一种能高效表达LL-37多肽的多拷贝重组表达载体及应用的制作方法
一种能高效表达ll
‑
37多肽的多拷贝重组表达载体及应用
技术领域
1.本发明涉及基因工程技术领域,尤其涉及一种能高效表达ll
‑
37多肽的多拷贝重组表达载体及应用。
背景技术:
2.ll
‑
37是一种人源抗菌肽,具有广谱抗菌性,不仅对革兰氏阴性菌有抑制效果,还能抑制革兰氏阳性菌。现在国内的奶业出口上还存在很大问题,因为牛奶的抗生素超标,而且现在抗生素的使用容易导致耐药菌的出现,从而出现“超级细菌”,因此这个问题亟待解决,并且添加到牛奶中的防腐剂超标也导致了牛奶的安全问题。而ll
‑
37本质属于蛋白质,会在体内分解成基本单元的氨基酸,不产生耐药性,无残留问题,是一种新型的生物防腐剂,有很好的研究前景。
3.现在用大肠杆菌来诱导表达ll
‑
37是比较成熟的原核表达系统,但是大部分为包涵体,需要变性和复性操作,此过程中ll
‑
37损失严重,大大限制了ll
‑
37的工业化生产。现有技术公开了利用基因工程技术的方法,通过构建融合了ll
‑
37编码基因的重组表达载体和重组工程菌来表达ll
‑
37,但是目前的方法仍然存在发酵耗时较长的问题。
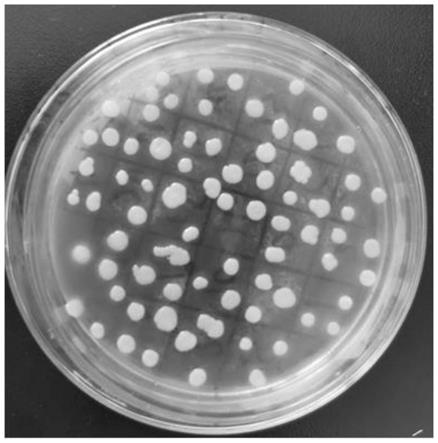
技术实现要素:
4.本发明的目的是要提供一种能高效表达ll
‑
37多肽的多拷贝重组表达载体及应用。本发明目的是让重组质粒能够在smd1168h毕赤酵母中高效表达,从而实现ll
‑
37在真核系统中的分泌表达,简化工序,提高表达量及避免了大肠杆菌会带来的安全性问题。
5.为达到上述目的,本发明是按照以下技术方案实施的:
6.本发明一种能高效表达ll
‑
37多肽的多拷贝重组表达载体,以ppiczα为基础载体插入目的片段得到重组表达载体,所述的重组表达载体ppiczα载体序列包括信号肽α因子、拷贝的ll
‑
37序列。
7.进一步,所述的能高效表达ll
‑
37多肽的多拷贝重组表达载体,其特征在于:所述目的片段插入在ppiczα的bgi ii和bamh i的限制性酶切位点之间。
8.本发明所述的重组毕赤酵母,采用以ppiczα为基础载体插入目的片段得到重组表达载体转化毕赤酵母感受态细胞,得到重组毕赤酵母。
9.进一步,所述重组毕赤酵母的原始菌株包括毕赤酵母smd1168h。
10.进一步,所述重组毕赤酵母的构建方法包括以下步骤:
11.s1:配置培养基:
12.ypd的配制:胰蛋白胨、酵母提取物用ddh2o溶解并定容,121℃灭菌20min;使用前再加入115℃灭菌15min葡萄糖溶液,固体则加热后再加琼脂粉;
13.ypds的配制:胰蛋白胨、酵母提取物、山梨醇用ddh2o溶解并定容,将溶液加热后加入琼脂粉,121℃灭菌20min;使用前再加入115℃灭菌15min的葡萄糖溶液;
14.山梨醇:山梨醇用ddh2o溶解并定容,过滤除菌或者121℃灭菌20min;
15.s2:制备感受态细胞:挑取毕赤酵母单菌落用ypd培养基培养至od值0.4
‑
0.6之间,离心加纯净水洗涤后再加纯水涡旋混匀,再离心弃上清,再加灭菌山梨醇混合离心一次,最后加入1ml山梨醇,制备感受态细胞;
16.s3:制备重组毕赤酵母:
17.重组毕赤酵母:用电击的方法电转化感受态,电击后立即加入1ml冷却的山梨醇吹打混匀;再30℃静置培养两小时后取150
‑
200ul涂布在含有zeocin的ypds平板上,倒置培养;剩余的液体静置后加入同体积的ypd培养基,30℃、200rpm培养2h,再分别涂布于ypds平板上,同上条件培养;
18.pcr筛选阳性克隆:将引物与pcr相关试剂混合后分装标记好,将长出来的乳白色菌落用牙签挑取到相应的pcr管中,进行pcr反应,再取跑电泳100v跑到三分之二的位置,将条带明显的菌落点到新的ypd固体培养基上,30℃倒置培养,再将菌落挑到液体ypd培养基中,加入相应体积zeocin溶液到液体培养基,并将液体培养基置于摇床上200rpm,30℃过夜培养,制备得到重组毕赤酵母。
19.本发明所述重组毕赤酵母的应用,所述重组毕赤酵母具有广谱抑菌性,能够抑制大肠杆菌和金黄色葡萄球菌。
20.本发明的有益效果是:
21.本发明是一种能高效表达ll
‑
37多肽的多拷贝重组毕赤酵母及应用,与现有技术相比,本发明具有如下技术效果:
22.1本发明提供了一种高效表达ll
‑
37多肽的多拷贝重组表达载体(ppiczα
‑
ll
‑
37)。本发明以ppiczα为骨架质粒来构建重组表达载体,所述的重组表达载体ppiczα载体序列包括信号肽α因子、拷贝的ll
‑
37序列。
23.2本发明将载体毕赤酵母与人源抗菌肽有机结合,其表达是通过酵母菌利用甲醇诱导产生目标蛋白;利用本发明的重组载体转化酵母宿主菌能够有序合成并在α
‑
信号肽的作用下将目标蛋白分泌到胞外,并且该过程α
‑
信号肽会自动被切除,同时多次串联重复序列释放为单体ll
‑
37多肽,产生与人源抗菌肽ll
‑
37相同功能的结构多肽。
24.3经过对比抑菌圈的大小和最小抑菌浓度的测量,检测ll
‑
37的活性。本发明的重组毕赤酵母得到的人源抗菌肽ll
‑
37具有广谱抗菌性,能够抑制大肠杆菌和金黄色葡萄球菌。
附图说明
25.图1是阳性克隆的重组酵母结果图;
26.图2是阳性克隆的多拷贝重组酵母结果图;
27.图2中:上面:17、16、15、14、13、12、11、10、ladder(1kb)、9、8、7、6、5、4、3、2、1下面:ladder(1kb)、负对照、、正对照、29、28、27、26、25、24、23、22、21、20、空18;
28.图3是电转化后pcr扩增结果图;
29.图3中:从左到右:第一块板:酵母,22、20、18、17、15、空、ladder、13、12、11、8、7、6、4、3、1、空;第二板:空*2、23、24、25、26、空、28、29、空、酵母、正对照、空、负对照;
30.图4是三种酶单酶切验证图的1%琼脂糖核酸电泳图;
31.图5是30℃、200rpm工程菌甲醇诱导表达结果图;
32.图5中:第一块胶:29号、宿主、marker、ll
‑
37标准品、21号、11号、9号、3号、13号、1号(丙酮沉淀);第二块胶:1号、3号、9号、11号、21号、宿主、marker、ll
‑
37标准品、12号、29号(三氯乙酸沉淀);
33.图6是25℃、250rpm工程菌甲醇诱导表达结果图;
34.图6中:marker、29号48h、13号48h、14号
①
48h、14号
②
48h、ll
‑
37标准品、14号
②
72h、11
②
72h、9
②
72h、3
②
72h;
35.图7是重组工程菌表达样品的抑菌图;
36.图7中(a)为:amp、11号、14号、21号、中间为空白;(b)为:3号、1号、9号、amp,中间为空白;
37.图8是重组工程菌表达样品对金黄色葡萄球菌的抑制图;
38.图8中:顺时针左上角开始:21号、21号多拷贝、14号、11号菌,中间为空白。
具体实施方式
39.下面结合附图以及具体实施例对本发明作进一步描述,在此发明的示意性实施例以及说明用来解释本发明,但并不作为对本发明的限定。
40.如图1
‑
8所示:本发明一种能高效表达ll
‑
37多肽的多拷贝重组表达载体,以ppiczα为基础载体插入目的片段得到重组表达载体,所述的重组表达载体ppiczα载体序列包括信号肽α因子、拷贝的ll
‑
37序列。
41.进一步,所述的能高效表达ll
‑
37多肽的多拷贝重组表达载体,其特征在于:所述目的片段插入在ppiczα的bgi ii和bamh i的限制性酶切位点之间。
42.本发明所述的重组毕赤酵母,采用以ppiczα为基础载体插入目的片段得到重组表达载体转化毕赤酵母感受态细胞,得到重组毕赤酵母。
43.进一步,所述重组毕赤酵母的原始菌株包括毕赤酵母smd1168h。
44.进一步,所述重组毕赤酵母的构建方法如下:
45.ypd的配制(100ml):胰蛋白胨2g,酵母提取物1g,用ddh2o溶解并定容至90ml,121℃灭菌20min;使用前再加入10ml 20%的葡萄糖溶液(115℃灭菌15min)。(固体则加热后再加2g琼脂粉)
46.ypds的配制(100ml):胰蛋白胨2g,酵母提取物1g,山梨醇。用ddh2o溶解并定容至90ml,将溶液加热后加入2g琼脂粉,121℃灭菌20min;使用前再加入10ml 20%的葡萄糖溶液(115℃灭菌15min)。
47.1m的山梨醇:1g山梨醇用ddh2o溶解并定容至100ml,(过滤除菌或者121℃灭菌20min)
48.制备感受态细胞:挑取毕赤酵母单菌落用ypd培养基培养至od值0.4
‑
0.6之间,离心加纯净水洗涤后再加纯水涡旋混匀,再离心弃上清,再加灭菌山梨醇混合离心一次,最后加入1ml山梨醇,制备感受态细胞;
49.制备重组毕赤酵母:取重组表达质粒,用电击的方法(1.5kv,25vf,200ω),电转化感受态,电击后立即加入1ml冷却的1m的山梨醇;再30℃静置培养两小时后,取200ul涂布在含有0.1%zeocin的ypds平板上,倒置培养;剩余的液体静置后加入同体积的ypd液体培养基,200rpm,30℃培养2h,再分别涂布于ypds平板上,同上条件培养。
50.pcr筛选阳性克隆:将引物与pcr相关试剂混合后分装标记好,将长出来的乳白色菌落用牙签挑取到相应的pcr管中,进行pcr反应(94℃预变性2min,98℃变性45s,再56℃退火30s,68℃延伸2min,步骤2
‑
4进行30个循环)。再取10ul跑电泳(1%的核酸电泳)100v跑到三分之二的位置。对照电泳结果图将条带明显的菌落点到新的ypd固体培养基上,30℃倒置培养。再将菌落挑到液体ypd培养基中,终浓度为100ug/100ml的zeocin溶液,并将液体培养基置于摇床上200rpm,30℃过夜培养,制备得到重组毕赤酵母。
51.引物的序列如下:
52.p1:gactggttccaattgacaagc
53.p2:gcaaatggcattctgacatcc
54.通过双酶切含aox1启动子、α信号肽和ll
‑
37基因的目标片段后进行试剂盒回收处理,乙醇沉淀后再用连接试剂盒构建两拷贝重组质粒,再用乙醇沉淀,重复前一步骤,得到多拷贝的质粒;同时对重组质粒进行线性化处理,用乙醇沉淀,然后去磷酸化处理,再次用乙醇沉淀后与多拷贝的重组质粒用连接试剂盒连接,再次用bamh i单酶切重组质粒ppiczαa
‑
ll
‑
37,使之线性化;乙醇沉淀法进行纯化,然后用去磷酸化试剂盒按照2.2ug/pmol的比例加入到质粒中,再次用乙醇沉淀,备用。
55.单酶切体系(共200ul体系)
56.名称体系质粒dna75ulbamh i(10u/ul)3ul10x l buffer20uldd h2o102ul
57.备注:此时ppiczαa
‑
ll
‑
37质粒的浓度为140ug/ml,酶切时需要37℃1h。
58.用bglⅱ和bamh i双酶切重组质粒ppiczαa
‑
ll
‑
37;用凝胶回收柱纯化目的片段;乙醇沉淀;用连接试剂盒构建多拷贝;再用乙醇沉淀后用bglⅱ和bamh i双酶切,再次沉淀后加入(1)步骤得到的样品,用连接试剂盒连接多拷贝片段与载体;利用电转化的方式转化到毕赤酵母中;涂ypd平板(含100ug/100ml zeocin);
59.双酶切体系
60.名称体系质粒dna10ul(140ug/ml)bgl
ꢀⅱ
1ulbamh i1ul10x m buffer2uldd h2o6ul
61.做菌落pcr鉴定:挑取有明显酵母特征的菌落做pcr鉴定,再挑选几个菌落培养后提取质粒,进行酶切验证是否是多拷贝。
62.挑取重组毕赤酵母单菌落到灭菌的新鲜冷却的含有博来霉素溶液(终浓度为100ul/100ml)的5ml ypd培养基中,250rpm,30℃先长到od值在2
‑
5之间时,转接1.5ml到50ml ypd培养基中分别于24、48、72、84时加入甲醇,待其诱导至96h时取出5000rpm,4℃离心5min,取上清进行后续操作。
63.用10ml lb液体培养基培养dh5α大肠杆菌和金黄色葡萄球菌,37℃180rpm过夜培养后,再转接到25ml lb液体培养基中,同样条件培养1.5h
‑
2h,od值在0.45
‑
0.55之间,取30ul菌液到100mllb冷却至不烫手的固体培养基中,混匀后倒平板;待其凝固后将牛津杯放在表面,分别加入50ul样品,同时用生理盐水做负对照,氨苄青霉素和标准品ll
‑
37做正对照,37℃过夜培养后测定其透明圈的大小。
64.本发明的实施例:
65.实施例一:
66.先制备感受态细胞,取酵母用ypd培养基培养至od值0.4
‑
0.6之间,离心加50ml纯净水洗涤后再加25ml纯水涡旋混匀,再离心弃上清,再加2ml 1mol/l的灭菌山梨醇混合离心一次,最后加入1ml山梨醇,制备感受态细胞(整个过程均需在冰上进行)。再制备工程菌,取重组表达质粒,用电击的方法(1.5kv,25vf,200ω),电转化感受态,电击后立即加入1ml冷却的1m的山梨醇;再30℃静置培养两小时后,取200ul涂布在含有%0.1zeocin的ypds平板上,倒置培养;剩余的液体静置后加入同体积的ypd培养基200rpm,30℃培养2h,再分别涂布于ypds平板上,同上条件培养。载体质粒进行单酶切处理构建,含有目标片段的质粒进行双酶切和连接反应构建多拷贝,表达量明显高于一个拷贝的重组菌。
67.将活化后的工程菌液(od值2
‑
5之间)按照2%的比例接种至50ml新鲜的含博来霉素的ypd培养基中,200rpm,30℃培养12、24、36、48、60、72、84、96h,1ml发酵液5000rpm离心5min后剩余的菌体重约为0.8g,用1.5ml的ep管取200ul上清液并加入四倍体积丙酮过夜沉淀后(加入后立即温和上下颠倒8
‑
10次),14000rpm离心15min,弃上清,于通风橱中开盖放置20min
‑
30min让丙酮挥发。制样时样液与5x的loading buffer为3:1的比例,97℃15min即可。用tricine
‑
sds
‑
page凝胶电泳检测目标蛋白的表达量,如图5所示。
68.用25ml lb液体培养基培养dh5α大肠杆菌和金黄色葡萄球菌,37℃180rpm过夜培养后,再转接到25ml lb液体培养基中,同样条件培养1.5h
‑
2h,od值在0.4
‑
0.6之间,取30ul菌液到100mllb冷却至不烫手的固体培养基中,混匀后倒平板;待其凝固后将牛津杯放在表面,分别加入50ul样品,同时用生理盐水做负对照,氨苄青霉素和标准品ll
‑
37做正对照,37℃过夜培养后测定其透明圈的大小。得出了ll
‑
37对金黄色葡萄球菌的抑制效果优于对dh5α大肠杆菌的抑制效果。
69.实施例二:
70.先制备感受态细胞,取酵母用ypd培养基培养至od值0.4
‑
0.6之间,离心加50ml纯净水洗涤后再加25ml纯水涡旋混匀,再离心弃上清,再加2ml 1mol/l的灭菌山梨醇混合离心一次,最后加入1ml山梨醇,制备感受态细胞(整个过程均需在冰上进行)。再制备工程菌,取重组表达质粒,用电击的方法(1.5kv,25vf,200ω),电转化感受态,电击后立即加入1ml冷却的1m的山梨醇;再30℃静置培养两小时后,取200ul涂布在含有0.1%zeocin的ypds平板上,倒置培养;剩余的液体静置后加入同体积的ypd培养基200rpm,30℃培养2h,再分别涂布于ypds平板上,同上条件培养。载体质粒进行单酶切处理,含有目标片段的质粒进行双酶切和连接试剂盒进行连接反应构建多拷贝,表达量明显高于一个拷贝的重组工程菌。
71.将活化后的工程菌液(od值2
‑
5之间)按照2%的比例接种至50ml新鲜的含博来霉素的ypd培养基中,250rpm,25℃培养12、24、36、48、60、72、84、96h,1ml发酵液5000rpm离心5min后剩余的菌体重约为0.8g,用1.5ml的ep管取200ul上清液并加入四倍体积丙酮过夜沉
淀后(加入后立即温和上下颠倒8
‑
10次),14000rpm离心15min,弃上清,于通风橱中开盖放置20min
‑
30min让丙酮挥发。制样时样液与5x的loading buffer为3:1的比例,97℃15min即可。用tricine
‑
sds
‑
page凝胶电泳检测融合蛋白的表达量,如图6所示。
72.用25ml lb液体培养基培养dh5α大肠杆菌和金黄色葡萄球菌,37℃180rpm过夜培养后,再转接到25ml lb液体培养基中,同样条件培养1.5h
‑
2h,od值在0.4
‑
0.6之间,取30ul菌液到100mllb冷却至不烫手的固体培养基中,混匀后倒平板;待其凝固后将牛津杯放在表面,分别加入50ul样品,同时用生理盐水做负对照,氨苄青霉素和标准品ll
‑
37做正对照,37℃过夜培养后测定其透明圈的大小。得出了ll
‑
37对金黄色葡萄球菌的抑制效果优于对dh5α大肠杆菌的抑制效果。
73.本发明的技术方案不限于上述具体实施例的限制,凡是根据本发明的技术方案做出的技术变形,均落入本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1