.4、fr.5、fr.6;
[0146]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得45mg黄色晶体即为式1化合物;步骤3)所得洗脱组 分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反相硅 胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30,ods柱,检 测波长为254nm)分离纯化,得25mg淡黄色油状物即为式2化合物。
[0147]
实施例3式1、式2化合物的制备
[0148]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用9倍量90%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0149]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至4.0,乙酸乙酯萃取4次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至9.5,氯仿萃取3次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物140.2g。
[0150]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,氯仿甲醇体积比1:1进行洗脱得到生物总碱;将所得生物总碱加适量甲 醇溶解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采用石油 醚
‑
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄层色谱 进行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、 fr.4、fr.5、fr.6;
[0151]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得43mg黄色晶体即为式1化合物;步骤3)所得洗脱组 分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反相硅 胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=50:50,ods柱,检 测波长为254nm)分离纯化,得24.2mg淡黄色油状物即为式2化合物。
[0152]
实施例4式1、式2化合物的制备
[0153]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用9倍量85%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0154]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.7,乙酸乙酯萃取5次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至9.2,氯仿萃取6次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物151.6g。
[0155]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,以体积比氯仿:甲醇=2:1洗脱收集得到生物总碱;将所得生物总碱加适 量甲醇溶解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采用 石油醚
‑
二乙胺溶剂体系(石油醚:二乙胺=6:1
‑
1:1)进行梯度洗脱,得目标洗脱液,经薄层 色谱进行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、 fr.4、fr.5、fr.6;
硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=60:40,ods柱, 检测波长为254nm)分离纯化,得21.7mg淡黄色油状物即为式2化合物。
[0167]
实施例7式1、式2化合物的制备
[0168]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用10倍量87%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0169]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.7,乙酸乙酯萃取1次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至9.1,氯仿萃取5次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物127g。
[0170]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,体积比氯仿:甲醇=1:1洗脱得到生物总碱;将所得生物总碱加适量甲醇溶 解,用40
‑
80目硅胶拌样,将拌好的样品采用200
‑
300目硅胶柱层析分离。采用石油醚
‑
二 乙胺溶剂体系(石油醚:二乙胺=6:1
‑
1:1)进行梯度洗脱,得目标洗脱液,经薄层色谱进行 检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、fr.4、 fr.5、fr.6;
[0171]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得41.5mg黄色晶体即为式1化合物;步骤3)所得洗脱 组分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反相 硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30,ods柱, 检测波长为254nm)分离纯化,得23.2mg淡黄色油状物即为式2化合物。
[0172]
实施例8式1、式2化合物的制备
[0173]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用15倍量75%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0174]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.3,乙酸乙酯萃取5次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至8.6,氯仿萃取2次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物123.5g。
[0175]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,体积比氯仿:甲醇=1:1洗脱得到生物总碱;将所得生物总碱加适量甲醇溶 解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采用石油醚
‑ꢀ
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄层色谱进 行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、fr.4、 fr.5、fr.6;
[0176]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得40.5mg黄色晶体即为式1化合物;步骤3)所得洗脱 组分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反 相硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30,ods柱, 检测波长为254nm)分离纯化,得22.1mg淡黄色油状物即为式2化合物。
[0177]
实施例9式1、式2化合物的制备
[0178]
1)醇提:以13kg干燥的无叶假木贼地上部分为原料,用8倍量95%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0179]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.8,乙酸乙酯萃取2次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至9.3,氯仿萃取6次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物129.7g。
[0180]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,体积比氯仿:甲醇=1:1洗脱得到生物总碱;将所得生物总碱加适量甲醇溶 解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采用石油醚
‑ꢀ
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄层色谱进 行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、fr.4、 fr.5、fr.6;
[0181]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得40.5mg黄色晶体即为式1化合物;步骤3)所得洗脱 组分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反相 硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30,ods柱, 检测波长为254nm)分离纯化,得20.8mg淡黄色油状物即为式2化合物。
[0182]
实施例10式1、式2化合物的制备
[0183]
1)提取:以13kg干燥的无叶假木贼地上部分为原料,用12倍量80%乙醇加热提取3 次,过滤,合并滤液,滤液减压浓缩至无醇味,得乙醇粗提物,备用;
[0184]
2)萃取:将步骤1)所得乙醇粗提物加适量蒸馏水加热溶解,制成水混悬液,水混悬 液先用10%的盐酸调节ph值至3.4,乙酸乙酯萃取6次,过滤,浓缩回收乙酸乙酯组分; 合并滤液,用10%的氨水调节ph值至8.9,氯仿萃取1次,过滤,合并萃取液,减压浓缩 至浸膏,得到氯仿萃取物131.1g。
[0185]
3)柱色谱层析:将步骤2)所得氯仿萃取物加适量甲醇和氯仿溶解,经sephadex lh
‑
20 凝胶柱层析分离,以体积比氯仿:甲醇=1:1洗脱得到生物总碱;将所得生物总碱加适量甲醇 溶解,用40~80目硅胶拌样,将拌好的样品采用200~300目硅胶柱层析分离。采用石油醚
‑ꢀ
二乙胺溶剂体系(石油醚:二乙胺=6:1~1:1)进行梯度洗脱,得目标洗脱液,经薄层色谱进 行检测,显色,合并显色相似的洗脱部位,浓缩,得到6段洗脱组分fr.1、fr.2、fr.3、fr.4、 fr.5、fr.6;
[0186]
4)精制:步骤3)所得洗脱组分fr.2经sephadex lh
‑
20凝胶柱层析,甲醇洗脱收集流 出液,浓缩后降温0~5℃重结晶,得39.5mg黄色晶体即为式1化合物;步骤3)所得洗脱 组分fr.3经sephadex lh
‑
20凝胶柱层析、氯仿和甲醇体积比为1:1进行洗脱后,再经反相 硅胶(rp
‑
18)柱层析和半制备型高效液相色谱仪(pre
‑
hplc,甲醇:水=70:30,ods柱, 检测波长为254nm)分离纯化,得22.3mg淡黄色油状物即为式2化合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1