一种合成2-氨基-6-溴吡啶的方法与流程
1.本发明涉及医药中间体合成领域,尤其是涉及一种合成2-氨基-6-溴吡啶的方法。
背景技术:
2.2-氨基-6-溴吡啶是合成药物和农用化学品分子中重要的结构之一,广泛存在于天然产物、药物以及发光材料和各种精细化学品的合成应用中。现有的合成方法有四种:第一种是以环氧氯丙烷为起始原料,经过开环取代以及环合等步骤完成,合成路线如下:,其中环氧氯丙烷作为一种三元环的环醚,性质活泼,容易在酸或碱的催化下开裂,能与各种不同的试剂发生反应而生成各种不同的产物,因此以环氧氯丙烷为起始原料进行合成会导致副反应较多,转化率低,收率低等不利因素制约着合成工艺的顺利生产化,同时,环合过程采用溴化氢的冰醋酸溶液为环合的无水供氢试剂,这种试剂非常不宜操作,一丁点的敞开空间就会导致浓烟滚滚,几乎无法放大生产,而且醋酸的酸度不高,供氢全部靠溴化氢,反应效率不高。
3.第二种以2,6-二溴吡啶为起始原料进行氨化制得,合成路线如下:,虽然合成步骤很短,但是起始原料比较昂贵,合成反应条件比较苛刻,而且反应选择性不好,容易生成2,6-二氨基吡啶。
4.第三种以6-溴-2-吡啶羧酸为起始原料的水解法,合成路线如下:;第四种以2-氨基-6-甲基吡啶起始原料,经溴化、kmno4氧化、酰化、霍夫曼水解后得到2-氨基-6-溴吡啶,合成路线如下:;
第三种和第四种的合成路线的反应步骤多,不利于生产操作,并且会降低总收率。因此亟需提供一种转化率高且易于操作的合成方法。
技术实现要素:
5.为了解决上述技术问题,本发明提供一种合成2-氨基-6-溴吡啶的方法。
6.本发明提供的一种合成2-氨基-6-溴吡啶的方法,采用如下技术方案:一种合成2-氨基-6-溴吡啶的方法,其特征在于:包括以下步骤:s1、向三口瓶中加入1,3-二氯丙醇和乙醇,搅拌后加入碘化钾和氰化钠,加热至70℃,保温反应完成后得到反应液一,合成路线如下:;s2、将上述反应液一进行浓缩,蒸出乙醇后加入乙酸乙酯,并用水洗涤,干燥后减压蒸干得到油状物;s3、将上述油状物加入三口瓶中,加入三氟乙酸后搅拌,在室温下通入溴化氢气体,反应后得到反应液二,合成路线如下:;s4、将上述反应液二用冰水浴进行冷却,加氢氧化钠调节ph至9.5-10.5,用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥,减压脱溶至不出馏,向残余物中加入甲醇,搅拌降温析晶,过滤,鼓风干燥,得到产品。
7.优选的,s1中的1,3-二氯丙醇、氰化钠、碘化钾和乙醇的摩尔之比为0.77:1.61:0.0077:8.68。
8.优选的,s1中的保温反应时间为5h。
9.优选的,s2中采用的干燥剂为无水硫酸钠。
10.优选的,s4鼓风干燥的温度为60℃。
11.综上所述,本发明具有的有益技术效果为:本发明仅通过两步反应得到目标产物,反应条件温和,操作简单,易于控制,且转化率高。
附图说明
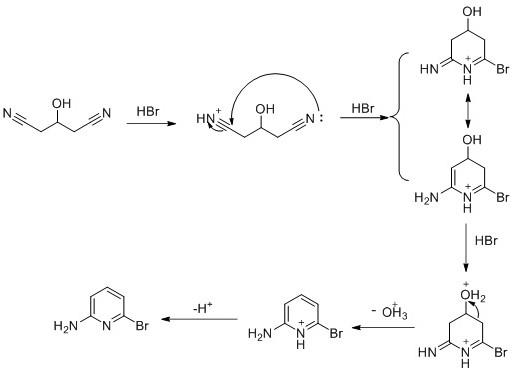
12.图1本发明的环合反应机理图。
13.图2是本发明的产品氢谱数据图。
具体实施方式
14.实施例1
15.向1000ml三口瓶中加入1,3-二氯丙醇100g,乙醇400g,搅拌,加入碘化钾1.27g,加入氰化钠79g,加热反应体系到70℃,保温反应5小时,监控反应完成,浓缩反应液,蒸出乙醇,加入乙酸乙酯500ml,分别用200ml水洗涤三次,有机相无水硫酸钠干燥,减压蒸干溶剂得到油状物90g,气相检测纯度80%,收率为85.19%,不需要纯化直接进行下一步反应。
16.将上步所得油状物加入到1000ml三口瓶中,加入三氟乙酸300g ,搅拌,室温缓慢通入溴化氢气体,反应5小时,取样检测反应完毕,此时气瓶中溴化氢气体减重约为160g左右,冰水浴冷却,30%氢氧化钠调ph至10左右,用二氯甲烷300ml萃取两次,合并有机相,无水硫酸钠干燥,减压脱溶至不出馏,残余物加入甲醇200ml,搅拌降温析晶,过滤,60℃鼓风干燥,得成品98g,收率85.76%。
17.实施例2
18.向1000ml三口瓶中加入1,3-二氯丙醇100g,乙醇400g,搅拌,加入碘化钾1.27g,加入氰化钠82.7g,加热反应体系到70℃,保温反应5小时,监控反应完成,浓缩反应液,蒸出乙醇,加入乙酸乙酯500ml,分别用200ml水洗涤三次,有机相无水硫酸钠干燥,减压蒸干溶剂得到油状物91g,气相检测纯度80%,收率为86.13%,不需要纯化直接进行下一步反应。
19.将上步所得油状物加入到1000ml三口瓶中,加入三氟乙酸300g ,搅拌,室温缓慢通入溴化氢气体,反应3小时,取样检测反应完毕,此时气瓶中溴化氢气体减重约为132g左右,冰水浴冷却,30%氢氧化钠调ph至10左右,用二氯甲烷300ml萃取两次,合并有机相,无水硫酸钠干燥,减压脱溶至不出馏,残余物加入甲醇200ml,搅拌降温析晶,过滤,60℃鼓风干燥,得成品95g,收率83.1%。
20.实施例3
21.向1000ml三口瓶中加入1,3-二氯丙醇100g,乙醇400g,搅拌,加入碘化钾1.27g,加入氰化钠86.5g,加热反应体系到70℃,保温反应5小时,监控反应完成,浓缩反应液,蒸出乙醇,加入乙酸乙酯500ml,分别用200ml水洗涤三次,有机相无水硫酸钠干燥,减压蒸干溶剂得到油状物92g,气相检测纯度80%,收率为87.08%,不需要纯化直接进行下一步反应。
22.将上步所得油状物加入到1000ml三口瓶中,加入三氟乙酸300g ,搅拌,室温缓慢通入溴化氢气体,反应4小时,取样检测反应完毕,此时气瓶中溴化氢气体减重约为145.5g左右,冰水浴冷却,30%氢氧化钠调ph至10左右,用二氯甲烷300ml萃取两次,合并有机相,无水硫酸钠干燥,减压脱溶至不出馏,残余物加入甲醇200ml,搅拌降温析晶,过滤,60℃鼓风干燥,得成品96.5g,收率84.45%。
23.以上均为本发明的较佳实施例,并非依此限制本发明的保护范围,故:凡依本发明的结构、形状、原理所做的等效变化,均应涵盖于本发明的保护范围之内。
技术特征:
1.一种合成2-氨基-6-溴吡啶的方法,其特征在于:包括以下步骤:s1、向三口瓶中加入1,3-二氯丙醇和乙醇,搅拌后加入碘化钾和氰化钠,加热至70℃,保温反应完成后得到反应液一,合成路线如下:;s2、将上述反应液一进行浓缩,蒸出乙醇后加入乙酸乙酯,并用水洗涤,干燥后减压蒸干得到油状物;s3、将上述油状物加入三口瓶中,加入三氟乙酸后搅拌,在室温下通入溴化氢气体,反应后得到反应液二,合成路线如下:;s4、将上述反应液二用冰水浴进行冷却,加氢氧化钠调节ph至9.5-10.5,用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥,减压脱溶至不出馏,向残余物中加入甲醇,搅拌降温析晶,过滤,鼓风干燥,得到产品。2.根据权利要求1所述的一种合成2-氨基-6-溴吡啶的方法,其特征在于: s1中的1,3-二氯丙醇、氰化钠、碘化钾和乙醇的摩尔之比为0.77:1.61:0.0077:8.68。3.根据权利要求1所述的一种合成2-氨基-6-溴吡啶的方法,其特征在于:s1中的保温反应时间为5h。4.根据权利要求1所述的一种合成2-氨基-6-溴吡啶的方法,其特征在于:s2中采用的干燥剂为无水硫酸钠。5.根据权利要求1所述的一种合成2-氨基-6-溴吡啶的方法,其特征在于:s4鼓风干燥的温度为60℃。
技术总结
本发明涉及医药中间体合成领域,尤其是涉及一种合成2-氨基-6-溴吡啶的方法,本发明采用1,3-二氯丙醇、碘化钾和氰化钠为原料进行反应生成3-羟基戊二腈,之后3-羟基戊二腈与溴化氢和三氟乙酸反应生成2-氨基-6-溴吡啶,与现有的合成方法相比,该制备方法简单,原料经济,反应条件温和,易于控制,且产品转化率高。且产品转化率高。且产品转化率高。
技术研发人员:刘庆贵 姚辉涛 刘凡振
受保护的技术使用者:济南泺合医药技术有限公司
技术研发日:2023.01.31
技术公布日:2023/3/14
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1