一种血液样本中病原菌DNA的制备方法和基于此方法的临床病原菌感染诊断试剂盒
一种血液样本中病原菌dna的制备方法和基于此方法的临床病原菌感染诊断试剂盒
技术领域
1.本发明属于病原菌感染的分子诊断学领域,具体涉及一种血液样本中病原菌dna的制备方法和基于此方法的临床病原菌感染诊断试剂盒。
背景技术:
2.临床微生物学实验室目前用于鉴定病人系统性感染常用的标准方法是血培养。然而,由于血中病原菌数量过少而导致血培养方法的灵敏度非常低下,甚至有些细菌是不可培养型的,无法在体外培养生长。另外,血培养方法周期长,通常需要3—7天的时间才有可能得到结果,极大的延误对致病菌的精准定性和对病人的有效用药,从而加速耐药菌的形成。
3.近年来,荧光定量pcr(qpcr)技术在传染性疾病的分子诊断领域逐渐得到广泛的应用。理论上讲,qpcr反应方法对病原微生物的检测既特异灵敏又快速便捷。然而,从实际操作来看,这些以pcr为基础的诊断方法目前还没有显示出比血培养方法有更大的优越性,甚至还不如传统的血培养方法有效。其主要原因之一是临床样本中含有大量的宿主dna,这些宿主dna的存在严重的干扰了qpcr方法的特异性和灵敏度。
4.qpcr诊断方法的专一性和灵敏度在很大程度上取决于检测样品dna的提取方法。目前的常规核酸提取试剂盒用于提取临床样本中dna的时候区分不开宿主dna和病原菌dna。因此,所得的dna实质上是病原菌dna和宿主dna(如人血液细胞dna)的混合物。而病原菌dna的占比非常小,这极大的降低了qpcr的灵敏度。另外,用于检测病原菌的引物可能会与宿主dna非特异性结合,导致假阳性或者假阴性结果的出现。临床样本中提取的dna中病原菌dna的占比越小,则产生假阳性或者假阴性结果的可能性越大。
5.通常用于病原菌鉴定的临床血液样本中存在的病原菌数量极少,如某些血流感染患者的血液样本中细菌含量仅有约10cfu/ml。从这样的临床样本中制备的dna仅有痕量的病原菌dna分子,而绝大多数的dna都是宿主dna。临床分子诊断的目标就是要求从这样的样品中检测证明是否有病原菌dna的存在,从而准确无误的做出诊断结果(鉴定导致感染的病原菌)。占绝大多数的宿主dna在临床样本中的存在,给qpcr反应在传染性疾病诊断上的应用带来了严峻的困难和挑战。
技术实现要素:
6.本发明的目的是提供一种能从临床血液样本中有效去除宿主血细胞dna,富集病原菌dna的制备方法。利用本方法制备的dna进行qpcr反应,能尽量避免占绝大多数的宿主dna对qpcr检测病原菌的干扰,在很大程度上提高了pcr反应的特异性和灵敏度。
7.本发明提供的一种临床血液样本中病原菌dna的制备方法,包括以下步骤:
8.(a)将临床血液样本与足量的皂素saponin充分混合,血液样本中的非病原菌细胞被saponin裂解;
9.(b)待血液样本由浑浊变透明后,离心富集病原菌,弃掉上清;
10.(c)离心沉淀中加入叠氮溴化丙锭(pma)溶液,震荡重悬,避光孵育,然后亮处光解;
11.(d)按照常规dna提取方法从光解后混合物中分离制备病原菌dna。
12.进一步,所述步骤(a)中皂素saponin先溶于水,再跟临床血液样本混合。
13.进一步,所述步骤(a)中皂素saponin在所述混合体系中的质量百分比浓度为0.1%
‑
0.5%,裂解温度为室温,裂解时间为30min。
14.或者,所述步骤(a)中皂素saponin在所述混合体系中的质量百分比浓度为1%
‑
2%,裂解温度为室温,裂解时间为5
‑
10min。
15.进一步,所述步骤(a)中皂素saponin在混合体系中的质量百分比浓度为1%
‑
2%,在室温下置于hula mixer上孵育5min完成裂解。
16.进一步,所述步骤(b)中待血液样本由浑浊变透明后,加入等体积5m的nacl溶液翻转混匀,室温下孵育1分钟后,20000
×
g离心15
‑
20分钟富集病原菌,弃掉上清,将沉淀用pbs缓冲液重悬。
17.其中加入nacl是为了增加宿主细胞裂解后dna的溶解度,防止其析出而沉淀,另一个作用是稀释溶液,使病原菌更容易沉淀。
18.进一步,所述步骤(c)中pma溶液加入后的终浓度为10
‑
20μm,光解时间为25min。
19.或者,所述步骤(c)中pma溶液加入后的终浓度为50μm,光解时间为5min。
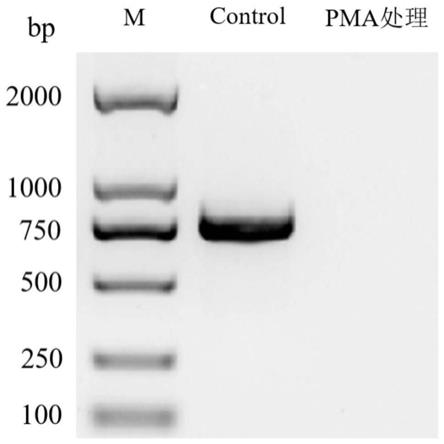
20.进一步,所述步骤(c)中震荡重悬混匀后的样品,室温避光孵育2
‑
10min后,在465
‑
475nm的特定光源曝光5
‑
30min,光源距离样品管10
‑
20cm,曝光期间样品经常轻柔震荡混匀。
21.所述特定光源采用pma
‑
lite
tm led photolysis device发光二极管光解装置发射。
22.进一步,所述步骤(c)中光解之后的样品20000
×
g离心15min,弃掉上清,用pbs重悬,然后按照常规方法提取dna。
23.所述常规dna提取方法是采用pathogen kit(qiagen,sp54104)提取dna。
24.本发明还提供一种临床血液样本中病原菌dna提取试剂盒,所述试剂盒包括saponin溶液、叠氮溴化丙锭(pma)溶液和常规dna提取试剂。
25.本发明还提供一种临床病原菌感染诊断试剂盒,所述试剂盒包括saponin溶液,叠氮溴化丙锭(pma)溶液,常规dna提取试剂盒、qpcr反应试剂和病原菌特异检测引物。
26.临床样本中的病原菌指的是耐saponin和pma的致病细菌,主要来源于以下属种:不动杆菌属(acinetobacter),拟杆菌属(bacteroides),柠檬酸细菌属(citrobacter),肠杆菌属(enterobacter),大肠杆菌(escherichia coli),梭形杆菌(fusobacterium),流感嗜血杆菌(haemophilus influenzae),克雷伯菌属(klebsiella),分支杆菌(mycobacterium),卟啉单胞菌(porphyromonas),普氏菌属(prevotella),变形杆菌(proteus),假单胞菌(pseudomonas),沙门氏菌(salmonella),志贺氏菌属(shigella),肠球菌(enterococcus)。
27.根据上述致病菌,本领域技术人员可以按照常规知识设计其特异性引物,再以本
发明的制备方法获得的致病菌dna为模板进行qpcr反应,检测样品中是否含有上述致病菌。
28.本发明采用的皂素saponin是一种天然表面活性剂,其中皂苷元与胆甾醇结合生成不溶性分子复合物,破坏血细胞的渗透性而发生崩解。
29.叠氮溴化丙锭(pma)是一种高亲和力的光敏dna结合染料,具有膜不通透性的特点,其倾向于结合双链dna,形成稳定的共价修饰,从而抑制正常的pcr反应。不仅如此,本发明人在实验中发现,其光解过程中使自身失去效力,同时将与其结合的dna裂解成片段。
30.与现有技术相比,本发明的有益效果在于:
31.1.本发明利用临床血液样本中病原菌细胞和非病原菌细胞的胞壁结构差异,使saponin只裂解非病原菌细胞而不破坏病原菌细胞从而仅使非病原菌细胞的dna释放,再加pma使释放的dna裂解,选择性的去除了非病原菌dna,使病原菌dna得到富集。然后用此dna进行qpcr反应,可以避免本占绝大多数的非病原菌dna对病原菌pcr检测的干扰,极大的提高了qpcr反应的特异性和灵敏度。
32.2.本发明采用的光敏dna结合染料pma,在暗培养时与游离的非病原菌dna结合,在光培养条件下发生光解,将与之结合的dna裂解成片段,同时多余的pma自身光解而失去效力。本发明者发现pma与裸露dna的结合,不仅是阻碍了pcr反应中dna的扩增,而是在光解过程中,将dna裂解。因此,在后续的实验操作中,既无非病原菌dna的干扰,也无pma的影响。无需洗涤离心,实验操作简单、方便。
33.3.pma去除非病原菌dna的过程无需特殊控温,比起需要37度控温的裂解dna的酶类更易于操作。而且pma去除非病原菌dna的作用不受saponin或其浓度的影响。
34.4.本发明将saponin先溶于水中,再与血液样本混合。这样使saponin在血液样本中分散性好,不会造成局部浓度过高。
35.5.本发明针对粘稠且成分复杂的临床血液样本的特殊性,将去除宿主dna后制备病原菌dna的过程精化到最简,极大的提高了临床诊断的效率、稳定性和可靠性。
附图说明:
36.图1从临床血液样本中富集病原菌dna的步骤图;
37.图2saponin对临床血液样本中非病原菌和病原菌选择性裂解;
38.图3光解对已结合pma的非病原菌dna的裂解作用,电泳图谱;
39.图4是应用qpcr反应对利用传统方法和本发明方法获得的宿主和细菌dna进行相对定量。
具体实施方式:
40.以下将对本发明的具体实施方式进行详细描述。通过实施例,科研人员可以对本发明有更清楚的了解,可以在此基础上对本发明做出一定的变更和修改,以获得不同的研究效果。下述实施例中的实验方法,如无特殊说明均为常规方法。实验过程中涉及到的试剂均为常规试剂,使用均参照产品使用说明书使用。实施例1:本发明方法从临床血液样本中富集病原菌dna,并提取dna的过程
41.包括以下步骤:
42.(a)将血液样本与足量的皂素saponin充分混合,血液样本中的非病原菌细胞被
saponin裂解;
43.(b)待血液样本由浑浊变透明后,离心富集病原菌,弃掉上清;
44.(c)离心沉淀中加入叠氮溴化丙锭(pma)溶液,震荡重悬,避光孵育,然后亮处光解;
45.(d)按照常规dna提取方法从光解后混合物中分离制备病原菌dna。
46.所述步骤(a)中皂素saponin先溶于水,再跟临床血液样本混合。
47.所述步骤(a)中皂素saponin在所述混合体系中的质量百分比浓度为0.3%,裂解温度为室温,裂解时间为30min。
48.所述步骤(b)中待血液样本由浑浊变透明后,加入等体积5m的nacl溶液翻转混匀,室温下孵育1分钟后,20000
×
g离心15分钟富集病原菌,弃掉上清,将沉淀用pbs缓冲液重悬。
49.所述步骤(c)中加入pma溶液后pma终浓度为20μm,光解时间为25min。
50.所述步骤(c)中震荡重悬混匀后的样品,室温避光孵育10min后,在465
‑
475nm的特
51.定光源照射下曝光30min。
52.实施例2:本发明方法从临床血液样本中富集病原菌dna,并提取dna的过程参见图1的步骤图。
53.实施例3:saponin对血细胞的裂解作用
54.saponin(皂素,sigma公司生产,货号为s7900)溶液是将一定量的saponin溶于水中,溶解完全过滤除菌即可。
55.取1ml新鲜人血液,添加109cfu的细菌(大肠杆菌和金黄色葡萄球菌各一组),分别用终浓度为0.1%,1%的saponin处理,反应5min后涂片,用瑞氏
‑
吉姆萨染色试剂盒(生工,e607315)染色,然后用显微镜观察血细胞和细菌细胞的裂解程度,阴性对照为添加等量无菌水的血液和细菌的混合液。
56.结果如图2所示,从图2可以看出,1%的saponin用时5min即可完全的将血细胞裂解而不会破坏细菌细胞。详细的讲,在无saponin的血液中,血细胞可以一直保持完整不会裂解,加入0.1%的saponin在5min内可以使部分裂解,加入1%的saponin作用5min之后会使血细胞完全裂解,saponin浓度越高,血细胞裂解的越快。两种浓度的saponin都不会使细菌细胞裂解。
57.实施例4:光解对已结合pma的非病原菌dna的裂解作用(电泳图谱)
58.首先利用pcr反应制备一份长度为750bp的dna,将其等分成两份,一份加入叠氮溴化丙锭(pma)溶液,震荡重悬,避光孵育,然后亮处光解;其中pma溶液浓度为50μm,光解时间为5min。
59.另一份添加等量无核酸酶的水进行同样的处理作为阴性对照。
60.然后将两份dna进行琼脂糖凝胶电泳。
61.结果如图3所示。
62.实施例5:采用不同方法获得的血液样本dna
63.一、模拟临床感染血液样本的制备
64.将大肠杆菌单菌落接种到5ml的lb液体培养基中,在37℃恒温摇床,180r/min,培养3小时,获得大肠杆菌培养液,转接50μl到5ml新的lb液体培养基中摇床培养3小时作为工
作菌液。取1ml菌液,洗去培养基,稀释至od
600
=0.5,然后进一步做10倍梯度稀释液。从稀释液中各取100μl涂板(lb固体培养基),37℃培养过夜,后数菌计数,以确定各稀释液中大肠杆菌的数量,将其加入正常血液样本中,得到模拟临床感染血液样本。
65.二、dna提取方法的比较
66.(1)传统dna制备方法:取200μl上述模拟临床感染血液样本,使用商品试剂盒pathogen kit(qiagen,货号sp54104),按试剂盒使用说明书的步骤提取样本dna。
67.(2)本发明方法1:将200μl模拟临床血液样本加入等体积的0.2%(终浓度0.1%)的saponin溶液,振荡混匀,室温放置15min,然后20000g离心5min以收集沉淀。用100μl pbs将沉淀物重悬混匀,加入10μl pma至pma终浓度为10μm,暗孵育5min,后光解25min,然后20000g离心3min收集沉淀。取100μlpbs将沉淀溶解重悬,震荡混匀,然后使用商品试剂盒pathogen kit(qiagen)按照试剂盒说明书的步骤分离提取dna(ex1)。
68.(3)本发明方法2:将200μl模拟临床血液样本加入等体积2%(终浓度1%)的saponin溶液,振荡混匀,室温放置5min,然后20000g离心20min以收集沉淀。用100μl pbs将沉淀物重悬混匀,加入10μl pma至pma终浓度为50μm,暗孵育2min,后光解5min,然后20000g离心15min收集沉淀。取100μl pbs将沉淀溶解重悬,震荡混匀,然后使用商品试剂盒pathogen kit(qiagen)按照试剂盒说明书的步骤分离提取dna(ex2)。
69.实施例6:血液样本中大肠杆菌dna的qpcr检测及应用qpcr对不同方法制备的dna进行人dna和细菌dna的分别定量
70.用于检测人血细胞dna和大肠杆菌基因dna的qpcr反应引物是:
71.宿主dna:
72.gapdh
‑
f:tggcaacaatatccactttaacaga
73.gapdh
‑
r:tcaacggatttggtcgtattagg
74.大肠杆菌dna:
75.e.coli
‑
f:ccagggctacacacgtgcta
76.e.coli
‑
r:tctcgcgaggtcgcttct
77.qpcr反应体系为20μl,各内容物体积见表1。
78.表1
79.[0080][0081]
反应条件是95℃,10min;95℃ 15s,60℃ 1min,40个循环;95℃ 15s,60℃ 1min,95℃ 15s。血液样本中大肠杆菌dna的qpcr检测结果如表2所示,由ct值可以看出(cutoff值为35),使用传统方法制备的dna,大肠杆菌qpcr反应灵敏度为10000cfu/ml,而使用本发明方法制备的dna,大肠杆菌qpcr反应灵敏度为10cfu/ml。由此表明,本发明的dna制备方法使大肠杆菌的检测灵敏度提升了1000倍。这主要是因为本发明的dna制备方法将干扰pcr检测灵敏度的大量宿主dna(血细胞dna)选择性去除的结果。
[0082]
表2
[0083][0084]
其中阴性对照是将反应体系中模板换成等量无核酸水。
[0085]
将实施例4中三种不同方法制备的dna提取物(control、ex1、ex2)进行实时qpcr反应,其结果如图4所示,其中gapdh是人的保守基因,16s是细菌基因。使用传统方法制备的dna提取物中含有大量宿主dna,而使用本发明方法制备的dna提取物则几乎检测不出宿主dna的存在。
[0086]
实施例7:pma对病原菌的影响
[0087]
实验方案:将pma加入大肠杆菌菌液中,pma终浓度设置三个梯度,分别为10μm,20μm,50μm;大肠杆菌菌液中含菌量为1000cfu/ml。对照为不加pma的菌液。暗处孵育2min,蓝光曝光5min,之后分别取50μl菌液涂板计数,两个重复。
[0088]
实验结果见表3:
[0089]
表3
[0090]
单位/cfu重复1重复2无pma858210μm pma979820μm pma8510050μm pma92104
[0091]
实验结论:由上表看出,10
‑
50μm的pma处理后菌落数与未pma处理的样本菌落数无显著差异,由此可得,10
‑
50μm的pma对菌活力无明显影响。
[0092]
实施例8:一种临床血液样本中病原菌dna富集提取试剂盒
[0093]
试剂盒包括组分a:saponin溶液,组分b:叠氮溴化丙锭(pma)溶液,组分c:nacl溶
液和组分d:常规dna提取试剂盒。
[0094]
操作步骤:
[0095]
1、分别配置浓度为5%、1mm和5m的saponin、pma和nacl母液,混匀,备用。
[0096]
2、取适量疑似细菌感染血样本,用终浓度1%的saponin处理5min至体系由浑浊变透明,加等量5m的nacl溶液,混匀。
[0097]
3、离心,去上清。后pbs重悬。
[0098]
4、pma处理5min。
[0099]
5、pbs洗一遍,用常规dna提取试剂盒进行病原菌dna提取。
[0100]
6、将提取的dna用nanodrop和qubit进行质量和浓度评估。
[0101]
7、将提取的dna置于
‑
20℃保存备用。
[0102]
实施例9:一种临床病原菌感染诊断试剂盒
[0103]
试剂盒包括组分a:saponin溶液,组分b:叠氮溴化丙锭(pma)溶液,组分c:nacl溶液,组分d:常规dna提取试剂盒,组分e:荧光定量pcr(qpcr)反应试剂和组分f:病原菌特异性检测引物。
[0104]
按照实施例8的方法提取临床血液样本中病原菌dna。
[0105]
qpcr反应体系与条件(20μl反应体系):
[0106]
反应体系
[0107][0108]
反应条件:
[0109]
[0110]
按照以上方法完成临床血液样本中特异性病原菌dna的qpcr检测,ct值≤35即为阳性。
[0111]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1