一组与小麦穗粒数显著关联的SNP位点及其在小麦遗传育种中的应用
一组与小麦穗粒数显著关联的snp位点及其在小麦遗传育种中的应用
技术领域
1.本发明涉及植物分子标记技术领域,具体涉及一组与小麦穗粒数相关基因位点显著关联的snp及其在小麦穗粒数的遗传及分子育种中的应用。
背景技术:
2.小麦是重要的粮食作物之一,不断选育高产新品种,持续提高小麦产量是国内外小麦育种的重要目标。小麦产量主要由单位面积穗数、穗粒数和粒重三要素构成,受品种遗传特性和环境条件的影响,穗粒数的变异幅度要大于单位面积穗数和粒重,因此在协调单位面积穗数、穗粒数、粒重三要素的基础上加强对穗粒数的选择具有较大的增产潜力。
3.穗粒数是小麦高产育种的一个重要选择指标。但穗粒数是复杂的数量性状,由多基因控制且易受环境条件影响,限制了通过表型选择育种对穗粒数进行改良的效果。全基因组选择(genomic selection,gs)育种是一种根据标记信息估计个体育种值进行选择育种的新方法,基本流程是利用覆盖整个基因组的分子标记,通过训练群体的标记基因型和表型构建预测模型,然后利用育种群体中个体所携带的标记信息对其未知的表型进行预测,即估计基因组育种值并进行选择。全基因组选择具有早代个体选择准确性高的优点,在提高小麦穗粒数改良的育种效率和遗传进度中具有重要应用价值。
4.小麦穗粒数的遗传研究对了解小麦穗粒数的遗传发育、基因作图与克隆,以及全基因组选择育种利用具有非常重要的作用。数量性状遗传位点(quantitative trait locus/loci,qtl)定位是剖析复杂数量性状遗传基础的常用方法。目前,利用双亲本杂交衍生的各种类型的遗传分离群体进行连锁分析或利用自然群体进行全基因组关联分析,在小麦21条染色体上都报道了结实性、穗粒数相关的qtl。左煜昕等(2020)对控制穗粒数的qtl进行了元分析,在10条染色体上鉴定到35个稳定一致的qtl。但是qtl定位区间往往较大,标记与目标qtl连锁不够紧密,应用于标记辅助选择育种效果较差,而且qtl定位利用的多是传统的分子标记,存在通量低、数量少、操作过程繁琐等局限,难以在小麦穗粒数qtl的分子标记辅助选择育种中广泛应用。
5.因此,鉴定与穗粒数qtl紧密连锁且易于进行高通量检测的分子标记是提高标记辅助选择育种效率的关键。相对于传统的分子标记,snp标记具有变异丰富、二态性、易于高通量自动化检测的特点,是功能基因组与遗传育种研究中最具有应用前途的分子标记技术。目前,常用的高通量snp位点鉴定技术主要有基因组测序和snp芯片两种。通过二代或三代测序能够获得高密度snp位点,但是对于具有庞大基因组的小麦(16g)而言成本较高,而且测序数据的处理、序列比对、基因分型等过程对数据分析的要求较高,需要有专业生物信息学背景的人员才能完成。
技术实现要素:
6.利用snp芯片进行基因分型,数据处理较为简单,更易于为育种家掌握和利用。基
于单个snp位点转化而成的kasp(kompetitive allele specific pcr)标记,即竞争性等位基因特异性pcr技术,能够对单个snp位点进行高通量的精准基因分型,具有高度稳定性和准确性、低成本、通量高的特点,可广泛应用于基因定位、精细作图、克隆以及大规模育种材料的高通量筛选,因此在遗传研究和分子标记辅助育种、基因组选择育种中具有重要应用价值。
7.针对现有技术存在的不足,本发明利用小麦中较为成熟的简化基因组测序(genotyping
‑
by
‑
sequencing,gbs)技术,对768份小麦材料进行大规模的简化基因组测序,并与小麦参考基因组进行序列比对,获得了全基因组高密度的snp位点;通过多点、多年表型性状鉴定和全基因组关联分析,鉴定出与穗粒数显著关联的snp位点,可广泛应用于snp芯片制备、kasp标记的开发、基因定位、标记辅助选择与全基因组选择,服务于小麦穗粒数的遗传研究和分子育种。
8.本发明提供了一套与小麦穗粒数显著关联的snp位点,可应用于小麦穗粒数的标记辅助选择和全基因组选择育种,并可用于开发单个kasp标记和snp芯片,方便在遗传和育种群体中利用。可广泛应用于小麦穗粒数相关基因的定位,精细作图与克隆,育种中的单个、多个控制穗粒数qtl的标记辅助选择、聚合及全基因组选择。
9.本发明所采用的技术方案是:
10.本发明提供了一组(50个)与小麦穗粒数显著关联的单核苷酸多态性位点(snp),包括snp侧翼序列、snp位点信息及碱基突变信息,这些snp位于普通小麦17条不同染色体上。
11.本发明提供的一组与小麦穗粒数显著关联的snp位点,包括50个snp位点,编号分别为snp01~snp50,它们的信息如下:
12.13.[0014][0015]
表中物理位置以中国春基因组iwgsc reference genome v1.1(iwgsc,2018)为参考序列;
[0016]
表中所列序列见序列表seq id no.1~seq id no.100。snp01~snp50每个snp为2个序列,即原始序列和变异位点各一条序列。
[0017]
本发明提供的一组与小麦穗粒数显著关联的snp位点及其碱基突变类型可以在小麦穗粒数qtl鉴定中应用。
[0018]
本发明提供的一组与小麦穗粒数显著关联的snp位点及其碱基突变类型可以在制备单个可检测的kasp标记或snp基因芯片中应用。
[0019]
本发明提供的一组与小麦显著关联的snp位点及其碱基突变类型可以在制备与穗粒数基因位点qgn4b.1连锁的kasp标记k4b28740074中应用。
[0020]
本发明提供的一组与小麦穗粒数显著关联的snp位点可以在小麦穗粒数分子标记辅助选择和全基因组选择检测方法中应用。
[0021]
检测方法中的应用,我们可以根据snp位点设计kasp引物,根据包含snp位点上下游各50bp的dna短序列设计kasp引物。具体利用网站polymarker(http://www.polymarker.info/)进行引物设计,采用网站默认的参数设置。引物前加接头,fam序列为“gaaggtgaccaagttcatgct”,hex序列为“gaaggtcggagtcaacggatt”。
[0022]
引物设计合成后,可以利用分离群体或者自然群体进行有效性检测,验证通过gwas鉴定到的qtl是否存在,其使用方法分别如下:
[0023]
(1)以小麦dna为pcr扩增模板,以设计合成的kasp引物,进行pcr扩增,反应体系为6μl。上述反应体系具体包括:20
‑
50ng/μl的dna 3μl,2
×
kasp master mix 3μl,kasp assay mix(上下游引物混合液)0.0825μl。置于384孔pcr仪扩增。
[0024]
(2)pcr扩增程序为:94℃预变性15min;94℃变性20s,65
‑
57℃复性60s(每循环降低0.8℃),10个循环;94℃变性20s,57℃复性60s,30个循环;10℃保存;
[0025]
(3)pcr结束后,置于omega snp分型仪检测pcr分型结果;
[0026]
(4)分析鉴定,根据分型结果分析基因型。
[0027]
本研究与现有技术相比有以下优点:
[0028]
(1)本发明鉴定了一组(50个)与穗粒数紧密连锁的显著关联snp,基本涵盖了目前在中国小麦种质中的穗粒数相关数量性状基因位点。
[0029]
(2)这些snp可进一步转化为单个可检测的snp标记(如kasp标记),用于穗粒数相关基因的定位、精细作图、克隆及大规模应用于育种材料的高通量分子标记辅助选择,提高分子育种的效率。
[0030]
(3)这些snp也可制作成基因芯片,应用于对育种材料进行穗粒数的分子标记辅助选择和全基因组选择,进一步提高分子育种的效率和准确性。
[0031]
(4)开发了与穗粒数基因位点qgn4b.1连锁的kasp标记k4b28740074不但证明我们鉴定到的snp是非常有效的,也为qgn4b.1的标记辅助选择提供了高效的检测标记。
附图说明
[0032]
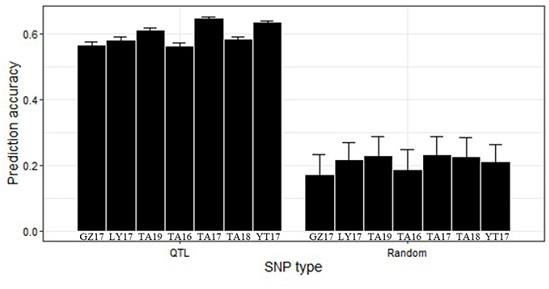
图1是利用标记预测穗粒数的准确度。左侧表示利用qtl关联的snp标记进行预测的结果,右侧表示全基因组随机筛选相同数目的snp位点进行预测的结果。gz17,ly17,ta19,ta16,ta17,ta18,yt17分别表示不同的环境。
具体实施方式
[0033]
实施例1通过简化基因组测序鉴定snp
[0034]
1.1测序材料
[0035]
选取了768份小麦品种(系)用于简化基因组测序,鉴定snp。这些材料主要是来自我国小麦主产区包括黄淮麦区、北方冬麦区、长江中下游麦区和西南麦区的小麦品种和优良品系。
[0036]
1.2研究方法
[0037]
1.2.1小麦基因组dna提取
[0038]
利用幼苗叶片提供基因组dna。dna提取采用改进的ctab(cetyl trimethyl ammonium bromide)法(stewart and via,1993)。具体操作:取小麦幼嫩叶片于2ml离心管中,利用液氮冷冻并于组织粉碎器上磨成粉末;(b)加入800μl ctab于2ml管中后,置于65℃水浴90min,水浴期间轻晃5
‑
8次,使dna充分裂解;(c)加入800μl氯仿异戊醇(体积比24:1)轻晃10min;(d)12000rpm条件下离心10min后取600μl上清液置于新的2ml管中(注意对应序号);(e)加入60μl 3m醋酸钠(ph=5.2)和600μl异丙醇(提前
‑
20℃冷冻),轻晃混匀,即可看
到白色dna絮状物产生,放于
‑
20℃冰箱1h以增加dna产量。(f)12000rpm离心10min后倒掉上清液,将沉淀用70%乙醇(提前放入
‑
20℃冰箱冷冻)清洗2
‑
3次,通风橱中静置风干;(g)加入200μl ddh2o溶解dna。
[0039]
1.2.2 dna样品质量检测
[0040]
用质量分数为1%的琼脂糖凝胶电泳检测,用凝胶成像系统查看电泳结果,保证基因组dna的完整性。基因组dna a260/280比值应在1.8
–
2.0之间,a260/230比值应在1.8
‑
2.2之间。将dna稀释到工作浓度20ng/ul。
‑
20℃保存备用。
[0041]
1.2.3 dna文库构建和gbs测序
[0042]
参照poland et al.(2012b)进行gbs dna文库的构建。基因组dna利用两个限制性内切酶psti和mspi(new england biolabs,inc.,ipswich,ma,united states)进行消化。利用t4(new england biolabs,inc.,ipswich,ma,united states)连接酶将条码序列连接到消化好的dna片段上。将每个平板的所有产物混合并使用qiaq快速pcr纯化试剂盒(qiagen,inc.,valencia,ca,united states)进行纯化。利用与条码序列互补的引物进行pcr扩增。pcr产物再次使用qiaquick pcr纯化试剂盒进行纯化,并利用qubit
tm
双链dna高灵敏度荧光定量试剂盒(life technologies,inc.,grand island,ny,united states)测定浓度。利用琼脂糖凝胶电泳(life technologies,inc.,grand island,ny,united states)筛选200
‑
300大小的dna片段,qubit 2.0荧光剂和qubit
tm
双链dna高灵敏度荧光定量试剂盒估计每个dna文库的浓度。使用ion chef instrument(ion pi hi
‑
q chef kit)将经片段大小筛选过的dna文库利用加载到p1v3芯片上并利用ion proton sequencer(life technologies,inc.,grand island,ny,united states,software version 5.10.1)进行序列测定。这个ion torrent系统可以产生各种读长的序列。
[0043]
1.2.4 snp位点鉴定
[0044]
对测序产生的序列,在其3’端加上80个poly
‑
a碱基,然后利用tassel 5.0进行序列分析,这样trait analysis by association,evolution and linkage(tassel)pipeline 5.0(tassel 5.0)(bradbury et al.,2007)就可以处理短于64碱基的序列而不仅仅是丢弃这些短序列。以中国春基因组iwgsc reference genome v1.1(iwgsc,2018)为参考序列,利用tassel 5.0(bradbury et al.,2007)进行序列比对鉴定snp位点。所有的参数都采用tassel 5.0的默认设置。共获得432,588个snp位点,覆盖全基因组约14gb,标记之间的平均距离为34.0kb。其中a基因组上150784个位点,平均间距32.9kb;b基因组182192个位点,平均间距28.9kb;d基因组99612个位点,平均间距40.3kb。各染色体上snp标记的数目10177到31149,标记间距变异范围是26.1
–
50.1kb。这些snp主要位于基因之间的区域,有364203个,占84.1,其次是cds区,39901个(9.2%)、内含子区22215个(5.1%)、5’utr区3543个(0.8%)和3’utr区3300个(0.8%)。
[0045]
表1.gbs鉴定的snp位点在小麦基因组中的分布
[0046][0047]
实施例2小麦穗粒数数量性状基因位点的全基因组鉴定
[0048]
2.1材料
[0049]
用于gbs技术鉴定基因型的768份小麦材料,同实施例1。
[0050]
2.2方法
[0051]
2.2.1穗粒数的鉴定
[0052]
为鉴定这些材料的穗粒数(grain number,gn),将这768份小麦材料于2017
‑
2018(ta17)、2018
‑
2019(ta18)、和2019
‑
2020(ta19)小麦生长年度种植于泰安山东农业大学实验站,并于2017
‑
2018年度,在烟台、洛阳、贵阳也进行了种植。每个材料种植1行,行长3米,行距25厘米,每行播种50粒饱满的种子,设置两次重复,常规大田管理。
[0053]
2.2.2全基因组关联分析
[0054]
对所有鉴定到的432588个snp位点,进一步筛选最小等位基因频率(maf)大于0.01,缺失率小于80%的snp位点,得到到的327609个snp。对穗粒数进行全基因组关联分析。采用gapit v.3程序包进行,它使用emma,压缩的混合线性模型(compressed mixed linear model,cmlm)和population parameters previously determined(p3d)提高gwas运行的效率。利用emma算法分析kinship,使用前3个主成分控制群体结构。显著性阈值设定为1.0
×
10
‑5。
[0055]
2.3结果
[0056]
2.3.1穗粒数的全基因组关联分析
[0057]
通过对多个环境下测定的穗粒数进行gwas,共鉴定到99个关联snp位点(marker trait association,mta),根据ld将其合并为49个穗粒数qtl。其中有38个qtl被定位在
1.0mb区间内,各含有不到10个注释基因。说明利用我们通过gbs鉴定到的高密度snp标记进行gwas能够将qtl定位在较窄的物理区间内,极大的方便了后续qtl的精细定位、基因克隆以及分子标记辅助育种。
[0058]
2.3.2鉴定出的与穗粒数关联的snp
[0059]
对通过gwas鉴定到的snp,每一个位点选取各环境下最显著关联的一个snp,共获得50个与穗粒数显著关联的snp位点,这些位点的信息见表2。表中“qtl”一列,表明snp连锁的qtl名称;表中“snp所在染色体及物理位置”一列,表明snp染色体所在的染色体和物理位置,物理位置参照以中国春基因组iwgsc reference genome v1.1(iwgsc,2018)为参考序列;表中“序列与snp变异位点”一列,“[]”中碱基表明变异的位点,有的位点仅有一个碱基,表示变异后缺失此碱基。
[0060]
表2与小麦穗粒数显著关联的snp位点信息
[0061]
[0062][0063]
实施例3小麦穗粒数qtl在基因组预测中的应用
[0064]
3.1材料
[0065]
同实施例1。
[0066]
3.2方法
[0067]
3.2.1穗粒数表型鉴定
[0068]
同实施例1。
[0069]
3.2.2预测模型构建
[0070]
采用r软件中的rrblup方法分别构建全基因组选择模型。利用鉴定到的50个显著
snp标记构建预测模型,以评估qtl的预测能力。同时,从全基因组中抽取相同数目的随机snp位点构建预测模型,评估随机标记的预测效果。
[0071]
3.2.3预测模型的交叉验证
[0072]
利用5折的交叉验证分析模型的预测准确性。即将整个群体随机分成5份,取其中的1份作为育种群体,另外4份作为训练群体。利用训练群体的表型和基因型估计标记效应值,根据育种群体的基因型预测育种群体的表型,计算预测表型和实际测量表型的相关性作为模型预测的准确性,重复进行100次,取平均值。
[0073]
3.3结果
[0074]
分别利用gz17,ly17,ta19,ta16,ta17,ta18,yt17等7个环境或年份下的数据构建了qtl预测模型以及随机snp位点的预测模型。利用鉴定到的qtl进行预测,各环境下的预测准确性为~0.60,显著的高于随机snp位点的预测准确性(各环境下的准确性仅为~0.2)(图1)。说明鉴定到的这些qtl及其连锁的snp标记对穗粒数具有较高的预测能力,可以应用于穗粒的标记辅助选择和全基因组选择育种。
[0075]
图1.利用标记预测穗粒数的准确度。左侧表示利用qtl关联的snp标记进行预测的结果,右侧表示全基因组随机筛选相同数目的snp位点进行预测的结果。gz17、ly17、ta19、ta16、ta17、ta18、yt17分别表示不同的环境。
[0076]
实施例4小麦穗粒数基因qgn4b.1的验证与作图
[0077]
4.1材料
[0078]
研究材料包括多花多实亲本烟农19、和亲本阳光5号及二者杂交杂交构建的f2分离群体,共有156个f2单株。
[0079]
4.2方法
[0080]
4.2.1分离群体穗粒数表型鉴定
[0081]
同实施例1。
[0082]
4.2.2 dna提取
[0083]
同实施例1。
[0084]
4.2.3 kasp引物设计、稀释:
[0085]
根据gwas分析结果,将与穗粒数qtl qgn4b.1显著关联的一个gbs
‑
snp(s4b28740074)根据snp位点信息设计了一组(三条)kasp引物k4b28740074。fam
‑
r引物序列cacgtacagtacgcgcaatggt,hex
‑
s引物序列acgtacagtacgcgcaatggc,反向引物序列cccgatcgtacaggcgcaggta。将三条引物用超纯水稀释到100μm后按照体积比正向引物
‑
fam
‑
r:正向引物
‑
hex
‑
s:反向引物:超纯水=6:6:15:23的比例进行混合后,保存至
‑
20℃备用。
[0086]
4.2.4 kasp pcr扩增体系和程序如下:
[0087]
kasp反应体系采用6μl体系:
[0088]
依下表在冰上配制体系,模板dna 3μl(20ng/μl左右),2
×
master mix3μl(lgc group uk),kasp化验引物0.0825μl(上海生工生物公司合成)。
[0089]
表3 kasp反应体系
[0090][0091]
pcr程序如下:
[0092]
1. 94℃预变性5min;
[0093]
2. 94℃变性20s;
[0094]
3. 65℃退火30s(每个循环降低0.8℃),步骤2
‑
3循环10次。
[0095]
4. 94℃变性20s;
[0096]
5. 57℃退火30s,步骤4
‑
5循环35次。
[0097]
6. 10℃保存。信号检测。
[0098]
4.3结果
[0099]
4.3.1分离群体中穗粒数的变异
[0100]
gwas分析结果表明,qgn4b.1是一个控制小麦穗粒数的qtl,位于4bs上。阳光5号与烟农19在这两个位点间存在多态性,烟农19的穗粒数显著高于阳光5号。为进一步验证这个qtl并开发与其连锁的kasp标记,利用阳光5号与烟农19杂交构建了f2分离群体,并自交至f4。
[0101]
对该群体进行穗粒数调查表明,f4代植株的穗粒数出现广泛的变异和明显的超亲分离的分离。
[0102]
4.3.2分离群体中穗粒数与标记基因型的关系
[0103]
将与该qtl关联最显著的一个gbs
‑
snp(s4b28740074)转换成kasp标记后(k4b28740074),对该群体进行基因分型,结果表明,这个kasp标记能将f2群体进行分型。将基因型与穗粒数表现进行anova分析,结果p值<0.001,表明该标记与穗粒数极显著关联,该snp标记基因型与烟农19相同的f4株系的穗粒数显著多于与阳光5号相同的株系,杂合株系的穗粒数介于二者之间,表明在该区间内存在一个控制小麦穗粒数qtl。
[0104]
以上所述仅是本专利的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本专利技术原理的前提下,还可以做出若干改进和替换,这些改进和替换也应视为本专利的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1