一种胺碘酮杂质C的合成方法与流程
一种胺碘酮杂质c的合成方法
技术领域
1.本发明属于医药技术领域,具体涉及一种盐酸胺碘酮杂质c的制备方法。
背景技术:
2.胺碘酮(amiodarone)又名乙胺碘呋酮,胺碘酮(或其盐酸盐cas:19774-82-4)是一个广泛用 于治疗及预防室性及室上性心律失常的第三类抗心律失常药物。现代社会心血管疾病的患者 大幅攀升,对该类药物的需求量也日益增加。基于稳定的疗效和较低的副作用,盐酸胺碘酮 的市场份额不断扩大。相应的对胺碘酮杂质的研究及合成方法也进行了广泛的报道,发现目 前对胺碘酮欧洲药典ep杂质c合成方法进行报道的文献并不多。因此,找到一种操作简单, 成本低廉、容易分离提纯,收率高的胺碘酮杂质c的合成方法具有重要意义。特提出如下发 明:
3.盐酸胺碘酮,其结构式如下:
[0004][0005]
盐酸胺碘酮杂质c结构式如下:
[0006][0007]
bioorganic & medicinal chemistry letters 18(2008)5920
–
5922公开报道了一种合成胺碘 酮杂质c的方法如下:
[0008][0009]
第一步化合物i加入甲醇溶解,加入碘化钠和氢氧化钠,降温到-5℃滴加次氯酸钠,反 应完用稀盐酸淬灭反应,后处理提纯得到中间体1,收率25%,上两个碘的副产物收率11%。 第二步在碱的作用下对接n,n-二乙基-β-氯乙胺盐酸盐,反应得到目标产物。文献中第一步反 应收率偏低,杂质较多,而且生成中间体1和上两个碘副产物极性类似,分离提纯困难。因 此找到一种收率高、成本低、操作简单、容易分离提纯的方法进行合成胺碘酮杂质c是十分 必要的,对提高药物质量标准有重要意义。
技术实现要素:
[0010]
为解决上述问题,本发明公开了一种,成本低、操作简单、收率高,易分离纯化的胺碘 酮杂质c的合成方法。
[0011]
为达到上述目的,本发明的技术方案如下:
[0012]
一种胺碘酮杂质c的合成方法,包括以下步骤:
[0013]
(1)将化合物ⅰ溶于溶剂中,在碱的作用下,与n,n-二乙基-β-氯乙胺盐酸盐进行取代 反应得到中间体1;
[0014]
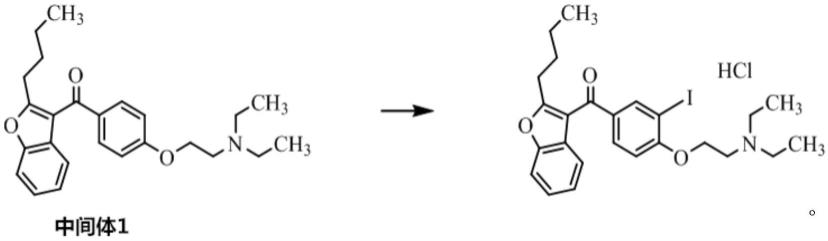
(2)中间体1溶于溶剂中,在碱的作用下,与碘单质反应得到胺碘酮杂质c。
[0015]
胺碘酮杂质c的具体合成工艺路线如下:
[0016][0017]
进一步地,步骤(1)中,所述化合物ⅰ与n,n-二乙基-β-氯乙胺盐酸盐的摩尔比为1:1.0-4.0, 优选地,所述化合物ⅰ与n,n-二乙基-β-氯乙胺盐酸盐的摩尔比为1:1.2。
[0018]
进一步地,步骤(1)中,所述碱与化合物i的摩尔比为1.0-4.0:1.0,优选地,所述碱与 化合物i的摩尔比为2.0:1.0;所述溶剂与化合物i的体积比为1.0-100.0:1.0,优选地,
所述溶 剂与化合物i的体积比为30.0:1.0。
[0019]
进一步地,步骤(1)中,所述碱为碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢 氧化锂中的一种或多种,优选地,所述碱为碳酸钾;所述溶剂为甲苯、水、乙腈、丙酮、四 氢呋喃、n,n-二甲基甲酰胺、二甲基亚砜中的一种或多种,优选地,所述溶剂为水和甲苯的 混合物。
[0020]
进一步地,步骤(1)中,所述取代反应的温度为30-90℃,优选地,所述取代反应的温 度为80-85℃,反应的时间为1-6小时,优选地,所述反应的时间为1小时。
[0021]
进一步地,步骤(2)中,所述间体1与碘单质的摩尔比为1:0.5-3.0,优选地,所述中间 体与碘单质的摩尔比为1:0.9。
[0022]
进一步地,步骤(2)中,所述碱与中间体1的摩尔比为1.0-5.0:1.0,优选地,所述碱与 中间体1的摩尔比为1.0:1.0;所述溶剂与中间体1的体积比为1.0-100.0:1.0,优选地,所述溶 剂与中间体1的体积比为20.0:1.0。
[0023]
进一步地,步骤(2)中,所述碱为碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢 氧化锂中的一种或多种,优选地,所述碱为氢氧化钠;所述溶剂为甲醇、乙醇、异丙醇、丙 酮、乙腈、四氢呋喃、乙酸乙酯中的一种或多种,优选地,所述溶剂为甲醇。
[0024]
进一步地,步骤(2)中,所述反应的温度为0-30℃,优选地,所述反应的温度为0-5℃; 反应的时间为2-12小时,优选地,所述反应的时间为6小时。
[0025]
本发明的有益效果为:
[0026]
本发明胺碘酮杂质c的合成方法,改变了原报道文献中先上碘,然后再对接n,n-二乙基
ꢀ‑
β-氯乙胺盐酸盐的合成路线,改为先对接n,n-二乙基-β-氯乙胺盐酸盐,然后再低温下和碘单 质反应上碘。本方法收率高,操作简单、成本低廉,容易分离提纯。为盐酸胺碘酮质量控制 提供了一种合格,低成本的杂质对照品。
附图说明
[0027]
图1为本发明实施例4所得产物的核磁氢谱图;
[0028]
图2为本发明实施例4所得产物的质谱谱图;
[0029]
图3为本发明实施例4所得产物的hplc谱图;
[0030]
图4为本发明合成工艺路线。
具体实施方式
[0031]
下面结合附图和具体实施方式,进一步阐明本发明,应理解下述具体实施方式仅用于说 明本发明而不用于限制本发明的范围。
[0032]
实施例1
[0033]
在500ml三口瓶中,加入10.0g(0.034mol)化合物i,加100ml水和200ml甲苯;加入 9.4g(0.068mol)碳酸钾,体系呈浑浊白色,升温到50-55℃,分批加入7.0g(0.041mol)n,n-二乙 基-β-氯乙胺盐酸盐,过程中体系渐溶清。升温到80-85℃反应1小时。tlc检测反应完全, 反应液降温,加入50ml水,分层分出有机相,水相加100ml*2乙酸乙酯萃取,合并有机相加 50ml饱和食盐水洗涤,加入20g无水硫酸钠干燥,过滤除去盐,浓缩除去溶剂;经柱层析(纯 ea)得到11.5g中间体1,收率85%,hplc纯度95%。
[0034]
实施例2
[0035]
在500ml三口瓶中,加入10.0g(0.034mol)化合物i,加100ml丙酮;加入1.4g(0.034mol) 氢氧化钠,体系呈浑浊白色,升温到50-55℃,分批加入11.7g(0.068mol)n,n-二乙基-β-氯乙 胺盐酸盐,过程中体系渐溶清。升温到80-85℃反应1.5小时。tlc检测反应完全,反应液降 温,加入50ml水,分层分出有机相,水相加100ml*2乙酸乙酯萃取,合并有机相加50ml饱 和食盐水洗涤,加入20g无水硫酸钠干燥,过滤除去盐,浓缩除去溶剂;经柱层析(纯ea) 得到10.7g中间体1,收率79.9%,hplc纯度94%。
[0036]
实施例3
[0037]
在500ml三口瓶中,加入10.0g(0.034mol)化合物i,加300ml乙腈;加入18.7g(0.136mol) 碳酸钾,体系呈浑浊白色,升温到50-55℃,分批加入23.4g(0.136mol)n,n-二乙基-β-氯乙胺 盐酸盐,过程中体系渐溶清。升温到80-85℃反应2小时。tlc检测反应完全,反应液降温, 加入50ml水,分层分出有机相,水相加100ml*2乙酸乙酯萃取,合并有机相加50ml饱和食 盐水洗涤,加入20g无水硫酸钠干燥,过滤除去盐,浓缩除去溶剂;经柱层析(纯ea)得到 10.2g中间体1,收率76.1%,hplc纯度90%。
[0038]
实施例4
[0039]
在500ml三口瓶中,加入230ml甲醇,加入11.5g(0.029mol)中间体1,冰浴降温到0-5℃ 加入1.2g(0.029mol)氢氧化钠。保持温度0-5℃,分10批次加入6.6g(0.026mol)单质碘。 加完后保温反应6小时,hplc检测反应显示原料剩余15%,单碘目标产物占比75%,二碘 副产物占比10%。体系中加入200ml水搅拌10分钟,低温浓缩除去大部分甲醇溶剂,加入 200ml*2乙酸乙酯萃取,分层合并有机相加入50ml饱和食盐水洗涤有机相,分层有机相加入 20g无水硫酸钠干燥抽滤,滤液浓缩至干,经柱层析(纯ea)得到粗品12.3g,用60ml乙腈 重结晶得到10.2g,加入100ml甲醇溶解,滴加盐酸甲醇调节ph到6,有大量白色固体析出, 搅拌0.5小时,抽滤,滤饼加少量甲醇淋洗,抽干得到9.4g目标产物,产率62.7%,hplc 纯度98%。
[0040]
实施例5
[0041]
在500ml三口瓶中,加入230ml甲醇,加入11.5g(0.029mol)中间体1,冰浴降温到5-10℃, 加入8.0g(0.058mol)碳酸钾。保持温度5-10℃,分10批次加入7.4g(0.029mol)单质碘。 加完后保温反应12小时,hplc检测反应显示原料剩余6%,单碘目标产物占比70%,二碘 副产物占比20%。体系中加入200ml水搅拌10分钟,低温浓缩除去大部分甲醇溶剂,加入 200ml*2乙酸乙酯萃取,分层合并有机相加入50ml饱和食盐水洗涤有机相,分层有机相加入 20g无水硫酸钠干燥抽滤,滤液浓缩至干,经柱层析(纯ea)得到粗品10.6g,用60ml乙腈 重结晶得到9.1g,加入100ml甲醇溶解,滴加盐酸甲醇调节ph到6,有大量白色固体析出, 搅拌0.5小时,抽滤,滤饼加少量甲醇淋洗,抽干得到8.6g目标产物,产率57.3%,hplc 纯度87%。
[0042]
实施例6
[0043]
在500ml三口瓶中,加入230ml甲醇,加入11.5g(0.029mol)中间体1,冰浴降温到5-10℃, 加入2.8g(0.116mol)氢氧化锂。保持温度5-10℃,分10批次加入6.6g(0.026mol)单质碘。 加完后保温反应6小时,hplc检测反应显示原料剩余30%,单碘目标产物占比60%,二碘 副产物占比10%。体系中加入200ml水搅拌10分钟,低温浓缩除去大部分甲醇溶剂,加
入 200ml*2乙酸乙酯萃取,分层合并有机相加入50ml饱和食盐水洗涤有机相,分层有机相加入 20g无水硫酸钠干燥抽滤,滤液浓缩至干,经柱层析(纯ea)得到粗品10.3g,用60ml乙腈 重结晶得到8.3g,加入100ml甲醇溶解,滴加盐酸甲醇调节ph到6,有大量白色固体析出, 搅拌0.5小时,抽滤,滤饼加少量甲醇淋洗,抽干得到8.1g目标产物,产率54.0%,hplc 纯度91%。
[0044]
需要说明的是,以上内容仅仅说明了本发明的技术思想,不能以此限定本发明的保护范 围,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干 改进和润饰,这些改进和润饰均落入本发明权利要求书的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1