具有多位点六甘醇修饰的两亲性BODIPY化合物NBDP及其制备方法和应用
具有多位点六甘醇修饰的两亲性bodipy化合物nbdp及其制备方法和应用
技术领域
1.本发明属于高分子材料技术领域,具体为一种具有多位点六甘醇修饰的两亲性bodipy化合物nbdp及其制备方法和应用。
背景技术:
2.bodipy是一类优良的光敏剂,其结构多样,易于制备,具有优异的光学性质,在生物功能材料领域表现突出。但是其结构具有一定的刚性,因而水溶性和生物相容性有限,这极大地限制了其在肿瘤诊断与治疗领域的应用。药物自我输送系统已被提议作为一种新的癌症治疗范例,其具有以下优点:具有特定的尺寸形状,易于在肿瘤部位富集;无需大量赋形剂,无载体可能诱发的毒性和免疫原性;优异的载药量;没有繁琐的纳米载体制备过程。[biomaterials,2017,112(10),234-247.]
技术实现要素:
[0003]
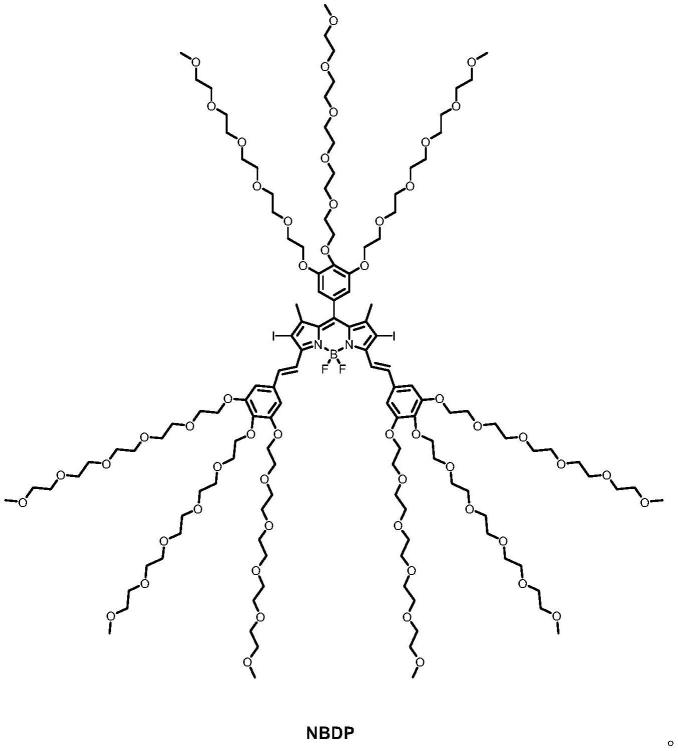
本发明的第一个目的是设计合成了一种具有多位点六甘醇修饰的两亲性bodipy化合物nbdp,六甘醇的多位点引入赋予其更优的水溶性,可以在水中自组装为球形纳米粒子,进一步提供了该分子作为载体,负载抗癌药物阿霉素(dox)作为肿瘤的光动力及化疗联合治疗剂中的应用。其化学结构式如下:
[0004][0005]
本发明的第二个目的是提供一种所述具有多位点六甘醇修饰的两亲性bodipy化合物nbdp的合成方法,其合成路线如下所示:
[0006][0007]
所述合成方法为:
[0008]
(1)将化合物1、二氯甲烷和少量三氟乙酸混合,氮气环境下搅拌10~15分钟。缓慢滴加2,4-二甲基吡咯,常温搅拌2.5~3.5小时。加入2,3-二氯-5,6-二氰基-1,4-苯醌,常温搅拌反应0.5~1.5小时。然后将反应置于冰水浴中,依次滴加三乙胺和三氟化硼乙醚,常温反应过夜。反应结束后,经后处理得到化合物2;
[0009]
作为优选,所述化合物1、2,3-二氯-5,6-二氰基-1,4-苯醌、三乙胺、三氟化硼乙醚的物质的量之比为1:1.15~1.25:9.5~10.5:19.5~20.5;
[0010]
作为优选,所述二氯甲烷与2,4-二甲基吡咯的体积比为300~320:1;
[0011]
作为优选,所述后处理的方法为:反应物用二氯甲烷和水萃取,收集有机相,干燥过滤后得到粗产物,然后以二氯甲烷/甲醇体积比为18~22:1的混合液为洗脱剂,通过硅胶层析柱分离纯化得到化合物2。
[0012]
(2)将化合物2与碘单质混合,加入乙醇作为溶剂得到溶液,将碘酸溶于水后缓慢滴加入上述溶液中,在常温下搅拌过夜。反应结束后,减压除去乙醇,经后处理得到化合物3;
[0013]
作为优选,所述化合物2、碘单质、碘酸的物质的量之比为1:1.08~1.22:2.15~2.25;
[0014]
作为优选,所述乙醇与水的体积比为3.5~4.5:1;
[0015]
作为优选,所述后处理的方法为:反应物用二氯甲烷和水萃取,收集有机相,干燥过滤后得到粗产物,然后以二氯甲烷/甲醇体积比为18~22:1的混合液为洗脱剂,通过硅胶层析柱分离纯化得到化合物3。
[0016]
(3)将化合物1、化合物3、对甲苯磺酸一水合物、哌啶、分子筛(即沸石)混合,加入无水甲苯作为溶剂,115~125℃下回流反应并点板监测。反应结束后,浓缩除去甲苯,经后处理得到两亲性bodipy化合物nbdp;
[0017]
作为优选,所述化合物1、化合物3物质的量之比为1:0.38~0.42;
[0018]
作为优选,所述后处理的方法为:反应物用二氯甲烷和水萃取,收集有机相,干燥过滤后得到粗产物,然后以二氯甲烷/甲醇体积比为18~22:1的混合液为洗脱剂,通过硅胶层析柱分离纯化得到两亲性bodipy(nbdp)。
[0019]
本发明的第三个目的是提供具有多位点六甘醇修饰的两亲性bodipy化合物nbdp在作为药物载体中的应用。
[0020]
本发明的第四个目的是提供一种肿瘤的光动力及化疗联合治疗剂,包括抗肿瘤药物和用于负载所述抗肿瘤药物的药物载体,所述药物载体采用所述两亲性bodipy化合物nbdp。所述抗肿瘤药物为盐酸阿霉素dox。
[0021]
作为优选,所述肿瘤的光动力及化疗联合治疗剂的制备方法包括如下步骤:
[0022]
(1)将两亲性nbdp溶于水,自组装为球形纳米粒子,作为载体水溶液备用;将盐酸阿霉素dox溶于水中,加入四氢呋喃,滴加三乙胺中和盐酸后得到阿霉素溶液;
[0023]
(2)在快速搅拌下,将阿霉素溶液缓慢滴加到nbdp水溶液中。在室温下继续搅拌16~24小时后,静置过夜;
[0024]
(3)经过滤去除过量的阿霉素悬浮物,滤液使用分子量为1000的透析袋在水中进行透析,去除未包裹的游离阿霉素。透析完毕后将纳米粒子溶液冻干,得到墨绿色干燥油状物,即nbdp/dox纳米粒子。
[0025]
作为优选,所述的载体水溶液中两亲性nbdp的浓度为3.0
×
10-4
~3.7
×
10-4
mol/l;所述的两亲性nbdp与dox的投料质量比为1:0.8~1.2。
[0026]
本发明的有益效果在于:
[0027]
本发明设计合成了一种具有多位点六甘醇修饰的两亲性bodipy化合物nbdp,含有多数具有亲水性的醚氧链,合成方法简单,反应条件温和。制备所得的nbdp具有良好的水溶性,可以在水中自组装为球形纳米粒子,能够作为载体负载抗癌药物阿霉素(dox),在肿瘤
的光动力治疗联合化疗中具有应用前景。
附图说明
[0028]
图1为实施例1中合成的化合物2的核磁共振氢谱谱图(氘代氯仿为溶剂);
[0029]
图2为实施例1中合成的化合物2的核磁共振碳谱谱图(氘代氯仿为溶剂);
[0030]
图3为实施例1中合成的化合物2的核磁共振氟谱谱图(氘代氯仿为溶剂);
[0031]
图4为实施例1中合成的化合物2的高分辨质谱谱图;
[0032]
图5为实施例1中合成的化合物3的核磁共振氢谱谱图(氘代氯仿为溶剂);
[0033]
图6为实施例1中合成的化合物3的核磁共振碳谱谱图(氘代氯仿为溶剂);
[0034]
图7为实施例1中合成的化合物3的核磁共振氟谱谱图(氘代氯仿为溶剂);
[0035]
图8为实施例1中合成的化合物3的高分辨质谱谱图;
[0036]
图9为实施例1中合成的化合物nbdp的核磁共振氢谱谱图(氘代氯仿为溶剂);
[0037]
图10为实施例1中合成的化合物nbdp的核磁共振碳谱谱图(氘代氯仿为溶剂);
[0038]
图11为实施例1中合成的化合物nbdp的核磁共振氟谱谱图(氘代氯仿为溶剂);
[0039]
图12为实施例1中合成的化合物nbdp的高分辨质谱谱图;
[0040]
图13为应用例1中合成的纳米粒子nbdp/dox nps的透射电镜图;
[0041]
图14为测试例1中合成的纳米粒子nbdp/dox nps培育细胞后的荧光显微镜图。
具体实施方式
[0042]
如前所述,鉴于现有技术的不足,本案发明人经长期研究和大量实践,提出了本发明的技术方案,其主要是依据至少包括:
[0043]
将六甘醇多位点修饰在bodipy分子上,使其具有良好的亲水性,制备过程简单快捷,反应条件温和;利用该分子良好的水溶性可将其应用于抗肿瘤药物载体,制备nbdp/dox纳米粒子以将其应用于肿瘤的光动力治疗联合化疗中。
[0044]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
[0045]
本发明的具体合成方法如下:
[0046][0047]
(1)将化合物1加到两颈烧瓶中,加入二氯甲烷作为溶剂,滴加少量三氟乙酸,氮气环境下搅拌10~15分钟。抽取2,4-二甲基吡咯,缓慢滴加到反应瓶中,常温搅拌2.5~3.5小
时,溶液颜色由无色逐渐转变为红色。接着将2,3-二氯-5,6-二氰基-1,4-苯醌加入反应瓶中,常温搅拌反应0.5~1.5小时。然后将反应置于冰水浴中,依次滴加三乙胺和三氟化硼乙醚,常温反应过夜。反应结束后,经后处理得到化合物2;化合物1、2,3-二氯-5,6-二氰基-1,4-苯醌、三乙胺、三氟化硼乙醚的物质的量之比为1:1.15~1.25:9.5~10.5:19.5~20.5,二氯甲烷与2,4-二甲基吡咯的体积比为300~320:1。
[0048]
(2)将化合物2与碘单质加到两颈烧瓶中,加入乙醇作为溶剂。再将碘酸溶解在水中,通过恒压滴液漏斗缓慢滴加到上述反应瓶中,在常温下搅拌过夜。反应结束后,减压除去乙醇,经后处理得到化合物3;化合物2、碘单质、碘酸的物质的量之比为1:1.08~1.22:2.15~2.25;乙醇与水的体积比为3.5~4.5:1。
[0049]
(3)将化合物1、化合物3、对甲苯磺酸一水合物、哌啶、分子筛分别加入到干燥的两颈烧瓶中,加入无水甲苯作为溶剂,115~125℃下回流反应并点板监测。反应结束后,浓缩除去甲苯,经后处理得到两亲性bodipy化合物nbdp;化合物1、化合物3物质的量之比为1:0.38~0.42。
[0050]
将该分子应用于制备抗肿瘤药物载体中的具体步骤为:
[0051]
(1)称取4.5~5.5mg两亲性nbdp,溶于4.5~5.5ml水中。称取4.5~5.0mg盐酸阿霉素溶解于0.8~2.0ml水中,加入1.0~1.5ml四氢呋喃,滴加15~20μl三乙胺中和盐酸,得到阿霉素溶液;
[0052]
(2)在快速搅拌下,将阿霉素溶液缓慢滴加到nbdp水溶液中。在室温下继续搅拌16~24小时后,静置过夜;
[0053]
(3)经过滤去除过量的阿霉素悬浮物,滤液使用分子量为1000的透析袋在水中进行透析,去除未包裹的游离阿霉素,透析完毕后将纳米粒子溶液冻干,得到墨绿色干燥油状物,即nbdp/dox纳米粒子。
[0054]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此。
[0055]
实施例1
[0056]
化合物2的制备:
[0057]
将化合物1(0.65g,0.756mmol)加到250ml两颈烧瓶中,加入60ml二氯甲烷作为溶剂,滴加少量三氟乙酸,氮气环境下搅拌十分钟。抽取2,4-二甲基吡咯(195μl),缓慢滴加到反应瓶中,常温搅拌3小时,溶液颜色由无色逐渐转变为红色。接着将2,3-二氯-5,6-二氰基-1,4-苯醌(0.21g,0.91mmol)加入反应瓶中,常温搅拌反应1小时。然后将反应置于冰水浴中,依次滴加三乙胺(1.0ml,7.56mmol)和三氟化硼乙醚(1.9ml,15.12mmol),常温反应过夜。反应结束后,经萃取得到粗产物。经色谱柱分离纯化(洗脱剂为二氯甲烷/甲醇,v/v=20/1),得到橙棕色油状化合物2(0.25g,31%)。
[0058]1h nmr(500mhz,cdcl3,298k)δ(ppm):6.52(s,2h),5.97(s,2h),4.30
–
4.18(m,2h),4.10
–
4.08(m hz,4h),3.81(m,6h),3.75
–
3.56(m,42h),3.56
–
3.45(m,6h),3.36(s,3h),3.35(s,6h),2.52(s,6h),1.49(s,6h).
13
c nmr(125mhz,cdcl3,298k)δ(ppm):155.6,153.7,143.0,141.2,139.0,131.3,129.9,121.2,107.7,77.3,77.1,76.8,72.7,71.9,70.8,70.7,70.6,70.5,70.5,69.7,69.2,59.0,14.6,14.3.
19
f nmr(471mhz,cdcl3,298k)δ(ppm):-146.21(dd,j=65.1,30.2hz,bf2).esi-hrms[m+na]
+
:calcd.for[c
52h85
n2o
18
bf2na]
+
1097.6251,found 1097.5751.
[0059]
化合物3的制备:
[0060]
将化合物2(0.10g,0.093mmol)与碘单质(0.03g,0.112mmol)加到100ml两颈烧瓶中,加入20ml乙醇作为溶剂。再将碘酸(0.04g,0.205mmol)溶解在5ml水中,通过恒压滴液漏斗缓慢滴加到上述反应瓶中,在常温下搅拌过夜。反应结束后,减压除去乙醇,经萃取得到粗产物。经色谱柱分离纯化(洗脱剂为二氯甲烷/甲醇,v/v=20/1),得到粉色油状化合物3(0.12g,93%)。
[0061]1h nmr(500mhz,cdcl3,298k)δ(ppm):6.50(s,2h),4.23
–
4.22(m,2h),4.11
–
4.09(m,4h),3.84
–
3.82(m,6h),3.73
–
3.62(m,42h),3.55
–
3.52(m,6h),3.37(s,3h),3.36(s,6h),2.63(s,6h),1.52(s,6h).
13
c nmr(125mhz,cdcl3,298k)δ(ppm):156.7,153.9,145.1,140.8,139.3,131.1,129.4,107.3,85.5,72.7,71.8,70.8,70.6,70.5,70.5,70.5,70.4,70.4,69.6,69.2,58.9,16.9,16.0.
19
f nmr(471mhz,cdcl3,298k)δ(ppm):-145.54(dd,j=64.0,31.2hz,bf2).esi-hrms[m+na]
+
:calcd.for[c
52h83
n2o
18
bf2i2na]
+
1349.3684,found 1349.4223.
[0062]
化合物nbdp的制备:
[0063]
将化合物1(324mg,0.377mmol)、化合物3(200mg,0.151mmol)、对甲苯磺酸一水合物(2mg)、哌啶(81μl)、分子筛分别加入到干燥的100ml两颈烧瓶中,加入无水甲苯作为溶剂,120℃下回流反应并点板监测。反应结束后,浓缩除去甲苯得到粗产物。经色谱柱分离提纯(洗脱剂为二氯甲烷/甲醇,v/v=20/1),得到绿色油状化合物nbdp(77mg,17%)。
[0064]1h nmr(500mhz,cdcl3,298k)δ(ppm):7.90(d,j=16.4hz,2h),7.40(d,j=16.4hz,2h),6.81(s,4h),6.51(s,2h),4.21
–
4.10(m,18h),3.82(s,18h),3.77
–
3.69(m,126h),3.48(s,18h),3.32(s,27h),1.56(s,6h).
13
c nmr(125mhz,cdcl3,298k)δ(ppm):153.8,152.6,150.5,145.8,139.5,139.2,138.6,132.9,132.1,129.7,118.0,107.5,107.3,72.6,72.3,71.7,71.7,71.7,70.7,70.5,70.4,70.3,70.3,70.2,69.5,69.4,69.1,68.8,58.8,31.7,31.3,30.0,29.5,29.2,22.5,17.4,14.0.
19
f nmr(471mhz,cdcl3,298k)δ(ppm):-138.98(dd,j=65.9,32.3hz,bf2).esi-hrms[m+3na]
3+
:calcd.for[c
132h223
n2o
54
bf2i2na3]
3+
1024.0864,found 1024.4562.
[0065]
应用例1
[0066]
纳米粒子nbdp/dox nps的制备:
[0067]
称取5.0mg两亲性nbdp,溶于5.0ml水中。称取5.0mg盐酸阿霉素溶解于0.8ml水中,加入1.2ml四氢呋喃,滴加18μl三乙胺中和盐酸,得到阿霉素溶液。在快速搅拌下,将阿霉素溶液缓慢滴加到nbdp水溶液中。在室温下继续搅拌24小时后,静置过夜。经过滤去除过量的阿霉素悬浮物,滤液在水中透析(透析袋分子量为1000),去除未包裹的游离阿霉素。透析完毕后将纳米粒子溶液用冻干机冻干,得到墨绿色干燥油状物,即nbdp/dox纳米粒子(nbdp/dox nps),用tem分析其形态,其透射电镜图如图13所示。
[0068]
测试例1
[0069]
nbdp/dox nps的细胞吞噬实验:
[0070]
将4t1细胞以每孔1
×
104个的密度种于24孔板中,培养24小时。吸除每孔培养基,加入材料dox
·
hcl(4.00μg/ml)、nbdp(40μg/ml)、nbdp/dox nps(40μg/ml,以nbdp浓度计)
的dmem溶液,孵育24小时。用pbs清洗去除材料,用4%多聚甲醛溶液固定细胞。10分钟后,再用pbs清洗,加入dapi染色10分钟。用pbs清洗后,用clsm观察,其荧光显微镜图如图14所示,该纳米粒子培育的细胞内都呈现着荧光,说明其释放的dox能被肿瘤细胞摄取从而起到治疗的效果,表明所制备的两亲性bodipy化合物nbdp在药物载体方面有着一定的应用。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1