一种加氢开环催化剂
1.本发明属于多相催化技术领域,尤其涉及以过渡金属磷化物作为金属活性组分的多环芳烃和环烷烃固体双功能加氢开环催化剂。
背景技术:
2.燃料油中的多环芳烃(pahs)本身是一类具有致癌、致畸和致突变性质的持久性有机污染物,并且pahs十六烷值低,其含量直接影响了柴油的品质和柴油机尾气污染物的排放。车用柴油国v标准规定pahs含量不大于11%,而国vi标准则进一步规定pahs含量不大于7%。因此,pahs的转化已经成为清洁柴油生产亟待解决的一个紧迫问题。柴油馏分油中烃类组分十六烷值大致按照以下顺序递减:链烷烃》环烷烃》芳烃,芳烃的十六烷值最低,且分子中芳环数越多十六烷值越低。以萘为例,pahs在加氢精制(改质)过程中可能发生加氢、开环、异构化以及加氢裂化和脱烷基等四类反应。pahs加氢生成环烷烃虽然能够提高十六烷值,但是与加氢开环的链烷烃产物相比,十六烷值增量较低、氢耗较高,经济性差。加氢裂化和脱烷基反应会生成小分子的芳烃和烷烃,降低柴油收率。因此,在不降低反应物分子量前提下将pahs加氢开环为链烷烃,是提高柴油十六烷值、降低其密度,全面改善其品质的一条重要途径。在多环芳烃和环烷烃的加氢开环反应中,催化剂是核心。
3.加氢开环催化剂为典型的双功能催化剂,同时包含金属活性中心和酸中心两种不同功能的活性中心。加氢反应主要发生在金属中心上,而酸中心和金属中心都可以催化环烷烃的开环反应。在酸中心上,除了开环反应外,还会发生缩环、裂化和异构化等反应。沸石分子筛酸性强并且可调,同时具有规整的孔道结构,是最受关注酸性载体。对于多环芳烃加氢开环反应,y、β和丝光沸石等大孔沸石作载体的催化剂上开环反应产物收率和选择性都高于中孔的zsm-5和mcm-22等沸石。在孔道尺寸较小的沸石分子筛中,可能严重限制了开环产物或中间体的生成和扩散。大孔沸石中,又以β沸石性能为佳。
4.目前加氢开环催化剂的金属组分主要有金属类和过渡金属硫化物两类。niw和nimo等双金属硫化物是主要的硫化物活性组分。硫化物催化剂主要问题是活性较低并且稳定性较差,在反应初期失活较快(arribas et al.,appl.catal.a,2002,230:203-217)。金属类组分可分为贵金属和非贵金属两类。ir、rh和pt等是主要的贵金属活性组分,其中ir的性能最优。但贵金属催化剂价格昂贵,并且不耐硫,易失活,亟待开发能够替代贵金属的高性能催化剂。以过渡金属碳化物和氮化物为代表的非贵金属催化剂表现出较高的活性。li等人报道hβ和usy担载的过渡金属氮化物ni2mo3n在pahs的加氢开环反应中于较为温和的条件下(330℃和3.0mpa)就表现出很高的开环转化率(》90%)(catal.commun.,2005,6:656-660)。mouli等人对比研究了十氢萘在hβ和hy担载的pt-ir贵金属及ni-mo碳化物上的开环反应,发现碳化物和贵金属催化剂性能相当(j.mol.catal.a,2009,304:77-84)。过渡金属碳化物和氮化物存在的主要问题是在柴油馏分油加氢过程中,由于存在含硫化合物和h2s,催化剂容易因硫化而快速失活(appl.catal.a.,2017,539:114-127)。
技术实现要素:
5.本发明的目的在于克服现有技术的不足,提供一种在加氢开环反应中活性和稳定性更优的催化剂。
6.实现本发明目的的技术方案为:
7.本发明提供了一种以过渡金属磷化物为金属活性组分的双功能加氢开环催化剂。具体说来,以酸性材料作为载体,以过渡金属磷化物作为金属活性组分,过渡金属磷化物担载量以过渡金属和磷的总质量分数计为1%~60%。优选的担载量为20~50%,进一步优选的担载量为15~30%。
8.所述的过渡金属磷化物为ni2p或mop或wp。这些过渡金属磷化物具有良好的加氢脱硫和加氢脱氮活性及耐硫稳定性,构成了一类新型非硫化物加氢精制催化剂。
9.酸性材料载体可以为无定形硅铝和沸石分子筛等酸性材料,进一步的优选沸石为β沸石。β沸石具有三维大孔结构及适宜的酸性,在针对大分子多环芳烃加氢开环反应中表现出良好的性能。
10.原则上,本发明并不特别限定催化剂制备方法。作为推荐的实施方式,本发明优选采用程序升温还原前驱体-载体复合物的方法制备催化剂。其中在前驱体中,磷和过渡金属的原子摩尔比为1.0。
11.在具体实施中,本发明所述的方法中的前驱体-载体复合物的制备方法是浸渍法,主要包括以下步骤:
12.(1)配制过渡金属盐和磷酸氢二铵的水溶液;
13.(2)将浸渍液按一定顺序滴加到载体上制得混合物;
14.(3)将混合物在室温下静置8~12小时后于100~120℃烘干,然后在马弗炉中400~600℃焙烧3~6小时后制得催化剂前驱体。
15.关于本发明的催化剂制备方法,在具体的实施方式中,所述的程序升温还原方法,其中还原气体为氢气,最终还原温度在400~800℃,氢气压力为0.1~10.0mpa,还原时间为1~12小时。
16.本发明催化剂用于多环芳烃和环烷烃加氢开环反应时,使用条件为:在氢气存在的条件下,于固定床反应器中使反应原料与催化剂接触发生反应。反应条件为:温度200~500℃,压力1.0~10.0mpa,氢油体积比700~10000,液时体积空速0.1~100小时-1
。
17.优选的反应条件为:温度300~400℃,压力3~5mpa,氢油体积比为800~1200nm3/m3,液时体积空速0.1~0.5小时-1
。
18.多环芳烃和环烷烃加氢开环反应如十氢萘加氢开环反应、萘的加氢开环反应、蒽和菲的加氢开环反应以及它们的混合物如重整重芳烃油的加氢开环反应,该类反应均适用。
19.本发明的优点和有益效果:
20.本发明制备的ni2p、mop和wp过渡金属磷化物作金属活性组分的催化剂在多环芳烃和环烷烃的加氢开环反应中活性和稳定性均显著优于过渡金属硫化物催化剂。由于过渡金属磷化物中的金属为ni、mo和w等非贵金属,价格远低于贵金属,经济性好,有利于工业应用。
附图说明
21.图1是实施例1制备的mop/hβ催化剂的xrd谱图。
22.图2是实施例2制备的wp/hβ催化剂的xrd谱图。
23.图3是实施例3制备的ni2p/hβ催化剂的xrd谱图。
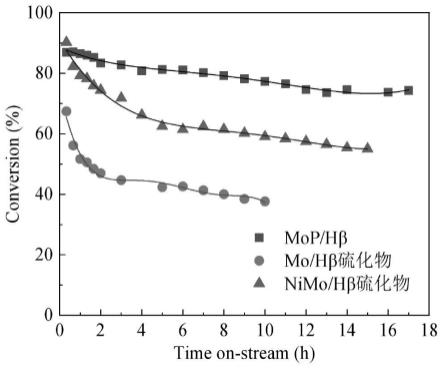
24.图4是十氢萘在实施例1制备的mop/hβ以及对比例1制备的mo/hβ和对比例2制备的nimo/hβ硫化物催化剂上转化率随时间的变化关系。
25.图5是重芳烃油在对比例1制备的mo/hβ硫化物催化剂上进行反应时产物组成随时间的变化关系。
26.图6是重芳烃油在对比例2制备的nimo/hβ硫化物催化剂上进行反应时产物组成随时间的变化关系。
27.图7是重芳烃油在实施例3制备的ni2p/hβ催化剂上进行反应时产物组成随时间的变化关系。
28.图8是重芳烃油在实施例1制备的mop/hβ催化剂上进行反应时产物组成随时间的变化关系。
29.图9是重芳烃油在实施例2制备的wp/hβ催化剂上进行反应时产物组成随时间的变化关系。
具体实施方式
30.下面结合附图并通过具体实施例对本发明作进一步详述。以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围。
31.实施例1
32.制备mop/hβ催化剂。
33.首先称取0.246克的四水合钼酸铵((nh4)6mo7o
24
·
4h2o)溶于1毫升去离子水中制备浸渍液a,再称取0.184克的磷酸氢二铵((nh4)2hpo4))溶于0.5毫升去离子水中制备浸渍液b。然后采用分步等体积浸渍法先后将a和b溶液逐滴滴加到1克的氢型β沸石(hβ)载体上。混合物室温下静置10小时,120℃下烘干12小时,最后在500℃下焙烧3小时得到mop/hβ催化剂的前驱体。其中mo和p的重量负载量为15%,p/mo原子摩尔比为1。
34.将前驱体压片并破碎为40~60目。称取1.7克前驱体置于固定床反应器中,程序升温还原反应条件为:氢气压力1.0mpa,氢气流量150毫升/分钟,从室温以2℃/分钟升温至400℃,再以1℃/分钟的升温速率升至650℃,并在还原终温下保持2小时。由图1的xrd谱图可以看出,mop/hβ的金属活性相为mop。
35.实施例2
36.制备wp/hβ催化剂。
37.首先称取0.202克的偏钨酸铵((nh4)6h2w
12o40
·
x4h2o)溶于0.5毫升去离子水中制备浸渍液a,再称取0.108克的磷酸氢二铵((nh4)2hpo4)溶于0.5毫升去离子水中制备浸渍液b。然后采用分步等体积浸渍法先后将a和b溶液逐滴滴加到1克的hβ载体上。混合物室温下静置10小时,120℃下烘干12小时,最后在500℃下焙烧3小时得到wp/hβ催化剂的前驱体。其中w和p的重量负载量为15%,p/w原子摩尔比为1。
38.将前驱体压片并破碎为40~60目。称取1.7克前驱体置于固定床反应器中,程序升
温还原反应条件为:氢气压力1.0mpa,氢气流量150毫升/分钟,从室温以4℃/分钟升温至120℃,保持60分钟,以10℃/分钟的升温速率升至400℃,再以1℃/分钟的升温速率升至650℃并在还原终温下保持2小时。由图2的xrd谱图可以看出,wp/hβ的金属活性相为wp。
39.实施例3
40.制备ni2p/hβ催化剂。
41.首先称取1.68克的六水合硝酸镍(ni(no3)2·
6h2o)溶于1毫升去离子水中制备浸渍液a,再称取0.381克的磷酸氢二铵((nh4)2hpo4)溶于1毫升去离子水中制备浸渍液b。然后采用分步等体积浸渍法先后将a和b溶液逐滴滴加到1克的hβ载体上。混合物室温下静置10小时,120℃下烘干12小时,最后在500℃下焙烧3小时得到ni2p/hβ催化剂的前驱体。其中ni和p的重量负载量为30%,p/ni原子摩尔比为1。
42.将前驱体压片并破碎为40~60目。称取1.7克前驱体置于固定床反应器中,程序升温还原反应条件为:氢气压力1.0mpa,氢气流量150毫升/分钟,从室温以4℃/分钟升温至120℃,保持60分钟,以10℃/分钟的升温速率升至400℃,再以1℃/分钟的升温速率升至500℃并在还原终温下保持2小时。由图3的xrd谱图可以看出,ni2p/hβ的金属活性相为ni2p。
43.对比例1
44.制备mo/hβ硫化物催化剂前驱体。
45.首先称取0.325克的四水合钼酸铵((nh4)6mo7o
24
·
4h2o)溶于1毫升去离子水中制备浸渍液,然后采用等体积浸渍法将上述浸渍液逐滴滴加到1克的hβ载体上,混合物室温下静置10小时,120℃下烘干12小时,最后在500℃下焙烧3小时得到mo/hβ硫化物催化剂的前驱体。
46.将mo/hβ硫化物催化剂的前驱体压片并破碎至40~60目。然后称取1.7克前驱体置于内径为10mm的高压固定床管式反应器恒温段,再于氢气气氛下用质量分数为5%的cs2的环己烷溶液硫化前驱体。具体条件为:氢气压力4.0mpa,氢气流量150毫升/分钟,从室温以10℃/分钟的速率升温至380℃,在此终温下硫化3小时得到mo/hβ硫化物催化剂。
47.对比例2
48.制备nimo/hβ硫化物催化剂前驱体。
49.首先称取0.325克的四水合钼酸铵((nh4)6mo7o
24
·
4h2o)溶于1毫升去离子水中制备浸渍液a,称取0.295克的六水合硝酸镍(ni(no3)2·
6h2o)溶于0.5毫升去离子水中制备浸渍液b。然后采用等体积浸渍法先后将a和b溶液逐滴滴加到1克的hβ载体上。混合物室温下静置10小时,120℃下烘干12小时,最后在500℃下焙烧3小时得到nimo/hβ硫化物催化剂的前驱体。
50.将nimo/hβ硫化物催化剂的前驱体压片并破碎至40~60目。然后称取1.7克前驱体置于内径为10mm的高压固定床管式反应器恒温段,再于氢气气氛下用质量分数为5%的cs2的环己烷溶液硫化前驱体。具体条件为:氢气压力4.0mpa,氢气流量150毫升/分钟,从室温以10℃/分钟的速率升温至380℃,在此终温下硫化3小时得到nimo/hβ硫化物催化剂。
51.应用实施例1
52.评价十氢萘分别在实施例1,对比例1,对比例2制备的催化剂上的反应活性。
53.采用内径为10mm的高压固定床管式反应器,温度调至反应温度(310℃),氢气压力为4.0mpa,然后用高压恒流泵将十氢萘泵入高压固定床反应器中,液体流量为0.1毫升/分
钟,氢/油体积比1000。原料和产物用thermo-fisher trace gc 1310气相色谱分析,色谱柱为tg-5ms毛细柱,氢火焰检测器。十氢萘在mop/hβ以及mo/hβ和nimo/hβ硫化物催化剂上转化率随时间的变化关系见图4。
54.由图4可以看出,实施例1制备的mop/hβ催化剂活性和稳定性均显著优于对比例1,对比例2制备的硫化物催化剂。硫化物催化剂在反应初期失活很快,十氢萘转化率在反应开始2~4小时之内快速下降。nimo/hβ硫化物催化剂的活性高于mo/hβ硫化物催化剂。反应10小时后,十氢萘在nimo/hβ硫化物催化剂上转化率为59%,高于其在mo/hβ硫化物催化剂上的转化率(38%)。
55.应用实施例2
56.评价重整重芳烃油分别在实施例1,实施例2,实施例3,对比例1,对比例2制备的催化剂上的反应活性。
57.采用内径为10mm的高压固定床管式反应器,将床层温度调整至反应温度(370℃),氢气压力调整为4.0mpa,然后用高压恒流泵将重芳烃油泵入高压固定床反应器中,液体流量为0.1毫升/分钟,氢/油体积比1000。原料和产物用thermo-fisher trace gc 1310气相色谱分析,色谱柱为tg-5ms毛细柱,氢火焰检测器,反应结果见图5-图9。
58.图5是重芳烃油在对比例1制备的mo/hβ硫化物催化剂上进行反应时产物组成随时间的变化关系。
59.图6是重芳烃油在对比例2制备的nimo/hβ硫化物催化剂上进行反应时产物组成随时间的变化关系。
60.图7是重芳烃油在实施例3制备的ni2p/hβ催化剂上进行反应时产物组成随时间的变化关系。
61.图8是重芳烃油在实施例1制备的mop/hβ催化剂上进行反应时产物组成随时间的变化关系。
62.图9是重芳烃油在实施例2制备的wp/hβ催化剂上进行反应时产物组成随时间的变化关系。
63.由图5和图6的对比可知,nimo/hβ的硫化物催化剂活性低于mo/hβ硫化物催化剂,但稳定性稍好。
64.由图7~图9与图5~图6的对比可知,过渡金属磷化物的性能优于硫化物催化剂。wp/hβ活性和加氢开环产物收率均高于硫化物催化剂。mop/hβ和ni2p/hβ催化剂上产物分布虽然与硫化物催化剂相当,但它们的稳定性特别是mop/hβ稳定性显著优于硫化物催化剂。
65.本发明经过上述的描述,已清楚地公开了本发明催化剂组成和制备条件。但是,本领域内的技术人员十分清楚,对本发明可以进行一些修改和改进。所以,只要不离开本发明的精神,对本发明所进行的任何修改和改进都应在本发明的范围内。本发明的范围在附属的权利要求书中提出。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1