一种前列腺癌增殖标志物tRF-Gly-TCC-2及检测引物和应用
一种前列腺癌增殖标志物trf-gly-tcc-2及检测引物和应用
技术领域
1.本发明涉及分子生物学检测技术领域,尤其涉及一种前列腺癌增殖标志物trf-gly-tcc-2及检测引物和应用。
背景技术:
2.2016年nature genetics杂志报道了trna是一种高丰度的、广泛存在的、被动参与的mrna解码器及蛋白翻译元件。trna通过其反密码子与mrna的密码子结合而显著影响生物学过程和疾病进展。同时,trna在体液中高丰度存在的特点使其成为临床应用的生物标志物,可被运用到肿瘤增殖、转移的检测中。
3.2019年pan jiang等在current medicinal chemistry杂志上综述报道了trf和tirna是在特定的细胞/组织中或者在细胞受到胁迫等特定条件下,由特定的核酸酶[如dicer、血管生成素(ang)]在trna的环上剪切,产生的特定大小的小片段rna。trf和tirna属于一类小的非编码rna,被统称为tsrna。trf分为trf-1,trf-2,trf-3,trf-5和i-trf;tirna分为5
′
tirna和3
′
tirna。
[0004]
成熟的trna经剪切后转变为trf和tirna,trf和tirna在抑制蛋白质合成、调节基因表达、启动病毒逆转录酶和调节dna损伤反应中发挥重要作用,可视为trna的功能单位。
[0005]
目前,对于前列腺癌细胞增殖检测的方法通常采取血清psa检测来评判。但在广泛应用的过程中发现此检测方法特异性差与敏感性不足,不能区分前列腺重度炎症及提示早期前列腺癌细胞增殖。虽然还有超声、核磁共振等检测方法作为补充,但这些方法仍具有上述不足之处。因此,寻找敏感性高、特异性强的标志物检测前列腺癌细胞增殖,已成为医学研究的热点。
技术实现要素:
[0006]
本发明的目的在于提供一种前列腺癌增殖标志物trf-gly-tcc-2及检测引物和应用,弥补了血清psa检测、超声、核磁共振等检测方法的不足之处。
[0007]
为了实现上述发明目的,本发明提供以下技术方案:
[0008]
本发明提供了一种前列腺癌增殖标志物trf-gly-tcc-2,所述标志物trf-gly-tcc-2的核苷酸序列如seq id no.1所示。
[0009]
本发明还提供了一种检测前列腺癌增殖标志物trf-gly-tcc-2的引物,所述引物的序列如seq id no.2~3所示。
[0010]
本发明还提供了一种检测前列腺癌增殖的试剂盒,所述试剂盒中包含序列为seq id no.2~3的引物。
[0011]
本发明提供了一种前列腺癌增殖标志物trf-gly-tcc-2及检测引物和应用,所述标志物trf-gly-tcc-2的核苷酸序列如seq id no.1所示。通过检测前列腺组织中trf-gly-tcc-2相对表达量,可以评判前列腺癌细胞的增殖水平。若检测样本中trf-gly-tcc-2相对含量明显高于对照样本中trf-gly-tcc-2相对含量,提示前列腺癌细胞处于增殖状态。本发
明的标志物敏感性高、特异性强,弥补了血清psa检测、超声、核磁共振等检测方法的不足之处。
附图说明
[0012]
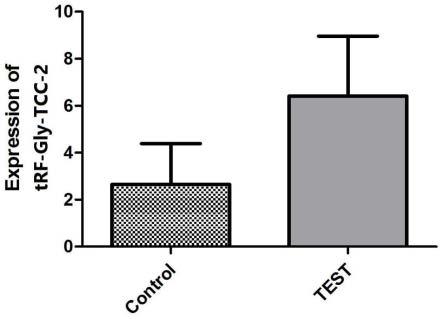
图1为trf-gly-tcc-2在前列腺癌组织和前列腺正常组织中的表达。
具体实施方式
[0013]
本发明提供了一种前列腺癌增殖标志物trf-gly-tcc-2,所述标志物trf-gly-tcc-2的核苷酸序列如seq id no.1所示;
[0014]
seq id no.1:gcgttggtggtatagtggtgagcatagctgcc。
[0015]
在本发明中,所述trf-gly-tcc-2全称是trf-1:32-gly-tcc-2,该tsrna片段来源于trna-gly-tcc-2,由该trna 5’端第1位至第32位核苷酸组成,属于trf-5c类型。
[0016]
本发明还提供了一种检测前列腺癌增殖标志物trf-gly-tcc-2的引物,所述引物的序列如seq id no.2~3所示;
[0017]
seq id no.2:acgatctcccacatggtctagc;
[0018]
seq id no.3:tccgatctccaggaatcctaac。
[0019]
本发明还提供了一种检测前列腺癌增殖的试剂盒,所述试剂盒中包含序列为seq id no.2~3的引物。
[0020]
下面结合实施例对本发明提供的技术方案进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
[0021]
实施例1样本选择
[0022]
试验组样本:新鲜前列腺组织(也可以使用前列腺细胞株、穿刺前列腺组织、新鲜尿液、新鲜前列腺按摩液、血液);
[0023]
对照组样品:人前列腺增生细胞系(bph-1细胞系,购自上海宾穗生物科技有限公司)。
[0024]
实施例2rna抽提及质量检测
[0025]
①
匀浆:
[0026]
将检测的新鲜前列腺组织研磨后,用蒸馏水溶解,振荡后,4℃7500转/分钟离心5分钟,得到沉淀细胞。加入tri reagent试剂反复吹打使细胞裂解,每克沉淀细胞使用1ml tri reagent试剂(避免在加入tri reagent试剂进行裂解前清洗细胞,因为清洗会很可能导致mrna的降解)。
[0027]
②
两相分离:
[0028]
匀浆后样本于25℃孵育5分钟,以便核酸蛋白复合体完全解离。每1ml的tri reagent试剂匀浆的样品中加入0.2ml的氯仿(上海化学试剂有限公司),盖紧管盖。手动剧烈振荡管体15秒后,25℃孵育3分钟。4℃下12000转/分钟离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层核上层的无色的水相。rna全部被分配于水相中。水相的体积为匀浆时加入的tri reagent试剂的60%。
[0029]
③
rna沉淀:
[0030]
将水相转移到新离心管中。水相与异丙醇(上海化学试剂有限公司)混合以沉淀其
中的rna,加入异丙醇的量为每个样本匀浆时加入1ml tri reagent试剂的此时加0.5ml的异丙醇。混匀后25℃孵育10分钟后,于4℃12000转/分钟离心10分钟。此时离心前不可见的rna沉淀将在管底部和侧壁上形成胶状沉淀块。
[0031]
④
rna清洗:
[0032]
移去上清液,每1ml tri reagent试剂匀浆的样本中加入至少1ml的75%乙醇(以depc水配制,depc水购自上海浦予工业科技有限公司),清洗rna沉淀。振荡后,4℃7500转/分钟,离心5分钟。
[0033]
⑤
重新溶解rna沉淀:
[0034]
去除乙醇溶液,空气中干燥rna沉淀10分钟,切勿真空离心干燥。注意rna沉淀不要完全干燥,否则将大大降低rna的可溶性。部分溶解的rna样品a260/280比值将小于1.6。溶解rna时,先加入无rna酶的水用枪反复吹打几次,然后60℃孵育10分钟。获得的rna溶液保存于-70℃。
[0035]
⑥
对照样本rna的抽提:
[0036]
从对照样本人前列腺增生细胞系(bph-1细胞系,购自上海宾穗生物科技有限公司)细胞中抽提rna:加入800μl的tri reagent试剂至样本中,样本裂解后,重复步骤
②
至步骤
⑤
操作。在加异丙醇沉淀rna前,加10μg的无rna酶的糖原作为水相载体。为降低溶液粘度,在加氯仿前先将样本两次过26号针头以剪切基因组dna。两相分离后糖原留在水相中并和rna共沉淀。只要糖原浓度不高于4mg/ml,不会影响cdna的合成,同样也不会对pcr过程产生影响。
[0037]
⑦
rna质量检测:
[0038]
采用紫外吸收测定法,使用nd-1000测定rna浓度和纯度,测量前先用溶解rna用的depc水1μl凋零处理测量基座表面,然后rna检测操作方法如下:
[0039]
a.滴加1μl rna样本至测量基座的表面。
[0040]
b.液滴会自动在上下基座之间形成液柱并自动完成测定,rna浓度及质量的各种参数将在电脑中自动生成文件。
[0041]
c.一次测定完成后,用柔软的擦镜纸擦去上下基座表面上的样本液,便可进行下一个样本的测量。
[0042]
d.测定结果(电脑自动生成的excel和jpeg文件附于实验报告文件夹中)。
[0043]
结果:
[0044]
浓度测定
[0045]
260nm处读值为1表示40ng rna/μl。样品rna浓度计算公式为:a260(读数)
×
40ng/μl。具体计算示例如下:
[0046]
rna溶于20μl depc水中,取1μl用于测定,测得a260=65.003
[0047]
rna浓度=65.003
×
40ng/μl=2600.12ng/μl
[0048]
取1μl用来测量以后,剩余样品rna为19μl,剩余rna总量为:19μl
×
2600.12ng/μl=49.4μg。
[0049]
纯度检测
[0050]
rna溶液的a260/a280的比值是一种rna纯度检测方法,比值范围1.8到2.1。即使比值超出这个范围,rna样品也一样可以用于一些普通实验中如northern杂交,rt-pcr和rna
酶保护实。
[0051]
实施例3rna前处理与cdna合成
[0052]
本实施例中的试剂除rtstar
tm
trf&tirna pretreatment kit(cat#as-fs-005)、rtstar
tm
first-strand cdna synthesis kit(3’and 5’adaptor)(cat#as-fs-003)均购自美国arraystar生物公司,其他相关试剂均购自赛默飞世尔科技(中国)有限公司。
[0053]
①3’
末端去乙酰化处理
[0054]
a.按照表1配置去乙酰化反应液:
[0055]
表1
[0056]
样本rna5μgdeacylation reaction缓冲液(5
×
)3ulsuperase
·
in
tm
rnase抑制剂1μl无核酸酶水变量总体积/样本加至15μl
[0057]
b.涡旋混合,37℃孵育40分钟;
[0058]
c.依次加入19μl deacylation stop缓冲液,涡旋混合,室温孵育5分钟,终止去乙酰化反应。
[0059]
②
去除3
’‑
cp与添加5
’‑
p
[0060]
a.将步骤
①
的反应液置于冰上,依次加入表2中的试剂:
[0061]
表2
[0062][0063][0064]
b.涡旋混合,37℃孵育40分钟;
[0065]
c.70℃孵育5分钟以终止反应;
[0066]
d.重新抽提rna。
[0067]
③
去甲基化处理
[0068]
a.融解除demethylase和reverse transcriptase两种酶外的所有试剂,涡旋混合,置于冰上备用。在使用前从冰箱将两种酶取出,简单离心,备用;
[0069]
b.按照表3配制去甲基化反应液:
[0070]
表3
[0071]
无核酸酶水变量demethylation reaction缓冲液(5
×
)10μldemethylase5μlsuperase
·
in
tm
rnase抑制剂1μl
input rna5μg总体积/样本加至50μl
[0072]
c.进行去甲基化反应;
[0073]
d.在37℃水浴锅中孵育2小时,然后加入40μl无核酸酶水与10μldemethylation stop缓冲液(5
×
),终止去甲基化反应;
[0074]
e.重新抽提rna。
[0075]
④
连接3
′
接头
[0076]
a.在200μl无rna酶的pcr管中依次加入表4的试剂:
[0077]
表4
[0078][0079][0080]
b.在热循环仪中70℃孵育2分钟,然后将pcr管移至冰上;
[0081]
c.添加表5的反应试剂;
[0082]
表5
[0083]3’
ligation reaction缓冲液(2x)5μl3’ligation enzyme mix1.5μl总体积加后变为10μl
[0084]
d.在热循环仪中25℃孵育1小时。
[0085]
⑤
反转录引物(reverse transcription primer)杂交
[0086]
此步反应对抑制接头二聚体的形成十分关键。反转录引物可与多余的3’接头杂交,从而将单链dna接头转化为双链dna分子。如果总rna起始量为100ng,将反转录引物用无酶水1:2稀释。
[0087]
a.向步骤
④‑
d的pcr管里加入表6的试剂;
[0088]
表6
[0089]
无核酸酶水2.3μlreverse transcription primer0.5μl总体积加后变为12.8μl
[0090]
b.在热循环仪中依次75℃孵育5min,37℃孵育15min,25℃孵育15min。
[0091]
⑥
连接5
′
接头
[0092]
a.在20μl无核酸酶水中重悬5’接头;
[0093]
注意:如果总rna起始量为100ng,将5
′
接头用无核酸酶水1:2稀释。
[0094]
b.在单独的无核酸酶的200μl pcr管中加入0.6μl的5’接头。(n为实验所处理的样本数目)在热循环仪中70℃孵育2分钟,然后立即在冰上冷却。
[0095]
注意:将剩余的5’重悬接头储存在-80℃冰箱。为了避免rna降解,请在接头变性后30分钟内使用接头。
[0096]
c.向步骤
⑤‑
b中的pcr管中依次加入表7的反应物,充分混合。
[0097]
表7
[0098]5′
adaptor(denatured)0.5μl5
′
ligation reaction缓冲液0.5μl5
′
ligation enzyme mix1.2μl总体积加后变为15μl
[0099]
d.热循环仪25℃孵育1小时。
[0100]
⑦
反转录反应
[0101]
a.在无核酸酶的200μl pcr管加入表8的反应物:
[0102]
表8
[0103]
adaptor ligated rna15μlfirst-strand synthesis reaction缓冲液4μlsuperase
·
in
tm
rnase抑制剂0.5μlreverse transcriptase0.5μl总体积加后变为20μl
[0104]
b.热循环仪50℃孵育1小时,然后立即在冰上冷却,反应产物可直接用于pcr扩增反应。
[0105]
注意:如果未打算立即进行pcr扩增,热循环仪70℃孵育15分钟以终止rt反应。然后将样本置于-20℃冰箱保存。
[0106]
实施例4合成的cdna用于实时定量pcr检测
[0107]
本实施例中的试剂2x pcr master mix(as-mr-006-5)购自美国arraystar生物公司;引物设计软件是primer 5.0;quantstudio
tm
5real-time pcr system(applied biosystems)购自美国应用生物系统公司。
[0108]
①
制备用于绘制标准曲线的梯度稀释dna模板
[0109]
1.针对每一需要测量的基因和管家基因,选择一确定表达该基因的cdna模板进行pcr反应:
[0110]
pcr反应的体系如表9所示:
[0111]
表9
[0112][0113][0114]
轻弹管底将溶液混合,5000转/分钟短暂离心,设置pcr反应:95℃,10min;40个pcr循环(95℃,10秒;60℃,60秒(收集荧光))。
[0115]
2.pcr产物与100bp dna ladder在2%琼脂糖凝胶电泳,溴化乙锭染色,检测pcr产物是否为单一特异性扩增条带。
[0116]
3.将pcr产物进行10倍梯度稀释:设定pcr产物浓度为1,分别稀释为1
×
10-1
,1
×
10-2
,1
×
10-3
,1
×
10-4
,1
×
10-5
,1
×
10-6
,1
×
10-7
,1
×
10-8
,1
×
10-9
,这几个梯度浓度的dna。
[0117]
②
进行realtime pcr反应
[0118]
1.将所有cdna样本分别配置realtime pcr反应体系。体系配置如表10所示:
[0119]
表10
[0120]2×
master mix5μl10um的pcr特异引物f0.5μl10um的pcr特异引物r0.5μl加无核酸酶水至总体积为8μl
[0121]
轻弹管底将溶液混合,5000转/分钟,离心5分钟。
[0122]
2.加样
[0123]
a.将8μl混合液加到384-pcr板对应的每个孔中;
[0124]
b.再加入对应的2μl cdna;
[0125]
c.粘上sealing film封口膜,并离心5分钟后混合;
[0126]
c.在设置pcr程序前将准备好的pcr板放在冰上。
[0127]
3.将上述384-pcr板置于realtime pcr仪上进行pcr反应。
[0128]
所有的指标均按以下程序进行:
[0129]
95℃,10min;40个pcr循环(95℃,10秒;60℃,60秒(收集荧光))。
[0130]
为了建立pcr产物的熔解曲线,扩增反应结束后,按(95℃,10秒;60℃,60秒;95℃,15秒);并从60℃加热到95℃(仪器自动进行-ramp rate为0.075℃/秒)。
[0131]
③
结果与计算
[0132]
各样本的目的基因和管家基因分别进行realtime pcr反应。根据绘制的梯度稀释dna标准曲线,各样本目的基因和管家基因的浓度结果直接由机器生成。每个样本的目的基因浓度除以其管家基因的浓度,即为此样本此基因的校正后的相对含量。
[0133]
④
待测基因以内参校正
[0134]
实时定量pcr时各样本加样量均为2μl,然而由于受rna浓度定量误差和rna逆转录效率误差等的影响,每个样品的2μl体积的cdna其含量并不完全相同,为校正此差异,使用管家基因u6(不同样品间表达量基本恒定)作为内参,以样品待测基因的值除以此样本内参的值,最终得到的比值为样品的待测基因相对含量。
[0135]
本实施例中所使用的基因和引物如表11所示。
[0136]
表11
[0137][0138]
结果:检测样本与对照样本经上述操作步骤处理后,均可得到组织中trf-gly-tcc-2的相对含量,若检测样本中trf-gly-tcc-2相对含量明显高于对照样本中trf-gly-tcc-2相对含量,提示前列腺癌细胞处于增殖状态。
[0139]
由以上实施例可知,本发明提供了一种前列腺癌增殖标志物trf-gly-tcc-2及检测引物和应用,所述标志物trf-gly-tcc-2的核苷酸序列如seq id no.1所示。通过检测前列腺组织中trf-gly-tcc-2相对表达量,可以评判前列腺癌细胞的增殖水平。若检测样本中trf-gly-tcc-2相对含量明显高于对照样本中trf-gly-tcc-2相对含量,提示前列腺癌细胞处于增殖状态。本发明的标志物敏感性高、特异性强,弥补了血清psa检测、超声、核磁共振等检测方法的不足之处。
[0140]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1