共聚聚酯树脂、成型品、热收缩性膜及纤维的制作方法
1.本发明涉及一种共聚聚酯树脂,其透明性、色调、再利用性和成型性优异,并且很少发生成型用模具、膜模头、纤维用模周围的污染及异物附着。
背景技术:
2.以聚酯、特别是对苯二甲酸(以下,有时简称为tpa)和乙二醇(以下,有时简称为eg)为原料制造的聚对苯二甲酸乙二醇酯(pet)由于化学物理性质优异,因此被广泛用于容器、膜、片材、纤维等用途。
3.近年来,在制造该聚对苯二甲酸乙二醇酯(pet)时,使二乙二醇(以下,有时简称为deg)共聚而得到的聚酯(以下,有时简称为共聚聚酯)由于透明性、抗冲击性、成型性、耐热性等优异而受到关注,被用于各种用途,特别是用作膜、片材、注塑成型体、异形成型体等成型体用原料聚合物。
4.另一方面,从加工性、环境稳定性、价格竞争力等出发,氯乙烯树脂被用作建材用等室外使用的成型体等,但是由于单体、增塑剂从这些成型体中溶出而有导致的致癌性及内分泌紊乱的作用的担忧,以及在焚烧时产生有毒气体的问题,对包含使上述的deg共聚得到的聚酯的聚酯树脂的替代需求不断提高。
5.提出了一种为了改善透明性及耐化学药品性或成型性等而将二乙二醇共聚量规定在特定的范围而得到的共聚聚酯及由上述共聚聚酯构成的成型品的技术(例如,参照专利文献1、2、3),然而,通过这些技术在连续生产膜及成型品时,存在在模附近等附着异物的问题。此外,对于在模附近等附着异物的问题,提出了一种通过控制聚对苯二甲酸乙二醇酯中的对苯二甲酸与乙二醇的环状三聚体来改善的技术(例如,参照专利文献4、5),然而,通过这些技术,在连续生产膜、成型品和纤维时,产生在模及模具附近等粘附低熔点异物,其粘附于膜的表面使商品价值下降,此外产生因树脂的劣化使着色及分子量下降的问题,而且产生再利用时等的问题,从而需要解决方案。现有技术文献专利文献
6.专利文献1:日本特开2016-141791号公报专利文献2:日本特开2006-206860号公报专利文献3:国际公布wo18/025801号专利文献4:日本特开平5-255491号公报专利文献5:日本特开2002-47340号公报
技术实现要素:
发明所要解决的问题
7.本发明是鉴于该现有技术中的问题而提出的,解决了如下课题:在连续地生产膜、成型品和纤维时发生模的污染及膜、成型品和纤维上的异物附着;共聚聚酯的热稳定性及
热氧化稳定性所需的再利用性;再利用时的树脂着色及分子量降低。解决问题的技术方案
8.对于上述的课题,发现通过制成特定的组成的共聚聚酯树脂中的由对苯二甲酸和二乙二醇构成的环状二聚体的含量为7000ppm以下和由对苯二甲酸、二乙二醇和三乙二醇构成的环状二聚体的含量为200ppm以下的共聚聚酯,从而可以提供一种可以利于赋予连续生产率提高而且透明性提高且商品价值高的成型品、膜和纤维的共聚聚酯树脂。而且,通过并用乙二醇、二乙二醇和三乙二醇,结晶性及聚合物粘度降低,膜的成膜性、纤维的成型性及透明性趋于提高。虽然本次共聚聚酯中使用的二乙二醇在成本方面非常优异,但判断为热稳定性及热氧化稳定性低,因此认为膜及成型品在再利用时的共聚聚酯的热稳定性及热氧化稳定性低,因此认为由于膜成膜时及成型品制造时的加热而引起树脂劣化,从而膜及成型品的特性粘度及颜色大幅降低。接着,本发明的发明人发现为了提高共聚聚酯树脂的热稳定性及热氧化稳定性,而在共聚聚酯树脂的羧基末端基浓度(av)为8~25eq/t的范围内趋于改善。而且,为了提高共聚聚酯树脂的进一步的热稳定性及热氧化稳定性,想到了变更在聚合中使用的催化剂的种类。并且,发现优选使用铝化合物和磷化合物的组合代替锑化合物、钛化合物、锗化合物用作聚合催化剂时,则可以提高树脂的热稳定性及热氧化稳定性,并且作为聚合催化剂的活性优异,因此即使为了提高成型品的成型速度、膜的成膜速度、纤维的生产率而降低树脂的数均分子量,得到的成型品及膜的数均分子量不会大幅地降低,由此成型品的成型性、膜成膜性、强度不会产生问题,能够降低成型品、膜和纤维的制造成本。
9.本发明是基于上述发现而完成的,具有以下(1)~(7)的构成:(1)一种共聚聚酯树脂,其特征在于,是以二羧酸和二醇为构成成分的共聚聚酯树脂,在全部聚酯树脂成分中,在二羧酸成分的主要成分为对苯二甲酸、二醇成分的主要成分为乙二醇并将全部的二醇成分设为100mol%时,二乙二醇的含量为7~30mol%和三乙二醇的含量为0.05~2mol%,由对苯二甲酸和二乙二醇构成的环状二聚体的含量为7000ppm以下,以及由对苯二甲酸、二乙二醇和三乙二醇构成的环状二聚体的含量为200ppm以下。(2)根据(1)所述的共聚聚酯树脂,其特征在于,颜色b值为-5.0~10.0。(3)根据(1)或(2)所述的共聚聚酯树脂,其特征在于,羧基末端基浓度av为8~25eq/t,共聚聚酯树脂中含有铝原子和磷原子,共聚聚酯树脂中的铝原子的含量为15~40ppm,共聚聚酯树脂中的磷原子与铝原子的摩尔比为1.8~2.6。(4)根据(1)~(3)中任一项所述的共聚聚酯树脂,其特征在于,将共聚聚酯树脂成型而得到的阶梯状成型板的厚度5mm处的雾度值为10%以下。(5)一种成型品,其特征在于,含有(1)~(4)中任一项所述的共聚聚酯树脂。(6)一种热收缩膜,其特征在于,含有(1)~(4)中任一项所述的共聚聚酯树脂。(7)一种纤维,其特征在于,含有(1)~(4)中任一项所述的共聚聚酯树脂。发明的效果
10.本发明的共聚聚酯树脂在连续地生产膜、成型品和纤维时,很少发生模的污染及膜和成型品上的异物附着,具有树脂的热稳定性及热氧化稳定性所需的再利用性,再利用时的树脂着色大幅降低。
11.另外,由本发明的共聚聚酯树脂得到的成型品、膜和纤维可以具有再利用性、耐热
性和出色的外观。
具体实施方式
12.以下,对本发明的共聚聚酯树脂进行具体说明。本发明的共聚聚酯树脂是以二羧酸成分和二醇成分为构成成分的聚酯树脂。在二羧酸成分的主要成分为对苯二甲酸、二醇成分的主要成分为乙二醇并将全部的二醇成分设为100mol%时,二乙二醇的含量为7~30mol%和三乙二醇的含量为0.05~2mol%。二羧酸成分的主要成分为对苯二甲酸,这是指在二羧酸成分中以mol%单位计含有最多成分的是对苯二甲酸。二醇成分的主要成分为乙二醇,这是指在二醇成分中以mol%单位计含有最多成分的是乙二醇。二乙二醇的含量优选为10~30mol%,更优选为15~25mol%。三乙二醇的含量优选为0.05~1.5mol%,更优选为0.1~1.2mol%。
13.在本发明中,由对苯二甲酸和二乙二醇构成的环状二聚体、由对苯二甲酸、二乙二醇和三乙二醇构成的环状二聚体表示以下的化合物。前者是以对苯二甲酸、二乙二醇、对苯二甲酸、二乙二醇的顺序键合成环状的环状二聚体(以下简称为t2d2),后者是以对苯二甲酸、二乙二醇、对苯二甲酸、三乙二醇的顺序键合成环状的环状二聚体(以下简称为t2d1te1)。其特征在于t2d2的含量为7000ppm以下和t2d1te1的含量为200ppm以下。在本发明中,“共聚聚酯树脂”不仅包括称为聚酯的化学物质,还包括称为t2d2和t2d1te1的低聚物成分及后述的催化剂成分。但在对称为聚酯的化学物质进行说明时,为方便起见,有时记载为“共聚聚酯树脂”。
14.通过使二乙二醇和三乙二醇的含量在上述范围内,能够得到透明性高、即非晶性的共聚聚酯树脂。由于二乙二醇和三乙二醇的含量在小于上述范围的下限时为结晶性,因此成型品及膜的透明性变差,无法达到充分的透明性,从而趋于丧失商品价值。当三乙二醇的含量超过2mol%时,则耐热性趋于降低。此外,从生产时的经济性出发,该含量的下限值为0.05mol%。予以说明,这里非晶性是指使用差示扫描量热计(dsc)将利用yamato dp63干燥机在120℃下放置120分的试料以20℃/min从-100℃升温至300℃,然后以50℃/min降温至-100℃,接着以20℃/min从-100℃升温至300℃的二次升温过程中,任一个升温过程中均未出现熔融峰。本发明的共聚聚酯树脂是非晶性的,由此特别是即使是厚壁的成型体,也可以具有能够优选使用的透明性。即,本测定条件下所说的“非晶性”表示可以高质量地保持膜的透明性,并且即使是制成厚壁的膜,也可以保持充分的透明性。而且,由于t2d2量也增加,在连续膜成膜及纤维的挤出成型机的模的树脂出口附近及注塑成型机的模具等的污染非常严重,所附着的异物附着于成型体表面使商品价值趋于下降。此外,当超过上述范围时,则游离的t2d1te1变多,在连续膜成膜、纤维挤出成型时,在挤出成型机的模的树脂出口附近及注塑成型机的模具等的污染非常严重,所附着的异物附着于膜、成型品和纤维的表面使商品价值趋于下降。虽然附着的机理尚不明确,但由于t2d2中的二乙二醇的影响,熔点及玻璃化转变温度低,因此表现出粘接性,成为粘接剂。而且,由于t2d1te1中的二乙二醇和三乙二醇的影响,熔点及玻璃化转变温度变得低于t2d2,因此表现出强粘接性,通过t2d2与t2d1te1的协同效果,在成型时及膜挤出成型机的模上附着增多。
15.二乙二醇也可以在共聚聚酯树脂聚合时通过乙二醇缩合而生成。由该缩合生成的
二乙二醇的量因聚合条件、制造的装置而改变,但相对于总二醇成分为0.5~2.0mol%左右。考虑该量,只要考虑作为原料添加的二乙二醇的量即可。另外,三乙二醇也可以在共聚聚酯树脂聚合时通过乙二醇与二乙二醇缩合而生成。由该缩合生成的三乙二醇的量也因聚合条件、制造的装置而改变,但在聚合开始时仅存在乙二醇的情况下,相对于总二醇成分为0.01~0.3mol%左右。考虑该量,只要考虑作为原料添加的三乙二醇的量即可,有时也可以特意作为原料不添加三乙二醇。由于三乙二醇也通过乙二醇与二乙二醇缩合来生成,因此控制在上述范围内对于保持商品价值很重要。
16.t2d2的含量优选为6000ppm以下,更优选为5000ppm以下。当超过7000ppm时,则在膜及纤维生产时挤出成型机的模的树脂出口附近的污染变得严重,所附着的异物附着于成型体表面而使表面状态变差,对透明性也产生影响使商品价值下降。此外,在成型品的连续注塑成型时注塑成型的模具排气口被堵塞,不能得到正常的成型品。此外,从生产时的经济性出发,该含量的下限值为1000ppm。t2d2的含量是通过后述实施例的测定方法进行定量得到的值。
17.t2d1te1的含量优选为150ppm,更优选为100ppm以下。当超过200ppm时,则在膜及纤维生产时挤出成型机的模的树脂出口附近的污染变得严重,所附着的异物附着于成型体表面而使表面状态变差,对透明性也产生影响使商品价值趋于下降。此外,在成型品的连续注塑成型时注塑成型的模具排气口被堵塞,不能得到正常的成型体。此外,从生产时的经济性出发,该含量的下限值为1ppm。t2d1te1的含量是通过后述实施例的测定方法进行定量得到的值。
18.本发明的共聚聚酯树脂主要的二羧酸成分是对苯二甲酸,对苯二甲酸成分相对于总二羧酸成分的比例优选为70mol%以上,更优选为80mol%以上,进一步优选为90mol%以上,最优选为100mol%。
19.作为能够与对苯二甲酸一起使用的其他二羧酸成分,可举出:(1)间苯二甲酸、邻苯二甲酸、2,6-萘二甲酸、二苯基-4,4
’‑
二羧酸、二苯氧基乙烷二羧酸等芳香族二羧酸及其功能性衍生物;(2)己二酸、癸二酸、琥珀酸、戊二酸、二聚酸、十二烷二羧酸、壬二酸等脂肪族二羧酸及其功能性衍生物;(3)六氢对苯二甲酸、六氢间苯二甲酸、环己烷二羧酸等脂环族二羧酸及其功能性衍生物等。
20.本发明的共聚聚酯树脂优选总二醇成分由乙二醇、二乙二醇和三乙二醇构成,但只要在不损害本发明目的的各种特性的范围内,为了赋予其他功能或改善特性,也可以使用其他的二醇成分。相对于总二醇成分,乙二醇、二乙二醇和三乙二醇的总量优选为85mol%以上,更优选为90mol%以上,进一步优选为95mol%以上,最优选为100mol%。
21.作为其他二醇成分,可举出:(1)四亚甲基二醇、五亚甲基二醇、六亚甲基二醇等脂肪族二醇类;(2)1,3-环己烷二甲醇、1,4-环己烷二甲醇等脂环式二醇类;(3)对苯二甲醇、间苯二甲醇等芳香族二醇类等。这些之中,优选1,4-环己烷二甲醇。此外,这些二醇成分可以单独使用任一种,也可以以任意的比例合用两种以上。
22.本发明的共聚聚酯树脂中,从提高异型挤出成型性的方面出发,优选含有共聚聚酯树脂的酸成分和/或二醇成分的0.001~5mol%的多官能化合物,所述多官能化合物具有3个以上的羧基、羟基或它们的酯形成性基(例如偏苯三酸、均苯四酸、甘油、三羟甲基丙烷
等)。
23.本发明的共聚聚酯树脂可以通过利用直接酯化反应和缩聚反应的制造法或利用酯交换反应和缩聚反应的制造法中的任一方法来生产。上述的反应可以用间歇式反应装置来进行,也可以用连续式反应装置来进行,但经济性和品质的稳定性方面考虑,优选利用连续式反应装置。
24.连续式反应装置(连续式缩聚法)中,酯化反应、酯交换反应和溶融缩聚反应分别可以通过一个步骤来进行,优选分为多个步骤来进行。在酯化反应或酯交换反应分为多个步骤来进行时,反应罐的数量优选为2个罐~3个罐。此外,在溶融缩聚分为多个步骤来进行时,反应罐的数量优选为3个罐~7个罐。
25.在通过连续式缩聚法制造本发明的共聚聚酯树脂时,制备浆料,该浆料中相对于总二羧酸或其酯衍生物1mol,含有1.02~1.5mol、优选为1.03~1.4mol的总二醇,将该浆料连续地供给于含有低聚物的酯化反应工序中。酯化反应温度通常为240~270℃,优选为250~265℃。此外,反应罐内的压力通常为0.2mpa以下,优选为0.01~0.05mpa。此外,缩聚反应的温度通常为265~285℃,优选为270~280℃,反应罐内的压力通常为1.5hpa以下,优选为0.5hpa以下。酯化反应的反应时间优选为5小时以下,特别优选为2~3.5小时。此外,缩聚反应的反应时间优选为3小时以下,特别优选为1~2小时。
26.在通过间歇式缩聚法制造本发明的共聚聚酯树脂时,酯化反应温度通常为220~250℃,优选为230~245℃。此外,反应罐内的压力通常为0.2~0.4mpa,优选为0.25~0.30mpa。此外,缩聚反应可以通过一个步骤来进行,也可以分为多个步骤来进行。在通过一个步骤来进行时,进行逐渐减压和升温,并将最终的温度设在260~280℃,优选为265~275℃的范围,最终的压力通常设为3hpa以下,优选设为0.5hpa以下。酯化反应的反应时间优选为4小时以下,特别优选为2~3小时。此外,缩聚反应的反应时间优选为5小时以下,特别优选为1~3小时。
27.接着,在通过连续式酯交换反应制造低缩聚物时,制备溶液,该溶液含有对苯二甲酸二甲酯和二醇,该二醇相对于对苯二甲酸二甲酯1mol为1.1~1.6mol,优选为1.2~1.5mol,将该溶液连续地供给于酯交换反应工序。酯交换反应温度通常为200~270℃,优选为230~265℃。在酯交换法的情况下,除了缩聚催化剂以外,还需要使用酯交换催化剂。使得到的低缩聚物与上述的连续式缩聚同样地进行反应。
28.另外,在通过间歇式酯交换反应制造低缩聚物时,在间歇式反应器中加入对苯二甲酸二甲酯和二醇,该二醇相对于对苯二甲酸二甲酯1mol为2.3~2.0mol,优选为2.2~2.0mol,并在酯交换催化剂存在下进行反应。使得到的低缩聚物与上述的酯化反应时同样地操作进行缩聚。
29.作为缩聚催化剂,可以使用锑化合物、锗化合物、钛化合物、铝化合物中的至少一种。作为上述锑化合物,例如可举出:三氧化二锑、五氧化锑、乙酸锑、乙二醇锑等。这些之中,优选为三氧化二锑、乙酸锑、乙二醇锑,特别优选为三氧化二锑。相对于所生成的共聚聚酯树脂,这些锑化合物优选含有50~400ppm,进一步优选为100~350ppm,特别优选为150~300ppm。
30.另外,作为上述锗化合物,例如可举出:结晶性二氧化锗、非晶性二氧化锗、四氧化锗、氢氧化锗、草酸锗、氯化锗、四乙氧基锗、四正丁氧基锗、亚磷酸锗等化合物等。这些之
中,进一步优选为结晶性二氧化锗、非晶性二氧化锗,特别优选为非晶性二氧化锗。相对于所生成的共聚聚酯树脂,这些锗化合物优选含有10~100ppm,进一步优选为30~70ppm,特别优选为30~50ppm。
31.另外,作为上述钛化合物,例如可举出:钛酸四乙酯、钛酸四异丙酯、钛酸四正丙酯、钛酸四正丁酯等钛酸四烷基酯及它们的部分水解产物;乙酸钛、草酸氧钛、草酸氧钛铵、草酸氧钛钠、草酸氧钛钾、草酸氧钛钙、草酸氧钛锶等草酸氧钛化合物;偏苯三酸钛、硫酸钛、氯化钛、卤化钛的水解产物、溴化钛、氟化钛、六氟化钛酸钾、六氟化钛酸铵、六氟化钛酸钴、六氟化钛酸锰、乙酰丙酮钛、与羟基多元羧酸或含氮多元羧酸的钛络合物;由钛和硅或锆构成的复合氧化物、钛醇盐与磷化合物的反应物等。这些之中,优选为钛酸四异丙酯、钛酸四丁酯、草酸氧钛钾,特别优选为钛酸四丁酯。相对于所生成的共聚聚酯树脂,这些钛化合物优选含有1~50ppm,进一步优选为2~20ppm,特别优选为3~10ppm。
32.作为缩聚催化剂,从再利用性(耐热性、热氧化稳定性)出发,优选铝化合物。铝化合物优选与磷化合物并用。作为构成在制造本发明的共聚聚酯树脂时所使用的聚合催化剂的铝化合物,可以没有限定地使用公知的铝化合物。
33.作为铝化合物,具体地,可举出:乙酸铝、碱性乙酸铝、乳酸铝、氯化铝、氢氧化铝、羟基氯化铝和乙酰丙酮铝、草酸铝等有机铝化合物及它们的部分水解产物等。这些之中,优选羧酸盐、无机酸盐和螯合物,这些之中,更优选乙酸铝、碱性乙酸铝、乳酸铝、氯化铝、氢氧化铝、羟基氯化铝和乙酰丙酮铝,进一步优选乙酸铝、碱性乙酸铝、氯化铝、氢氧化铝和羟基氯化铝,最优选乙酸铝、碱性乙酸铝。
34.聚合催化剂中使用的铝化合物的使用量,相对于得到的聚酯树脂的总质量,作为铝原子优选残留15~40ppm,更优选为17~38ppm,进一步优选为20~35ppm。在铝原子的残留量在小于上述范围时,有可能催化活性不良。另一方面,当铝原子的残留量超过上述范围时,则存在热稳定性和热氧化稳定性的降低而成为问题的情况、由铝引起的异物产生及着色的增加而成为问题的情况。予以说明,如上所述,即使铝化合物在聚酯聚合时置于减压环境下,作为聚合催化剂的使用量基本100%残留,因此可以认为使用量是残留量。在可以稍微牺牲共聚聚酯树脂的特性的情况下,共聚聚酯树脂中的铝原子的含量也可以为9~42ppm。
35.聚合催化剂中使用的磷化合物没有特别限定,但当使用膦酸系化合物、次膦酸系化合物时,催化剂活性的提高明显,因此优选,这些之中,当使用膦酸系化合物时,催化剂活性的提高特别明显,因此优选。
36.这些磷化合物中,当使用在同一分子内具有酚部分的磷化合物时,树脂的热稳定性和热氧化稳定性的提高明显,因此优选。只要是具有酚结构的磷化合物,就没有特别限定,但当使用在同一分子内具有酚部分的选自膦酸系化合物、次膦酸系化合物中的1种或2种以上的化合物时,催化剂活性的提高效果和树脂的热稳定性和热氧化稳定性的提高效果这两者均明显,因此优选。这些之中,当使用1种或2种以上的在同一分子内具有酚部分的膦酸系化合物时,催化剂活性的提高效果和树脂的热稳定性和热氧化稳定性的提高效果这两者均特别明显,因此优选。
37.另外,作为在同一分子内具有酚部分的磷化合物,可举出:下述通式(1)、(2)所示的化合物等。
38.[化1]p(=o)r1(or2)(or3)
ꢀꢀꢀ
通式(1)
[0039]
[化2]p(=o)r1r4(or2)
ꢀꢀꢀ
通式(2)
[0040]
(通式(1)~(2)中,r1表示包含酚部分的碳原子数1~50的烃基、羟基或卤素基或烷氧基或氨基等取代基和包含酚部分的碳原子数1~50的烃基。r4表示包含氢、碳原子数1~50的烃基、羟基或卤素基或烷氧基或氨基等取代基的碳原子数1~50的烃基。r2、r3分别独立表示包含氢、碳原子数1~50的烃基、羟基或烷氧基等取代基的碳原子数1~50的烃基。但,烃基可以包含支化结构、环己基等脂环结构、苯基或萘基等芳香环结构。r2和r4的末端彼此可以键合。)
[0041]
作为上述的在同一分子内具有酚部分的磷化合物,例如可举出:对羟基苯基膦酸、对羟基苯基膦酸二甲酯、对羟基苯基膦酸二乙酯、对羟基苯基膦酸二苯酯、双(对羟基苯基)次膦酸、双(对羟基苯基)次膦酸甲酯、双(对羟基苯基)次膦酸苯酯、对羟基苯基苯基次膦酸、对羟基苯基苯基次膦酸甲酯、对羟基苯基苯基次膦酸苯酯、对羟基苯基次膦酸、对羟基苯基次膦酸甲酯、对羟基苯基次膦酸苯酯等。另外,可举出下述通式(3)所示的磷化合物。
[0042]
[化3]
[0043]
通式(3)中、x1、x2分别表示氢、碳原子数1~4的烷基、或1价以上的金属。另外,可以是x1的金属为2价以上,不存在x2。而且,还可以对磷化合物配置相当于金属的剩余价数的阴离子。作为金属,优选li、na、k、ca、mg、al。
[0044]
当在聚酯的聚合时添加这些在同一分子内具有酚部分的磷化合物时,铝化合物的催化活性提高,同时聚合得到的聚酯树脂的热稳定性和热氧化稳定性也提高。其理由认为是磷化合物中的受阻酚部分使聚酯树脂的热稳定性和热氧化稳定性提高。这种情况下,当磷化合物的残留量少于31ppm时,上述的热稳定性和热氧化稳定性的提高效果较弱,作为结果,有时未发现本发明的聚酯树脂的热稳定性和热氧化稳定性的改善效果及着色的改善效果。
[0045]
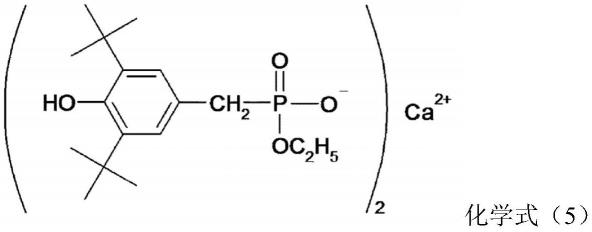
上述之中,作为缩聚催化剂优选使用的磷化合物是选自下述化学式(4)、化学式(5)所示的化合物中的至少一种的磷化合物。
[0046]
[化4]
[0047]
[化5]
[0048]
作为上述的化学式(4)所示的化合物,市售有irganox1222(basf株式会社制造)。此外,作为化学式(5)所示的化合物,市售有irganox1425(basf株式会社制造),均可以使用。
[0049]
聚合催化剂中使用的磷化合物的使用量,相对于得到的共聚聚酯树脂的总质量,作为磷原子优选残留31~119ppm,更优选为39~105ppm,进一步优选为48~92ppm。当超过上述的上下限量的磷原子残留时,则有可能使聚合活性降低。予以说明,如上所述,磷化合物在聚酯树脂聚合时置于减压环境下时,作为催化剂最初添加到体系的使用量的一部分被去除到体系外,但该除去量基本为恒定的比例,因此可以认为考虑到除去比例以残留量来规定也是适当的。在可以稍微牺牲共聚聚酯树脂的特性的情况下,共聚聚酯树脂中的磷原子的含量也可以为19~125ppm。
[0050]
另外,如上所述,本发明中磷化合物与铝化合物的比率也很重要。具体地,本发明中,聚酯树脂中的磷原子与铝原子的摩尔比(p/al比)优选为1.8~2.6,更优选为2.0~2.4,进一步优选为2.1~2.3。铝化合物即使单独作为聚合催化剂来使用,也无法充分发挥催化剂活性。除了铝化合物之外,磷化合物也作为聚合催化剂并以特定的比率并用,由此可以充分地提高催化活性。聚酯树脂中的磷原子与铝原子的摩尔比在上述范围外时,有可能无法充分地发挥作为聚合催化剂的功能。
[0051]
本发明中,在不损害本发明的效果范围内,除上述铝化合物和磷化合物以外,还可以合用钛化合物、锡化合物、锗化合物等含金属的缩聚催化剂,以进一步提高催化活性。这种情况下,锗化合物中相对于得到的聚酯树脂的质量,作为锗原子选为10ppm以下,钛化合物中相对于得到的聚酯树脂的质量,作为钛原子优选为3ppm以下,锡化合物中相对于得到的聚酯树脂的质量,作为锡原子优选为3ppm以下。但是,出于本发明的目的,优选尽可能不使用这些钛化合物、锡化合物、锗化合物等含金属的缩聚催化剂。此外,通常作为聚合催化剂使用的锑化合物对于上述的树脂的热稳定性和热氧化稳定性的提高效果较差,因此避免在本发明中使用。
[0052]
另外,在本发明的共聚聚酯树脂的制造中,可以合用碱金属化合物或碱土金属化合物。作为碱金属化合物或碱土金属化合物,可举出:这些元素的乙酸盐等羧酸盐、醇盐等,
作为粉体、水溶液、乙二醇溶液等添加到反应体系中。
[0053]
在直接酯化法的情况下,上述缩聚催化剂可以在从酯化反应开始前或在加压酯化反应结束后至初始缩聚反应开始前的任意时间点添加。但将锑化合物或钛化合物作为缩聚催化剂使用时,优选在酯化反应前添加。此外,其他缩聚催化剂、热稳定剂、添加物优选在酯化反应后添加。
[0054]
另外,在酯交换法的情况下,上述缩聚催化剂可以在从酯交换反应开始前至初始缩聚反应开始前的任意时间点添加。但钛化合物不仅具有作为缩聚催化剂的功能,而且还具有作为酯交换催化剂的功能,因此优选在酯交换反应开始前添加。此外,其他缩聚催化剂、热稳定剂、添加物优选在从酯交换反应结束后添加。作为酯交换催化剂,优选乙酸锰、乙酸镁、四丁醇钛等钛化合物等。酯交换催化剂需要在酯交换反应开始前添加。
[0055]
另外,在使用上述铝化合物以外的催化剂时,作为稳定剂,可以使用磷化合物。作为磷化合物,例如可举出:磷酸、亚磷酸、膦酸及它们的衍生物等。作为优选的具体例子,可举出:磷酸、磷酸三甲酯、磷酸三丁酯、磷酸三苯酯、磷酸单甲酯、磷酸二甲酯、磷酸单丁酯、磷酸二丁酯、亚磷酸、亚磷酸三甲酯、亚磷酸三丁酯、甲基膦酸、甲基膦酸二甲酯、乙基膦酸二甲酯、苯基膦酸二甲酯、苯基膦酸二乙酯、苯基膦酸二苯酯。这些之中,特别优选磷酸三甲酯、磷酸。相对于所生成的共聚聚酯,这些磷化合物优选含有1~100ppm,进一步优选为3~70ppm,特别优选为5~50ppm。
[0056]
为了改善共聚聚酯树脂的色调,可以配入钴化合物。通过添加该钴化合物,特别是可以减小颜色b值。钴化合物中作为钴原子相对于共聚聚酯树脂优选含有0.5~30ppm,进一步优选为1~20ppm,特别优选为1~15ppm的范围。当钴原子的含量超过上述范围时,则由于钴金属的还原,共聚聚酯树脂发黑或发蓝变强,颜色l值小于50,或颜色b值小于-5,商品价值下降。作为钴化合物,可举出:乙酸钴、氯化钴、苯甲酸钴、铬酸钴等。这些之中,优选乙酸钴。
[0057]
通过上述的连续式缩聚法或间歇式缩聚法得到的共聚聚酯树脂,通常从设置于反应罐底部的抽出口以绞线状抽出并水冷后,切成小片状或片材状。
[0058]
在控制本发明的三乙二醇量的同时,游离的t2d2及t2d1te1的含量少的共聚聚酯树脂可以通过以下方法制造。优选通过在酯化反应或酯交换反应期间、或者在反应后追加添加特定的二醇成分来进行缩聚反应的方法。特定的二醇成分优选为二乙二醇、三乙二醇,更优选为二乙二醇。例如,先使考虑了追加添加量的量的原料单体进行酯化反应或酯交换反应,在该反应后添加二乙二醇,搅拌5分钟以上后进行缩聚。追加添加的二乙二醇成分优选为总二乙二醇成分的7.5~30摩尔%。另外,先使考虑了追加添加量的量的原料单体进行酯化反应或酯交换反应,在其反应期间添加二乙二醇,进一步在反应后追加添加二乙二醇,搅拌5分钟以上后进行缩聚。追加添加的二乙二醇成分优选为总二乙二醇成分的5~20mol%。进一步,先使考虑了追加添加量的量的原料单体进行酯化反应或酯交换反应,在其反应期间添加二乙二醇和三乙二醇,进一步在反应后追加添加二乙二醇,搅拌5分钟以上后进行缩聚。追加添加的二醇(二乙二醇+三乙二醇)成分优选为总二醇(二乙二醇+三乙二醇)成分的7.5~30mol%。
[0059]
由于三乙二醇也由乙二醇与二乙二醇的缩合生成,因此,认为通过在酯化反应或酯交换反应的中途、或者在反应后追加添加特定的二醇成分并搅拌,从而控制由酯化反应或酯交换反应生成的三乙二醇的量,进而在缩聚反应中,使已经由酯化反应或酯交换反应生成的环状的t2d2或t2d1te1开环,减少t2d2、t2d1te1。进一步,通过使以绞线状抽出并水冷后并切成小片状或片状的切成物在二乙二醇或三乙二醇的蒸气中进行一定时间的接触,也能使t2d2、t2d1te1减少,虽然不清楚其详细机理,但认为通过二乙二醇、三乙二醇的蒸气使环状的t2d2、t2d1te1开环。
[0060]
如上所述得到的共聚聚酯树脂的末端羧基浓度以每吨聚合物计优选为8~25当量。末端羧基浓度更优选为23当量/吨以下。通过使末端羧基浓度在上述范围内,能够有助于抑制共聚聚酯树脂的着色。在共聚聚酯树脂的着色即使稍差也被允许时,末端羧基浓度可以为32当量/t以下。如果不考虑生产率(反应时间),则末端羧基浓度的下限为0当量/t。
[0061]
本发明的共聚聚酯树脂的数均分子量优选为15000~30000,更优选为17000~28000,进一步优选为18000~27000。当数均分子量小于上述范围时,结晶性提高而使雾度增高,由于树脂凝聚力不足,因此有时成型品的强伸度不足,变脆而不能使用。另一方面,当超过上述范围时,则溶融粘度过高,各种成型加工中的最适温度也升高,热稳定性变差,片材成膜性降低,而且上述的t2d2和t2d1te1增加,结果是成型体的透明性变差。
[0062]
本发明的共聚聚酯树脂的玻璃化转变温度优选为40℃以上且小于120℃,更优选为45℃以上且小于115℃,进一步优选为50℃以上且小于110℃,特别优选为50℃以上且小于70℃。这里玻璃化转变温度是指使用差示扫描量热仪(dsc),以20℃/min升温进行测定的值。在玻璃化转变温度在小于上述范围时,异形挤出的成型品在夏季在室外使用时,在夏季以密闭状态进行的产品输送及仓库保管时,经常发生膜及异形挤出的成型品发生热变形的问题。此外,在玻璃化转变温度超过上述范围时,片材的成膜性及透明性趋于下降,有可能根据用途而无法使用。
[0063]
本发明的共聚聚酯树脂的颜色b值优选为-5.0~10.0,下限更优选为-3.0,进一步优选为-2.5,上限更优选为8,进一步优选为7。当颜色b值超过10.0时,则共聚聚酯树脂的黄度变强,在色调方面不优选。另一方面,当颜色b值在负值方向数值相比于-5.0越来越大,共聚聚酯树脂蓝色色调变得明显,有时根据用途而无法使用。
[0064]
本发明的共聚聚酯树脂在模具温度10℃下成型而得到阶梯状成型板,在该阶梯状成型板的厚度5mm部位处的雾度值优选为15%以下,进一步优选为10%以下,特别优选为7%以下。当雾度值超过上述值时,成型品及膜的透明性变差,有时在严格要求透明性的用途中无法使用。
[0065]
本发明的共聚聚酯树脂中,也可以根据用途适当添加其他的成分。例如,可以添加抗冲击性提高剂、填充剂、紫外线吸收剂、表面处理剂、润滑剂、光稳定剂、颜料、抗静电剂、抗菌剂、交联剂、硫系酸化防止剂、阻燃剂、增塑剂、加工助剂、发泡剂等。本发明的共聚聚酯可通过用一直以来的pet及聚氯乙烯等通常使用的挤出吹塑成型、拉深成型、注塑成型、异形挤出成型、砑光加工成型等,适宜地成型为各种成型体。实施例
[0066]
以下,通过实施例具体地说明本发明,但本发明并不限于此。予以说明,根据以下方法测量共聚聚酯树脂的特性。
[0067]
1)数均分子量通过使用氯仿/六氟异丙醇混合溶剂(体积比=9/1)作为溶剂以及使用聚苯乙烯作为测试标准的waters(沃特世)凝胶渗透色谱法进行测定。将氯仿/六氟异丙醇混合溶剂(体积比=9/1)作为洗脱液得到聚苯乙烯换算的测定值。
[0068]
2)共聚聚酯树脂的特性粘度(iv)将在60℃下干燥24小时的样品精确称重0.1g,并使其溶解于25ml苯酚/四氯乙烷(3/2(质量比))的混合溶剂中,使用奥氏(ostwald)粘度计在30℃下进行测定。
[0069]
3)t2d2的含量将50mg共聚酯树脂溶解于1ml六氟异丙醇/氯仿混合溶液(体积比=1/9)中,并进一步添加4ml氯仿进行稀释。向其中加入10ml甲醇以使聚合物沉淀,然后进行离心分离。将离心分离后的上清液浓缩至干固,再溶解于0.4ml的二甲基甲酰胺中,通过高效液相色谱法测定t2d2的含量。装置:waters acquity uplc色谱柱:waters beh-c182.1
×
150mm(waters公司制造)
[0070]
4)t2d1te1的含量通过与上述t2d2含量测定相同的方法,通过高效液相色谱法测定t2d1te1含量。
[0071]
5)共聚聚酯树脂的组成比将约5mg的共聚聚酯树脂试料溶解在0.7ml的氘代氯仿和三氟乙酸的混合溶液(体积比9/1)中,并使用1h-nmr(varian公司制造,unity 50)来求出。
[0072]
6)色调使用色度计(日本电色株式会社制造,型号1001dp)测定共聚聚酯树脂的小片的颜色,求出b值。
[0073]
7)雾度值使用注塑成型机(株式会社名机制作所制造,m-150c-dm),在280℃下使共聚聚酯树脂熔融,在模具温度为10℃下成型厚度为2~11mm的阶梯状成型板,厚度为5mm的位置通过雾度计(日本电色株式会社制造,型号ndh2000)测定雾度值(%)。
[0074]
8)成型试验(片材成膜评价)将经过干燥的共聚聚酯树脂试料加入到带有片材用模的挤出机中,并在2天期间连续成型具有约0.5mm厚度的片材。根据以下标准通过肉眼评价模出口的污染附着状況及片材表面的状态。(评价标准)
◎
:几乎不存在模出口污染物的附着,片材表面状态良好;
○
:仅存在微量的模出口污染物的附着,片材表面状态良好;
△
:存在少量的模出口污染物的附着,片材表面上存在少量附着异物;
×
:模出口污染物的附着非常严重,片材表面上存在很多附着物。
[0075]
9)共聚聚酯树脂的dsc测定将在yamato dp63干燥机中在120℃下放置120分钟的样品,使用差示扫描量热仪(dsc)确认在下述二次升温过程中确认是否出现熔融峰,所述二次升温过程为以20℃/min从-100℃升温至300℃,然后以50℃/min降温至-100℃,接着以20℃/min从-100℃升温至
300℃,在二次升温过程中的任一过程中均没有出现熔融峰的共聚聚酯树脂设为
“○”
,在任一过程中出现熔融峰的共聚聚酯树脂设为
“×”
。
[0076]
10)热稳定性评价:热氧化分解参数(td)将经干燥的共聚聚酯树脂的小片3g放入玻璃试管中,在氮气气氛下,在280℃的油浴中浸渍120分钟以使其熔融。通过下式计算。测定加热后的[iv]
f1
。td如下求出。其中,[iv]i及[iv]
f1
分别指加热试验前和加热试验后的iv(dl/g)。td=0.245{[iv]
f1-1.47-[iv]
i-1.47
}共聚聚酯树脂的热氧化分解参数(td)的值小表示热稳定性高。
[0077]
(实施例1)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)、二乙二醇(deg)和三乙二醇(teg)作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.2而制成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为3小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为1小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使其与目标组成一致,搅拌15分钟,使其反应。
[0078]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为19,000。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。
[0079]
(实施例2)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)和二乙二醇作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.2而制成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为3小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为1小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使其与目标组成一致,搅拌15分钟,使其反应。
[0080]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化
合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为20,000。
[0081]
将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。
[0082]
(实施例3~5)与实施例2同样地进行反应,得到共聚聚酯树脂。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。
[0083]
(实施例6)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)和二乙二醇(deg)作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为1.6而制成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为3小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为1小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使其与目标组成一致,搅拌15分钟,使其反应。
[0084]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。得到数均分子量为20,000、聚合物羧基末端基浓度为24eq/t的共聚聚酯树脂。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型来评价时,结果成型性良好,但发现树脂颜色为4.5、热稳定性评价为0.10略有降低。
[0085]
(实施例7)在具有搅拌器和蒸馏冷凝器的容积10l酯化反应槽中加入对苯二甲酸(tpa)2414质量份、乙二醇(eg)和二乙二醇(deg),以使相对于生成共聚聚酯树脂含有锑金属为250ppm和钴金属为10ppm地添加三氧化二锑和乙酸钴的乙二醇溶液作为催化剂。然后,将反应体系内逐渐升温至最终240℃,并在压力0.25mpa下进行180分钟的酯化反应。在确认蒸馏水不再从反应体系内出来后,使反应体系内恢复至常压,在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使二乙二醇与目标组成一致,搅拌约15分钟,使其反应。添加磷酸三甲酯的乙二醇溶液以使相对于生成共聚聚酯含有残留磷原子为50ppm。
[0086]
将得到的低聚物转移至缩聚反应槽,在逐渐升温的同时减压,使最终温度为270℃、压力为0.2hpa。进行反应直到对应于特性粘度的搅拌叶片的扭矩值达到期望值,停止缩聚反应。将得到的溶融共聚聚酯树脂从缩聚槽下部的抽出口以绞线状抽出,在水槽中冷却后切成小片状。数均分子量为19000。与实施例1同样地进行加热处理。结果如表1所示。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。阶梯状成型板的雾度和热稳定性虽然趋于稍差,但没有问题。
[0087]
(实施例8、9)与实施例2同样地进行反应,得到共聚聚酯树脂。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。阶梯状成型板的雾度虽然趋于稍差,但没有问题。
[0088]
(实施例10)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)和二乙二醇(deg)作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.2而制成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为4小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为2小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使其与目标组成一致,搅拌15分钟,使其反应。
[0089]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为19,000。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。
[0090]
(实施例11)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)和二乙二醇(deg)作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.2而制成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为2.5小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为0.8小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。在生成的低聚物中添加二乙二醇的量为相当于酯化反应前所添加的量的20%,以使其与目标组成一致,搅拌15分钟,使其反应。
[0091]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为19,000。将评价结果示于表1。将本实施例中得到的共聚聚酯树脂通过8)的方法以片材成型进行评价,成型性良好。色调、阶梯状成型板的雾度和热稳定性虽然趋于稍差,但没有问题。
[0092]
(比较例1)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是通过将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.0而制成的。接着,在搅拌下并在罐内压力0.17mpa、255℃的条件下,以平均保留时间为3小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力0.05mpa并在搅拌下、261℃的条件下,以平均保留时间为1小时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、266~267℃的条件下进行酯化反应。在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在268℃、4.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在270℃、0.57kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在274℃、0.17kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为19000。将评价结果示于表1。环状低聚物量的测定未实施。
[0093]
(比较例2)使用与实施例2同样的合成方法将deg量调整为5mol%进行合成,并进行评价。当deg较少,则在所有的评价中均为较差。将评价结果示于表1。
[0094]
(比较例3)使用与实施例2同样的合成方法将deg量调整为34mol%进行合成,并进行评价。当deg较多,则片材成膜评价、热稳定性均为较差。将评价结果示于表1。
[0095]
(比较例4)使用与实施例2同样的合成方法将deg量调整为19mol%、将teg量调整为2.5mol%进行合成,并进行评价。当teg较多时,在所有的评价中均为较差。将评价结果示于表1。
[0096]
(比较例5)在预先残留有反应物的第一酯化反应罐中,连续地供给浆料,该浆料是将高纯度对苯二甲酸(tpa)作为二羧酸成分,将乙二醇(eg)和二乙二醇作为二醇成分,将总二醇成分相对于二羧酸成分的摩尔比(g/a)配制为2.2而成的。接着,在搅拌下并在罐内压力为0.15mpa、257℃的条件下,以平均保留时间为3小时地进行酯化反应。将该反应物转移至第二酯化反应罐,在罐内压力为0.05mpa并在搅拌下、257℃的条件下,以平均保留时间为1小
时地进行酯化反应。接着,将该酯化反应物转移至第三酯化反应罐,在搅拌下并在罐内压力0.05mpa、257℃的条件下进行酯化反应。
[0097]
在该酯化反应产物中,添加一定量的铝化合物(碱性乙酸铝)的乙二醇溶液和磷化合物(irganox1222:上述化学式(4)的化合物)的乙二醇溶液,连续地供给到第一缩聚反应罐中,在搅拌下并在261℃、6.7kpa下用1小时进行缩聚反应,接着在第二缩聚反应罐中在搅拌下并在272℃、0.6kpa下用1小时进行缩聚反应,进一步在最终缩聚反应罐中在搅拌下并在275℃、0.10~0.20kpa下用1小时进行缩聚反应。缩聚反应后,使反应物通过聚合物过滤器,将溶融状态的共聚聚酯以绞线状从模头喷嘴中抽出,在冷却浴中水冷,然后切成小片状。数均分子量为20,000。t2d1te1、t2d2变多,则在片材成膜评价、雾度的评价中均为较差。将评价结果示于表1。
[0098]
[表1]
工业上的可利用性
[0099]
本发明的共聚聚酯树脂的透明性、色调和成型性优异,很少发生模的污染及成型品和膜上的异物附着,因此可以有利地赋予经济性优异、商品价值高的成型体,对工业界贡献很大。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1