计算蛋白质设计的方法与流程
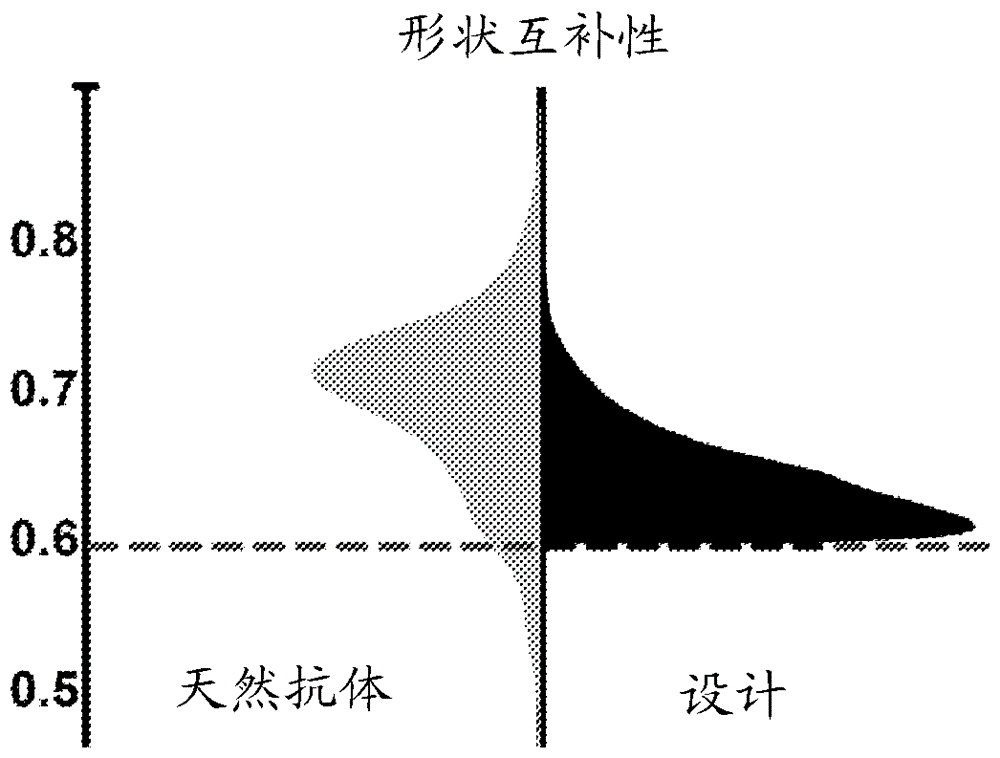
在一些实施方案中,本发明涉及计算化学和计算蛋白质设计,并且更特别地,但不排他地,涉及计算构建具有共同结构折叠的氨基酸序列文库的方法;以及设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法。这些方法可用于例如设计针对预定分子靶标具有结构稳定性和高结合亲和力的结合蛋白。在其一些实施方案中,本发明进一步涉及计算化学,并且更特别地,但非排他地,涉及产生对目标目标分子表面具有所需亲和力的氨基酸序列的方法,以及对目标目标分子表面具有所需亲和力的氨基酸序列。分子识别是许多中心生物过程的基础,因此,设计新的蛋白质相互作用的能力具有创造用于化学工业以及治疗、诊断和研究探针中的高度特异性和有效的分子的巨大前景。蛋白质结合剂设计中的近期策略已使用天然存在的蛋白质作为在其上设计结合表面的支架,同时依靠单个蛋白质支架或几百个不同的支架来实现结合所需的结构特征。在所有情况下,设计的支架作为主链自由度具有最小扰动的刚性结构元件处理。这些策略中的一些导致同低聚物、抑制剂和蛋白质亲和纯化试剂的实验验证的设计。设计具有例如约220个天然存在的氨基酸(大致等价于抗体Fv结构域)的序列的从头蛋白的计算练习将需要20220=10286个独特的氨基酸排列来限定一种蛋白质的氨基酸序列;当前的计算机器尚未配备以处理这一技术。当前的计算设计方法使用天然存在的刚性支架来设计从头分子功能;然而,这些方法从根本上受到已知三维结构的合适支架数目的限制。此外,已根据上述策略对成功设计的结合表面作出了几个一般限制:1.它们包含富含二级结构含量(α螺旋和β-折叠)的表面;2.与靶的相互作用主要由疏水性氨基酸侧链介导;和3.结合后的包埋表面积等于或小于天然存在的蛋白质-蛋白质相互作用的平均值,估计为1600Å2。对于使计算结合剂设计一般化必要的大型和极性表面的设计仍然是未被满足的挑战。已在许多蛋白质中鉴定的一些共同的蛋白质折叠,其中一些在系谱学、生物和功能方面看起来彼此不相关,也称为保守的结构域折叠,提供了研究序列-结构-功能关系的基本原理的独特机会,即使共享共同折叠但发挥无关功能的几个蛋白质的观察仍挑战现代科学。但是,一些研究仍尝试利用共同的结构折叠来帮助计算蛋白质设计。最引人注目的保守蛋白质折叠之一被称为TIM桶,或α/β蛋白质折叠。由许多蛋白质和许多生物共享的这种折叠的观察已帮助开发了与不同谱系物种中的类似特征有关的趋同进化理论。同样地,TIM桶已视为用于从头蛋白质设计的合适支架。Offredi,F.等人[JMolBiol.,2003,325(1),第163-74页]使用来自TIM桶折叠蛋白质的晶体结构的结构数据来限定具有4折叠对称性的“理想”折叠的几何规则,并且遵循主链几何结构的定义,尝试序列搜索以找到将稳定构象的序列。Figueroa,M.等人[PLoSOne,2013,8(8),第e71858页]使用Rosetta套件使用称为“OctarellinV”的模型作为起始主链模型来设计TIM桶蛋白,并使用Rosetta环构建方案构建了展示选择二级结构模式的PDB蛋白的六残基片段的环区域。基于在β链-α螺旋界面处的包装的β链之间的氢键和Rosetta全原子能量函数来估计最终结构。在蛋白质结合相互作用、结构预测和分子设计的背景下,研究最多的蛋白质家族之一是抗体家族。抗体包含称为轻链和重链的两类多肽。轻链和重链由具有相似结构的不同结构域组成,轻链包含两个此类结构域,并且重链包含四个此类结构域。每个结构域包含特征在于由反向平行β链组成的两个β折叠的“三明治”,具有连接两个β折叠的二硫键。在重链和轻链各自的N末端处的结构域在氨基酸序列上是可变的。这些“可变结构域”提供了广泛多样的不同抗体。其他结构域构成重链和轻链的“恒定区”。抗体的抗原结合区由一个轻链可变结构域与一个重链可变结构域组合形成。在可变结构域中,氨基酸序列的可变性主要限于由相对保守的“构架区”分隔的3个“互补决定区(CDR)”(也称为“高变区”,并且个别地称为CDR1、CDR2和CDR3)。因此,抗原结合区含有三个轻链CDR(称为L1、L2和L3)和三个重链CDR(称为H1、H2和H3)。每个结构域中的三个CDR在抗体的靶结合表面处聚簇,每个CDR与连接两条β链的环结合。保守的构架区形成特征在于结构同源性的刚性结构,其为抗体提供稳定性并影响CDR构象刚性。CDR中的许多可变性是V(D)J(可变、不同和连接基因区段)重组的结果,其中免疫细胞基因组经历重组,使得约44个V基因区段之一与6个J基因区段之一随机组合。另外,在重链基因中,27个D基因区段之一位于选择的V和J基因区段之间。V基因区段是最大的,编码CDR1和CDR2以及CDR3的一部分,而D和J基因片段编码CDR3的部分(在J片段的情况下L3或H3,在D片段的情况下H3)。V(D)J重组允许广泛多样的轻链和重链序列。另外的可变性起因于不同的重链和轻链的组合以及过程,其导致轻链和重链基因中的核苷酸或其他突变的添加和/或缺失。尽管其存在巨大的多样性,但CDR(除了H3CDR之外)落入被称为“规范构象”的少量离散构象中。例如,在数百个抗体分子结构中,对于L2CDR仅观察到七个构象变体。规范构象的特征在于维持主链构象的关键保守残基同一性。在关于功能的主链片段设计中的关键挑战在于所设计的表面需要起作用(结合其靶)和构象稳定两者。如上文提及的,抗体由高度保守与高度可变区段交替的序列块构建,并且抗体的分子结构显示保守区段属于称为构架的结构上同源且刚性的结构,其为抗体提供必要的稳定性,而可变区段在靶结合表面处聚簇,并且因此称为互补决定区(CDR)。抗体工程的关键吸引力在于抗体的模块结构,其表明可以挖掘良好折叠主链的大的组合复杂性。早在20世纪80年代,对抗体的结构模块性的观察提出可以通过组合天然存在的抗体的片段来构建合成抗体。从这个见解,研究人员已设计了用于抗体人源化的方法,其中来自小鼠抗体的CDR被移植到人抗体构架上以生成人源化功能抗体,开辟了安全的治疗性抗体工程的途径。这些早期进展激发了来自第一原理的抗体的完整设计是可实现的,但直到近期,用于蛋白质设计的计算工具仍未充分成熟以实现该目的。关于计算抗体设计的近期工作旨在增加结合亲和力[Clark,LA.等人,ProteinSci.,2006,15(5),第949-60页;Lippow,SM.等人,Nat.Biotechnol.,2007,25(10),第1171-6页;Clark,LA.等人,ProteinEngDesSel.,2009,22(2),第93-101页],鉴定用于实验性随机诱变的有利位置[Barderas,R.等人,Proc.Natl.Acad.Sci.USA,2008,105(26),第9029–34页],修饰结合特异性[Farady,C.J.等人,Bioorg.Med.Chem.Lett.,2009,19(14),第3744–7页],并增加热阻[Miklos,A.E.等人,Chem.Biol.,2012,19(4),第449–55页]。Pantazes等人提出了从头抗体设计策略,其利用了抗体CDR显示出规范构象的观察。Pantazes和Maranas[Protein.Eng.Des.Sel.,2010,23,849–858]描述了通过下述用于设计抗体的结合部分的通用计算方法(“OptCDR”):首先确定规范结构的哪些组合最可能有利地结合所选抗原,并且随后执行CDR主链的同时精制和关于每个位置的最佳氨基酸选择。Pantazes和Maranas[BMCBioinformatics2013,14:168]也描述了通过使用实验测定的抗体结构来编译929个模块化抗体部分(MAP)的数据库来预测抗体结构的方法,其可以组合以产生2.3·1010个独特抗体。MAPS被描述为与V、D和J基因片段类似。WeitznerB.D.等人[Proteins,2014年2月12日电子公开],教导了通过将个别抗体CDR移植到链特异性构架上构建初始模型,其中使用Rosetta对接算法对刚体取向取样的同时从头建模H3。ShiraiH.等人[Proteins,2014年4月22日电子公开],教导了基于H3亚型鉴定抗体Fv结构域构架模板,构建包括与位置特异性评分矩阵(PSSM)相关的H3的所有规范环的构象数据库,基于其PSSM得分选择关于给定序列最适合的簇,并且随后构建对模板模型的调和主链约束最小化的模型。然而,即使可以理论上包含并且有效地和系统地对可以通过在基因水平上的排列生成的所有构象组合空间取样的方法,此类方法也不能解释在天然存在的抗体中观察到的无数的随机突变。另外的
背景技术:
:包括美国专利申请号20030059827、20110224100、20130244940、20130296221和20140005125,以及Smadbeck,J.,Peterson,M.B.,Khoury,G.A.,Taylor,M.S.,Floudas,C.A.“ProteinWISDOM:AWorkbenchforInsilicoDenovoDesignofBioMolecules”,J.Vis.Exp.,(77),e50476,以及通过Khoury,G.A.,Smadbeck,J.,Kieslich,C.A.和Floudas,C.A.,TrendsinBiotechnology,2014,32(2),第99–109页的综述“Proteinfoldinganddenovoproteindesignforbiotechnologicalapplications”,所述参考文献整体引入作为参考,如同它在本文中完全阐述。发明概述根据本发明的一些实施方案的一个方面,提供了计算构建具有共同结构折叠的氨基酸序列文库的方法,所述方法包括下述步骤:(i)提供具有共同结构折叠的一个或多个源结构,而所述共同结构折叠具有限定结构构架的保守结构区和多样化结构区,并且所述结构构架具有最高结构保守性位置;(ii)将源结构中的全部或一些分段成结构同源区段,所述结构同源区段各自由最高结构保守性位置中的两个位置限定,以便获得多组结构同源区段,所述组各自由具有最高结构保守性的两个位置限定;(iii)选择具有共同结构构架并具有对应于最高结构保守性位置的特定位置的模板结构;对于区段组中的每个:(iv)在结构同源区段内的位点处拆分结构同源区段的至少一个结构同源区段,以便获得拆分区段;(v)将拆分区段的分别位置叠加到对应于所述位置的特定位置的相应位置上,并且置换模板结构中的相应区段;(vi)将用于区段闭合的拆分区段进行权重拟合,以便在模板结构中获得权重拟合和重新闭合的区段;(vii)任选重复步骤(iv)-(vi),同时在不同位点拆分或中止结构同源区段的进一步操作;(viii)对于至少一个另外的结构同源区段重复步骤(iv)-(vii),以便获得对应于每个组的至少一个附加的权重拟合和重新闭合的区段;和(ix)组合使用对应于每个组的多个权重拟合和重新闭合的区段用于重建多个重建结构,所述重建结构各自具有共同结构折叠,其具有限定结构构架的保守结构区和多样化结构区以及对应于最高结构保守性位置的位置;从而计算构建具有共同结构折叠的氨基酸序列文库。根据本发明的一些实施方案,进一步包括在多个源结构中任选限定末端区段,所述末端区段各自由最高结构保守性位置中的一个位置和共同结构折叠的末端位置限定,以便限定末端区段;并且在步骤(v)中将末端区段中的至少一个视为拆分区段。根据本发明的一些实施方案,拆分位点在多样化结构区中。根据本发明的一些实施方案,随机选择拆分位点。根据本发明的一些实施方案,该方法进一步包括在步骤(ix)之前,将对应于每个组的每个权重拟合和重新闭合区段分选到至少一个结构簇内,并且选择关于结构簇的代表性权重拟合和重新闭合区段,其中步骤(ix)的权重拟合和重新闭合区段各自是代表性权重拟合和重新闭合区段。根据本发明的一些实施方案,分选通过区段长度进行。根据本发明的一些实施方案,分选通过比对的多肽主链位置的均方根偏差进一步进行。根据本发明的一些实施方案的一个方面,提供了设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法,所述方法包括根据如本文呈现的方法计算构建具有共同结构折叠的氨基酸序列文库,并且进一步包括:(x)将重建结构中的至少一个匹配到目标分子表面上,以便设计各自具有匹配得分的多个重建结构-分子表面复合物,并且对于复合物中的重建结构任选重复步骤(x);(xi)任选用另一个相应的权重拟合的重新闭合区段取代复合物中至少一个的重建结构中的权重拟合的重新闭合区段中的至少一个,并且重复步骤(x),以便设计具有匹配得分的取代重建结构-分子表面复合物;和(xii)基于匹配得分,选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列。根据本发明的一些实施方案的一个方面,提供了产生对目标分子表面具有所需亲和力的氨基酸序列的方法,所述方法包括:在其实施方案的任何一个中,根据如本文呈现的方法设计和选择对目标分子表面具有所需亲和力的氨基酸序列;和在表达系统中表达所述氨基酸序列,从而产生对目标分子表面具有所需亲和力的氨基酸序列。根据本发明的一些实施方案的一个方面,提供了通过如本文呈现的方法在其实施方案的任何一个中产生的对目标分子表面具有所需亲和力的氨基酸序列。根据本发明的任何实施方案和方面的一些实施方案,本文呈现的任一方法中使用的位置是在多肽主链位置上的位置。根据本发明的任何实施方案和方面的一些实施方案,多肽主链位置选自原子和原子之间的预定位置。根据本发明的任何实施方案和方面的一些实施方案,原子选自α碳、主链羰基碳、主链羰基氧和主链氮。根据本发明的任何实施方案和方面的一些实施方案,权重拟合包括选自下述的至少一种操作:主链二面角的调节、氨基酸侧链包装和氨基酸的改变。根据本发明的任何实施方案和方面的一些实施方案,拆分区段的主链二面角的调节受对应于拆分区段的结构同源段的至少一个二面角约束。根据本发明的任何实施方案和方面的一些实施方案,氨基酸的改变由对于拆分区段的氨基酸以及至少部分在围绕拆分区段的壳内部的模板结构中的氨基酸实现。根据本发明的任何实施方案和方面的一些实施方案,氨基酸的改变由规则指定。根据本发明的任何实施方案和方面的一些实施方案,规则包含位置特异性评分矩阵。根据本发明的任何实施方案和方面的一些实施方案,匹配程序包括选自刚体取向、主链二面角的调节、氨基酸侧链包装和氨基酸的改变的至少一种操作。根据本发明的任何实施方案和方面的一些实施方案,刚体取向通过简化表示对接操作和/或完全表示对接操作来实现。根据本发明的任何实施方案和方面的一些实施方案,氨基酸的改变对于另一个相应的拟合的重新闭合区段的氨基酸,以及至少部分在围绕另一个相应的拟合的重新闭合区段的壳内部的模板结构中的氨基酸实现。根据本发明的任何实施方案和方面的一些实施方案,匹配得分选自结合能、包埋表面积、形状互补及其任何组合。根据本发明的任何实施方案和方面的一些实施方案,所述方法进一步包括折叠稳定性评分,并且在步骤(xii)中选择结构也基于折叠稳定性评分。根据本发明的任何实施方案和方面的一些实施方案,共同结构折叠是抗体的。根据本发明的任何实施方案和方面的一些实施方案,共同结构折叠选自免疫球蛋白或其部分、锚蛋白重复、犰狳重复、β抓握(BetaGrasp)、β三叶草(BetaTrefoil)、希腊钥匙(GreekKey)、胶冻卷(Jellyroll)、Keyroll、Plait、罗斯曼折叠、三角四肽重复和TIM桶。根据本发明的一些实施方案的一个方面,提供了预测具有氨基酸序列的靶蛋白的结构的方法,其包括:(i)提供具有共同结构折叠和长度与靶蛋白长度相同的氨基酸序列的多个源结构,所述共同结构折叠具有限定结构构架的保守结构区和多样化结构区,所述结构构架具有最高结构保守性位置;(ii)用靶蛋白的氨基酸序列替换多个源结构各自的氨基酸序列,并将多个源结构的结构分段成结构同源区段,所述结构同源区段各自由最高结构保守性位置的两个位置限定,以便获得多组结构同源区段,每个组由最高结构保守性位置的两个位置限定;(iii)选择具有共同结构构架并具有对应于所述位置的特定位置的模板结构;对于组中的每个:(iv)在结构同源区段内的位点处拆分结构同源区段的至少一个结构同源区段,以便获得拆分区段;(v)将拆分区段的分别位置叠加到对应于所述位置的特定位置的相应位置上,并且置换模板结构中的相应区段;(vi)将用于区段闭合的拆分区段进行权重拟合,以便在模板结构中获得权重拟合和重新闭合的区段,同时维持靶蛋白的氨基酸序列;(vii)任选重复步骤(iv)-(vi),同时在不同位点拆分或中止结构同源区段的进一步操作;(viii)对于至少一个另外的结构同源区段重复步骤(iv)-(vii),以便获得对应于每个组的至少一个附加的权重拟合和重新闭合的区段;和(ix)组合使用对应于每个组的多个权重拟合和重新闭合的区段用于重建多个重建结构,所述重建结构各自具有共同结构折叠,其具有限定结构构架的保守结构区和多样化结构区以及对应于最高结构保守性位置的位置;和(x)计算关于重建结构各自的能量得分,其中所述靶蛋白的结构具有低能量得分。除非另有定义,否则本文使用的所有技术和/或科学术语具有与本发明所属领域的普通技术人员通常理解的相同的含义。尽管下文描述了示例性方法和/或材料,但与本文所述的那些相似或等价的方法和材料可以用于本发明的实施方案的实践或测试中。在冲突的情况下,以本专利说明书(包括定义)为准。另外,材料、方法和实施例仅是举例说明性的,并不预期是必然限制性的。附图的几个视图的简述本发明的一些实施方案在本文中参考附图仅以示例的方式描述。现在详细地具体参考附图,强调的是所示出的细节是作为示例并且用于本发明的实施方案的举例说明性讨论的目的。在这方面,结合附图进行的描述使得本发明的实施方案可以如何进行实践对于本领域技术人员是显而易见的。在附图中:图1是根据本发明的一些实施方案,用于执行计算构建具有共同结构折叠的氨基酸序列文库的方法的示例性算法的示意性流程图;图2呈现了主链原子位置的结构相似性中的变化的图示,显示了作为不同厚度和阴影的管的主链的痕迹,对应于作为结构相似的蛋白质家族成员的一组蛋白质的3D结构中的主链原子位置的变化,其中所述结构彼此叠加以便提供最佳的总体结构拟合(比例尺显示在范围为0Å(白色)至1Å(黑色)的Cα原子位置中的RMSD);图3是根据本发明的一些实施方案,用于执行设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法的示例性算法的示意性流程图;图4呈现了目标函数、稳定性的乘积值和如由方程1定义的结合S形的3D图,其中在设计期间取样的主链构象通过由受抗体稳定性和结合亲和力两者约束的目标函数估计;图5A-B呈现了根据本文呈现的方法的一些实施方案,比较在精制之前和之后所设计的抗体的稳定性(图5A)和结合能(图5B)之间的散点图;图6A-D呈现了显示在本文呈现的方法的最后步骤中用于过滤所设计的抗体结构的能量和结构标准的曲线图,其中基于四个参数过滤所设计的抗体:预测的结合能(图6A)、包埋表面积(图6B)、抗体结构和配体之间的形状互补性(图6C)、以及可变轻和重结构域结构域与配体之间的包装质量(图6D),而截断值由虚线表示,并且衍生自303种天然蛋白质结合抗体的集合,而通过所有过滤器的抗体设计(黑色曲线)与天然蛋白质结合抗体(灰色曲线)进行比较;图7呈现了酵母表面展示的散点图,其中细胞就表达水平和结合进行染色,其中指示为“4m5.3”的模板结构由灰色散点标记,并且指示为“设计#1”(SEQIDNO.1)的所设计的结构由黑色散点标记;图8A-B呈现了在引入点突变之前和之后的抗ACP设计的滴定曲线(设计#1-5;SEQIDNOs.1-5,图8A),以及抗ACP设计(设计#1SEQIDNO.1)由其设计的底物ACP(在图8B中由圆圈标记)和阴性对照(TEM,在图8B中由正方形标记)的滴定曲线;和图9呈现了与充当设计模板的原始酶的序列(来自硫磺矿硫化叶菌(Sulfolobussolfataricus)的内酯酶;SEQIDNO:8)相比较的,具有设计的改变的底物特异性的5个TIM桶折叠设计(SEQIDNOs.8-12)的序列比对。发明的具体实施方案的详述在其一些实施方案中,本发明涉及计算化学和计算蛋白质设计,并且更特别地但不排他地,涉及计算构建具有共同结构折叠的氨基酸序列文库的方法;以及设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法。这些方法可用于例如设计针对预定分子靶具有结构稳定性和高结合亲和力的结合蛋白,或具有从头功能的从头设计的酶。在其一些实施方案中,本发明进一步涉及计算化学,并且更特别地但非排他地,涉及产生对目标分子表面具有所需亲和力的氨基酸序列的方法,以及对目标分子表面具有所需亲和力的氨基酸序列。参考实例和伴随的描述可以更好地理解本发明的实施方案的原理和操作。在详细解释本发明的至少一个实施方案之前,应理解本发明在其应用中不一定限制于在下述描述中阐述的或由实施例示例的细节。本发明能够具有其他实施方案或者以不同方式实践或进行。如上文讨论的,用于从头设计抗体的先前方法依赖于已知的规范构象,并且随后对接并设计为通过在表位结合位点(主要是称为CDR的区域)处的氨基酸序列排列来结合靶表位。尽管此类计算方法可以提供在计算机模拟上对靶显示出高亲和力的抗体结构,但这些方法未能解决抗体稳定性的问题,所述抗体稳定性受抗体的整个主链的贡献的影响,并且对主链构象以及在主链各处的氨基酸序列的微小和微妙的差异敏感。可以推断,解决设计大型和极性结合表面的挑战的关键在于蛋白质主链的设计,因为主链提供了许多另外的构象自由度,其迄今为止仍未被结合剂设计策略利用。然而,为了功能(例如,稳定性和结合)而设计的主链是未解决的问题,这是由于正确平衡来自极性基团的自由能的贡献的固有复杂性和由于对蛋白质主链开放的大构象空间。显而易见的是,在当前的计算资源下,即使通过在基因水平上设置的规范构象和氨基酸序列排列限制缩小,并且随后尝试根据结合亲和力评定空间的每个存在(inhabitant),定义整个构象组合空间乘以序列置换空间的壮举也是不切实际的,如果并非不可能的。此外,即使可以在理论上包含并有效地和系统地对可以通过在基因水平上的排列生成的所有构象组合空间取样的方法变成实际,此类方法也不能解释在抗体中观察到或可能出现的无数的随机突变。在构想本发明的同时,本发明人已考虑了从所有天然折叠(例如抗体)取样主链构象和序列信息的方法,包括高度同源的折叠,对于其可获得一组三维原子坐标即3D结构,以便将设计基于已进化了几千年的结构,并且从而改善具有高结合亲和力和高结构稳定性的折叠(例如,抗体)的设计。这种方法的根本假设是,自然具有进化时间尺度和测试场地来取样和选择最有效的支架的优势,其甚至超过目前可行的最雄心勃勃的强大计算技术。再者,依赖于天然存在的结构不仅将使用可预测的支架和序列,而且还将使用在天然抗体中出现的不可预测的随机突变。在将本发明付诸实践和作为例证的同时,本发明人已开发了依赖于天然存在的免疫系统抗体的模块结构来构建从头分子结合剂的方法,并且已使用基准重现测试测试了这种方法,其结果显示这种方法能够获得这样的结构,其类似于天然存在的结构,并且与使用先前的设计方法已构建的结构相比较改善所有的结合和结构参数。本文公开的方法已通过下述对具有已知三维结构的天然存在的高亲和力抗体结合复合物的多样化集合应用且验证:从该集合的成员中去除所有序列和主链构象信息,并且重现其天然结合模式、天然主链构象和序列,尤其是在其中天然结合表面很大的情况下。本发明人已进一步设想可以实施这种方法以设计其为结构相似蛋白(FSSP)家族的成员的分子结合蛋白,即属于家族的蛋白质至少在其部分中显示出一种或多种天然重现的,因此,保守的折叠,例如但不限于抗体及其结构域、α/β水解酶、TIM桶蛋白质等等。根据本发明的一些实施方案,本文提供的方法可用于设计高度稳定的蛋白质,所述蛋白质原则上可以以高亲和力结合以任何给定构象的任何靶分子。在抗体的示例性情况下,该方法利用了下述观察:高变区片段的主链通常可彼此互换以生成新的主链-片段组合,理论总复杂度在1013级别上;因此,该方法使用由抗体可变结构域的V(D)J基因区段编码的主链构象,其彼此组合以产生高度多样化的抗体支架集合。该预计算步骤创建了前所未有的主链组合空间和大序列数据集,其被预测为稳定的并且对其靶具有高亲和力。随后将支架针对靶分子对接。设计的抗体通过迭代过程进行精制,所述迭代过程用在天然抗体中观察到的主链构象片段替换主链构象片段,并且设计用于最佳结合和抗体稳定性的氨基酸序列。在设计过程自始至终,根据本发明的一些实施方案,使用对于每个构象区段衍生的序列约束来实施序列结构规则。最后,通过衍生自天然抗体结合复合物集合的能量和构象标准来选择模型。当应用于抗体时,本文呈现的蛋白质设计方法解决了几个相关的挑战,包括:1.合并来自构象和序列数据库的知识来约束设计选择;2.编码在可变区段(其大部分缺乏稳定的二级结构元件)和构架(其形成紧密包装且稳定的结构基础)之间的远程残基相关性;3.抗体的大主链和序列组合空间的有效取样;和4.设计最优抗体稳定性和靶分子结合两者的抗体构象和序列。在下文部分中,描述了算法的不同要素,以及它们如何解决当前的设计挑战。计算构建具有共同结构折叠的氨基酸序列文库的方法:因此,根据本发明的一些实施方案的一个方面,提供了计算构建具有共同结构折叠的氨基酸序列文库的方法,其根据下述步骤执行。根据本发明的一些实施方案,文库设计为有效和简洁地对共享该共同结构折叠的天然存在的蛋白质家族的成员占据的巨大构象空间取样,并且因此可以用于提供如下文所述的从头蛋白质设计的基础。图1呈现了根据本发明的一些实施方案,用于执行计算构建具有共同结构折叠的氨基酸序列文库的方法的示例性算法的示意性流程图。下文的描述涉及图1中呈现的算法中的一些操作。如由下文描述中可以理解的,算法中的一些操作可以以可替代的次序和可替代的周期数来执行。根据本发明的一些实施方案,该方法在其步骤(i)中包括提供具有共同结构折叠的多个源结构(图1中的框1)。在本发明的实施方案的上下文中,术语“源结构”指在其至少一个结构域中共享共同的多肽主链折叠的蛋白质的实验阐明的3D结构集合,而与其在该结构域中与彼此的序列同源性无关。一般而言,源结构的集合可以由一些或全部实验阐明的蛋白质结构形成。可替代地,源结构可包括所有可用3D结构的子集,或甚至其小部分。因此,根据本发明的一些实施方案,源结构的数目大于500、大于100、大于50、大于10、大于5或大于2。根据本发明的一些实施方案,共同结构折叠的特征在于具有限定结构构架区的保守结构区和多样化结构区,其中结构构架区通常显示出高结构保守性,而通常结构构架区中的一些位置显示出最高结构保守性。这些位置在本文中被称为“最高结构保守性位置”。结构保守性可以经由图2概念化。图2呈现了主链原子位置的结构相似性中的变化的图示,显示了作为不同厚度和阴影的管的主链的痕迹,对应于作为结构相似的蛋白质家族(FSSP)成员的一组蛋白质(例如,该示例性图示中的40个非冗余抗体Fv结构)的3D结构中的主链原子位置的变化,其中所述结构彼此叠加以便提供最佳的总体结构拟合(比例尺显示在范围为0Å(白色)至1Å(黑色)的Cα原子位置中的RMSD)。如由图2可见的,沿大多数结构的主链的一些区域在其3D结构中发散,在平均路径周围填充更厚的信封结构,而其他区域在平均路径周围填充更紧密、更薄的信封结构。在源结构的集合中,根据本发明的一些实施方案,“较薄”区域被说成显示出高结构保守性。通常,较薄区域对应于结构构架区,并且较厚区域对应于多样化结构区。根据本发明的一些实施方案,沿叠加的多肽主链的集合的最薄位置在本文中称为“最高结构保守性位置”。源于图1中呈现的图示的术语“薄”和“厚”也可以以通常以埃(Å)单位给出的多个比对的多肽主链位置的均方根偏差(RMSD)表示,其中“薄”与“厚”相比较具有相对小的值。当提及多个叠加结构中的特定结构时,最高结构保守性位置对应于特定位置,并且反之亦然,最高结构保守性的特定位置对应于最高结构保守性位置。根据本发明的一些实施方案,可以在施加选择标准的同时进行源结构的选择和/或区段的选择,以便使方法的结果偏向提供更可能类似显示出一种或多种性状的序列。选择标准可以基于源生物(以确保例如与表达系统或所需最终宿主的相容性)、氨基酸序列长度(整个或每个区段或环长度等)。在实施方案5中证明了基于源结构的特异性酶促活性的选择标准的使用,其中在构象文库的构建中施加偏向以选择具有PTE酶同源物源的叶片7构象。根据本发明的一些实施方案,多肽主链中的“位置”由主链原子或主链原子之间的预定位置表示,其中主链原子通常是α碳原子、主链羰基碳原子、主链羰基氧原子或主链氮原子。根据本发明的实施方案,源结构用于构建由多个多肽链区段填充的主链构象数据库。不受任何特定理论的束缚,假设蛋白质构象的稳定性依赖于阳性和阴性设计要素两者。属于共享共同折叠的非常多样化的家族的蛋白质的计算设计的关键优点在于可以提取关于根据位置偏向的氨基酸选择的统计学,所述位置偏向编码这些要素中的至少一些以指导设计过程。此外,通过使天然主链构象和序列关联,可以将折叠成特定构象类别的天然区段序列的集合分类。根据本发明的实施方案,多肽链的分段涉及构成在其上发生识别和结合的结构域的区域。根据本发明的实施方案,该方法基于实验确定的3D结构的结构比对和基于结构保守区(因此,对结构同源区段的分段)的考虑的多肽链分段。这不同于先前已知的计算方法,其基于氨基酸序列同源性比对和基于保守区的考虑的多肽链分段。因此,根据本发明的一些实施方案,该方法在其步骤(ii)中包括将源结构的结构各自分段成结构同源区段,其中每一个由最高结构保守性的两个位置限定,以便获得多组结构同源区段。因此,结构同源区段的每个组由最高结构保守性的两个位置限定(图1中的框2)。任选地,分段进一步包括一个或多个末端片段,其包括每个结构的多肽链的末端(末端或尾部),其中多肽链末端在本文中称为“末端位置”。因此,每组末端区段(在本文中也称为尾部区段)由最高结构保守性的一个位置和一个末端位置限定。应注意末端位置不一定共享高结构同源性。因此,在本发明的实施方案的上下文中,术语“区段”指源结构中的任何一个的多肽链的连续段,其在两个位置处或附近开始和结束。“结构同源区段”定义为在最高结构保守性的两个位置处或附近开始和结束的区段。“末端区段”定义为在最高结构保守性的一个位置处或附近开始/结束,并在一个末端位置结束/开始的区段。在步骤(ii)的上下文中,根据本发明实施方案中任一的一些实施方案,每个源结构的多肽链被分段成“结构同源区段”。任选地,在步骤(ii)的上下文中,根据本发明实施方案中任一的一些实施方案,每个源结构的多肽链被分段成“结构同源区段”,并且进一步分段为“末端区段”。根据本发明的一些实施方案,所提供的方法的示例性用途已应用于设计抗体的从头Fv片段,并且在随后的实施例部分中证明了基准重现实验。在这些示例性实施方案中,仅具有轻κ链的Fv结构域已包括在源抗体(即,源结构)的集合中。具体地,788个可变轻κ链和785个可变重链结构在基准重现实验中用作源抗体,用于测试通过本文呈现的方法获得的结果。在使用抗体的本文呈现的方法的示例性证明中,源抗体包括其为Fv结构域的结合结构域以及构成所有源抗体的Fv结构域的两条链中的每一条,即轻链和重链各自通过鉴定最高结构保守性的两个连续位置分成两个结构同源区段;例如形成结构上保守的链内二硫键的可变结构域的两个半胱氨酸残基,以及接近第二半胱氨酸的第三结构保守位置,从而对于每个Fv结构域形成四个结构同源区段。具体地,在本文呈现的方法的示例性证明中,根据本发明的一些实施方案,在随后的实施例部分中对于抗体的Fv所证明的,分段遵循四个区段的划分:L1-L2(称为“VL”)和H1-H2(称为“VH”),其各自跨越轻和重可变结构域L3和H3的两个结构上保守的半胱氨酸残基之间的所有氨基酸,各自起始于第二半胱氨酸之后的第一个氨基酸并结束于可变轻κ结构域的位置100和可变重结构域的位置103,使用Chothia位置编号方案(参见下表2)。根据本发明的一些实施方案,主链构象数据库的构建涉及将所有区段置于统一的相对坐标系统内。根据本发明的一些实施方案,该方法在其步骤(iii)中包括选择具有与源结构相同的共同结构构架的模板结构,并且因此具有对应于最高结构保守性位置的特定位置,并且将所有结构上同源的区段置于模板结构的坐标系统内(图1中的框4)。在本发明的任意实施方案的一些中,从源结构任意地和/或随机地选择模板结构。根据本发明的一些实施方案,模板结构作用于对其移植来自源结构的结构同源区段。根据本发明的一些实施方案,在从头蛋白质设计期间,模板结构用作使用一些结构特点作为参考与之比较设计的蛋白质的参考结构。根据本发明的一些实施方案,使所有结构同源区段共享共同的起点和终点,因为结构同源区段中的一些可以具有不同的构象并且通常具有不同的长度。可替代地,基于最高结构保守性的一些或所有位置的空间比对,所有结构在结构上进行比对。根据这些实施方案中的一些,根据本发明的一些实施方案,进行下述程序以使所有结构同源区段共享共同的起点和终点,并且任选进一步共享末端区段中的一个、一些或全部的最高结构保守性位置:根据本发明的一些实施方案,该方法在其步骤(iv)中包括在其中的位点处拆分至少一个结构同源区段,以便获得拆分区段(图1中的框3);任选地,如果该方法进一步包括一个或多个末端区段,则末端区段不被拆分,相反末端区段作为拆分区段进入程序,而程序中的所有其他步骤如本文呈现的应用,即末端区段在步骤(v)中视为拆分区段;根据本发明的一些实施方案,该方法在其步骤(v)中包括对于结构同源区段的每个组,将拆分区段的最高结构保守性的分别位置叠加到模板结构的相应特定位置上,并且置换模板结构中的相应区段(图1中的框5);并且,根据本发明的一些实施方案,该方法在其步骤(vi)中包括权重拟合拆分区段以便实现区段闭合,同时允许区段的主链形成具有很少内张力或无内张力的化学合理构象,并且从而在模板结构中获得权重拟合和重新闭合区段(图1中的框6)。对于末端区段,权重拟合不包括闭合。根据本发明的任意实施方案的一些实施方案,术语“权重拟合”指一个或多个计算结构精制程序或操作,旨在通过基于预定权重,与例如精制结构的序列同源性得分、主链二面角和/或原子位置(变量)有关的限制和约束(常量)来最小化多项式函数,最优几何形状、空间和/或能量标准。根据一些实施方案,权重拟合程序包括下述的一个或多个:主链二面角的调节、拟合区段长度的改变(增加或减少)、氨基酸侧链包装和氨基酸序列的改变,而术语“主链二面角的调节”、“氨基酸侧链包装”和“氨基酸序列的改变”在本文中也用于尤其指众所周知的最优程序和操作,其广泛用于计算化学和生物学领域。关于一般最优方法的综述,参见例如通过ChristodoulosA.Floudas和PanosM.Pardalos,SpringerPub.,2008的“EncyclopediaofOptimization”。根据本发明的一些实施方案,示例性最优程序是下文讨论的循环坐标下降(CCD),其在本文中与在用于大分子建模的Rosetta软件套件中实施的缺省全原子能量函数一起使用。在本发明的一些实施方案中,权重拟合中的限制和约束(权重)视为指定计算程序的规则。例如,当精制具有第一构象的任何给定多肽区段的主链原子位置和二面角,以便在尝试尽可能多地保存在第二构象中观察到的二面角的同时,驱动朝向不同的第二构象,该计算程序将使用偏向例如Cα位置的调和限制,以及使主链-二面角偏向自由地偏离在第二构象中观察到的那些的调和限制,因此允许在驱动整体主链变成第二构象的同时,每个结构决定簇发生最小的构象变化。约束也可以在改变蛋白质的氨基酸序列的程序中应用。这些约束还可以用于至少在某种程度上保存从前导序列继承的序列的某些部分。用于氨基酸序列改变的最常见约束之一源于在特定位置处的高度保守序列模式(定位),这通常在FSSP中显示出。根据本发明的一些实施方案,在权重拟合过程中通过其指定氨基酸变化的规则包括位置特异性评分矩阵值或PSSM。“位置特异性评分矩阵”(PSSM)在本领域中也称为位置权重矩阵(PWM)或位置特异性权重矩阵(PSWM),是生物序列中重复模式的常用表示,基于在沿序列的给定位置中的特征(单体;氨基酸;核酸等)的出现频率。PSSM通常衍生自比对序列的集合,所述比对序列被认为在结构和功能上相关,并且已广泛用于用于计算基序发现的许多软件工具中。在氨基酸序列的上下文中,PSSM是在蛋白质BLAST搜索中使用的一类评分矩阵,其中对于蛋白质多重序列比对中的每个位置分开给予氨基酸取代得分。因此,在比对的位置A处的Tyr-Trp取代可以接受与在位置B处的相同取代非常不同的得分,经受在两个位置处不同水平的氨基酸保守性。这与位置不依赖性矩阵例如PAM和BLOSUM矩阵形成对比,其中Tyr-Trp取代不管其在什么位置处出现均接受相同的得分。PSSM得分一般显示为正整数或负整数。正得分指示给定的氨基酸取代在比对中比预期偶然发生的更频繁,而负得分指示取代比预期的更不频繁地发生。大的正得分通常指示关键的功能残基,其可以是活性位点残基或者其他分子间或分子内相互作用所需的残基。可以使用位置特异性迭代基本局部比对搜索工具(PSI-BLAST)创建PSSM,其找到与查询序列相似的蛋白质序列,并且随后由所得到的比对构建PSSM。可替代地,PSSM可以从美国国家生物技术信息保守结构域数据库(NationalCenterforBiotechnologyInformationConservedDomainsDatabase)(NCBICDD)数据库中检索,因为每个保守结构域由编码在种子比对中观察到的取代的PSSM表示。这些CD记录可以通过下述来发现:Entrez保守结构域(EntrezConservedDomains)中的文本搜索,或使用反向位置特异性BLAST(ReversePosition-SpecificBLAST)(RPS-BLAST)(也称为CD搜索)在输入蛋白质序列上定位这些结构域。如上文讨论的,根据本发明的一些实施方案,在两种方式下,在不同权重拟合以及其他精制和设计计算期间,在本文呈现的方法中使用PSSM得分。首先,根据PSSM,设计序列选择仅限于高于某一保守阈值的标识。对于结合表面(对于具有在分子实体的目标分子表面的10Å距离截断内的具有Cβ的所有蛋白质残基,大于或等于0的示例性PSSM得分)、多样化结构区(大于或等于1)以及结构构架区、定位和位置(大于或等于2)分开测定截断;有效地,与结合相关的位置允许比蛋白质及其构架的核心中的位置更多的自由度,以与共有区不同。第二,在序列设计中使用的全原子能量函数被修改为包括根据PSSM使序列偏向更可能的标识的术语,并且在这些情况下,朝向序列共有区的偏向更远离结合位点。根据本发明的一些实施方案,将结构同源区段拆分成两个亚区段,每个亚区段被移植到对应于拆分区段的分别位置的模板结构中的特定位置上,同时替换模板结构中的相应区段,并且其后在模板结构的上下文中使拆分和叠加区段经历权重拟合。权重拟合包括精制具有第一构象的拆分区段的主链二面角,以便将其朝向闭合驱动,这意指其随后将具有略微不同的构象。如上所述,在末端区段的情况下,权重拟合不包括闭合。权重拟合过程尝试尽可能多地保存原始二面角,因此计算程序将二面角的可变性约束至在源结构中观察到的那些,从而允许每个二面角发生最小变化,同时驱动拆分区段闭合,从而在模板结构的上下文中获得权重拟合和重新闭合的区段。该过程在改变经受PSSM衍生的约束的拆分区段的氨基酸序列的同时迭代实现。氨基酸序列中的改变允许通过允许氨基酸侧链包装连同主链二面角一起最优来发生主链构象中的小变化。根据本发明的一些实施方案,权重拟合过程还改变设计结构中的氨基酸,其至少部分地存在于围绕拆分区段的壳内部。根据一些实施方案,壳的特征在于2Å至20Å的半径,或可替代地2Å、3Å、4Å、5Å、6Å、7Å、8Å、9Å、10Å、11Å、12Å、15Å或20Å的半径。根据一些实施方案,壳半径为6Å。下文是根据本发明的一些实施方案的示例性权重拟合过程的简要描述,其在本发明的上下文中关于“权重拟合”的定义是非限制性的。结构同源区段通过长度分选,并且对于每组结构同源区段的每个长度亚组,从源结构(“原始”主链构象)中提取主链二面角(Φ,Ψ和Ω),并且替换具有源二面角的模板结构中的相应区段中的那些,同时在移植区段的随机选择的位置中引入主链拆分位点。应注意在末端区段中不引入主链拆分。换言之,来自所有非模板结构的区段在远离结构构架区以及远离最高结构保守性位置(区段的末端)的任意位置处被切割,通过叠加相应的位置置于模板结构而不是模板结构中的相应区段上,并且随后允许拆分区段的两个部分的二面角根据约束增量移位,同时改变区段的氨基酸序列,经受PSSM衍生的约束,如本文所述。通过将替换相应的末端区段的保守位置叠加在模板结构上,将末端区段置于模板结构上。根据一些实施方案,使用循环坐标下降(CCD),随后为如例如在Rosetta软件套件中实现的“CCD移动器”中实施的小和剪切移动,将拆分和移植区段各自权重拟合到模板结构上。仅使用Rosetta软件套件中实施的小的和剪切移动,使用脚本例如在下文实施例部分中呈现的“TailSegmentMover”脚本,将末端区段权重拟合到模板结构上。在精制期间,由范德华包装、氢键和隐性溶剂化所支配的标准全原子能量函数通过下述进行修饰:加入促进主链拆分位点闭合的调和项,以及使Cα位置偏向的调和限制,判罚相对于源结构中的原始位置在权重拟合区段中的主链Cα位置中的差异,以及主链-二面角的调和项,判罚相对于源结构中观察到原始角度在二面角中的差异。调节区段构象使主链移动与组合氨基酸侧链包装交替。在包装步骤期间,该程序允许整个建模区段以及围绕该段的6Å壳中的氨基酸序列变化,经历区段的PSSM衍生的约束。取决于可用的计算能力和其他实际考虑,每个CCD步骤或TailSegmentMover重复几百次,并且在每次CCD程序结束时,计算来自源区段的建模区段的均方根偏差(RMSD),并且如果它超过1Å,或如果例如在拆分位点处的主链间隙得分(在Rosetta软件套件内限定的关于主链连续性的示例性标准;这不适用于末端区段)太大(例如大于或等于0.5),则对于该区段再一次重复该程序。如果在几百次之后得分仍然是无法接受的,则使用不同的任意和随机选择的拆分位点来重新运行该程序用于另一次闭合试验。根据本发明的一些实施方案,该方法在其步骤(vii)中包括任选重复步骤(iv)-(vi)(图1中的框7),同时在不同位点处拆分该区段(图1中的框8a)或中止对该结构同源区段的进一步操作(图1中的框8b)。因此,在预选数目的权重拟合循环(达到闭合时的尝试)之后未能正确关闭的区段从进一步考虑中放弃(图1中的框8b)。已发现在模板结构的上下文中,权重拟合程序在调节源区段主链构象方面是高度有效的。在下文实施例部分呈现的情况下,用于区段闭合程序的该权重拟合在不超过6次拆分位点尝试中成功地关闭长高达74个氨基酸的区段,具有1.2的平均迭代次数。根据本发明的一些实施方案,该方法在其步骤(viii)中包括对于至少一个另外的结构同源区段重复步骤(iv)-(vii),以便获得对应于每个组的至少一个另外的权重拟合和重新闭合的区段。一旦拆分区段通过权重拟合程序达到闭合,则它称为权重拟合和重新闭合区段。根据本发明的一些实施方案,其新的主链二面角被记录在构成文库部分的扭曲数据库中。上述过程的产物是已移植在模板结构上的权重拟合和重新闭合区段的集合;因此,来自任何区段组的每个区段均可以与来自所有其他区段组的区段组合,以形成完整的重建结构(图1中的框9)。根据本发明的一些实施方案,结构同源区段通过最高结构保守性的两个连续位置限定,而末端区段通过一个末端位置和最高结构保守性的一个位置(其可以是最接近连续多肽链中的末端位置的最高结构保守性位置)限定。在此类实施方案中,在区段之间基本上不存在氨基酸的重叠;即两个连续区段不共享氨基酸位置。在一些实施方案中,选择具有最高结构保守性的两个位置以具有多个氨基酸残基的一些重叠,其范围为两个连续区段之间的1-20、1-10或1-5个氨基酸重叠,即接近邻近区段的端部的一些氨基酸由两个邻近区段共享。如上文讨论的,主链构象取样在计算上是非常苛刻的,并且尽管一些成功,但主链重新设计已导致偏离原始计算模型的构象。通过设计构象高度多样化家族中的蛋白质,可以利用每个主链区段的天然存在的构象变体,其中构象在宿主蛋白质折叠内很可能是稳定的。为了计算高效利用在天然蛋白质中观察到的主链构象的丰度,将源结构的所有结构上同源的、权重拟合和重新闭合的区段的构象存储在数据库中,用于在蛋白质设计过程期间使用。根据本发明的一些实施方案,该方法在其步骤(ix)中包括任选使用对应于每个组的权重拟合和重新闭合的区段,用于组合地重建多个重建结构,每个重建结构具有共同结构折叠,其具有限定结构构架的保守结构区和多样化结构区以及对应于最高结构保守性的定位的位置。这是在本文中被称为具有共同结构折叠的氨基酸序列文库的这种多个重建结构(图1中的框12)。在该任选步骤中,文库是详尽的,并且基本上包含用于组合在该方法的先前步骤中生成的所有权重拟合和重新闭合区段的所有可能组合。根据本发明的一些实施方案,通过选择与目的区域重叠的一个或多个特定区段,该方法可以用于仅生成对给定蛋白质的结构中的特定区域的变化。即,不是创建所有可能的区段组合的多个重建结构,而是通过选择蛋白质中的一个或多个目标区段来使用该方法,以创建仅在目标区段中的构象中不同的多个重建结构。该方法的这种使用模式在下文的实施例4(一个区段)和实施例5(4个区段)中例证。为了简化在执行本文呈现的方法中的计算负荷,根据本发明的任何实施方案的一些实施方案,可以进行取样和还原程序。根据本发明的一些实施方案,计算构建具有共同结构折叠的氨基酸序列文库的方法进一步包括在步骤(ix)之前,将对应于每组结构同源区段的权重拟合和重新闭合的区段各自分选到至少一个结构簇内(图1中的框10),并且从结构簇中选择代表性权重拟合和重新闭合的区段,由此步骤(ix)的权重拟合和重新闭合的区段各自是代表性权重拟合和重新闭合的区段(图1中的框11)。根据本发明的一些实施方案,在本文呈现的方法的这个任选步骤(其在本文中也称为“取样和还原程序”)中,每组结构同源区段的内容物由代表性区段集合表示。随后将这些代表性区段重组成所有排列,以形成具有共同结构折叠的氨基酸序列文库(图1中的框12)。根据本发明的一些实施方案,在计算构建具有共同结构折叠的氨基酸序列文库的方法的上下文中,术语“分选”指通过其在每组结构同源区段中的权重拟合和重新闭合的区段中至少一些通过长度进行分选。根据本发明的一些实施方案,术语“分选”进一步包括通过比对的多肽主链位置的均方根偏差(RMSD),将长度分选的权重拟合和重新闭合的区段聚簇。根据本发明的一些实施方案,对于包含当前取样的构象区段的整个重建结构指定PSSM,从而使序列约束与当前取样的主链构象同步。该程序是有利的,因为在权重拟合步骤期间,在2-20Å壳内的权重拟合段外部的残基也可以改变,因此使PSSM与所有构象区段包括未经受在该步骤处的拟合的区段一致。这些序列相关的PSSM约束相当大地减少了组合序列最优问题。虽然减少了,但通过本文呈现的方法产生的文库可以含有多个重建结构,其超过具有相同的共同结构折叠的实验可用结构的数目;并且此外,与实验可用的结构相比较,文库的重建结构在构象上更多样化。设计和选择对目标分子表面具有所需亲和力的一个或多个氨基酸序列的方法:根据本文呈现的方法汇编的具有共同结构折叠的氨基酸序列文库,其包含已最优以对已知结构的构象空间取样的重建结构,用于测试与分子实体的目标分子表面的相互作用。使用用于设计和选择设计用于对目标分子表面的所需亲和力和分子稳定性的合格氨基酸序列的方法,进一步设计文库的成员并评价对目标分子表面的亲和力并进一步评价结构活力。根据本发明的一些实施方案的另一个方面,提供了设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法。图3是根据本发明的一些实施方案,用于执行设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法的示例性算法的示意性流程图。下文的描述涉及图3中呈现的算法中的一些操作。如由下文描述中可以理解的,算法中的一些操作可以以可替代的次序和可替代的周期数来执行。根据本发明的一些实施方案,该方法在其步骤(x)中包括将形成上文呈现的文库(图2中的框12)的重建结构中的至少一个匹配到目标分子表面上(图2中的框13),以便设计每个具有匹配得分的多个重建结构-分子表面复合物。根据一些实施方案,步骤(x)可以进一步包括步骤(x)的任选重复,使用导致先前迭代的复合物。如本文在本发明的实施方案的上下文中使用的,术语“匹配”指包括在任何给定的网格分辨率下的刚体取向最优(也称为对接或表面互补性刚体取向精制)的程序,并且可以进一步包括一个或多个另外的操作,例如但不限于,主链二面角的调节(最优)、氨基酸侧链包装最优和氨基酸的改变或序列最优。根据一些实施方案,匹配程序包括定义主体的任何原子子集的刚体取向最优的操作,其最优在重建结构和分子表面之间的界面处的表面互补性,将复合物配对物各自作为刚体处理。该刚体表面互补性最优在预定的原子子集处执行,所述原子子集可以是代表主链和代表侧链原子的质心的虚拟原子的减少原子集合(在本文中称为“简化表示对接”),或代表两个主体的整个结构的原子集合(本文称为“完全表示对接”)。根据本文所述的本发明的一些实施方案,匹配程序包括简化表示对接操作,其搜索重建结构与目标分子表面的最佳匹配,将两者作为具有不变的氨基酸序列的刚体处理(刚体取向精制)。此类匹配程序在本文中被称为“粗略匹配程序”(图2中的框14)。可替代地,在本文所述的本发明的一些实施方案中,匹配程序包括基于完全表示对接操作的刚体取向精制,并且进一步包括结构最优操作,其包括但不限于最优氨基酸序列(序列设计)、最优主链和侧链构象并最优原子位置,基本上旨在提供就目标分子表面而言具有最佳结构互补性的结构。此类匹配程序在本文中被称为“有利匹配程序”。可以在使用用于最优的规则(例如上文讨论的约束和限制,例如关于原子位置的移动的调和限制和关于氨基酸的改变的PSSM值)的同时,进行匹配程序的操作中的任何一个。另外,因为一些最优操作改变了在不同区域处的重建结构的氨基酸序列,所以可以将一组新的序列约束规则(例如PSSM)重新指定给整个所得到的结构。刚体取向操作通常导致通过重建结构和目标分子表面之间的相对取向而彼此不同的多个重建结构-分子表面复合物,其构成化学实体的部分(图2中的框15)。根据本发明的一些实施方案,匹配程序是粗略匹配程序,其包括简化表示对接操作。根据一些实施方案,起因于粗略匹配程序的所得到的多个匹配复合物可以进一步通过RMSD(通常通过以Å指示的范围标准聚簇)分类成相对取向相似性的簇,并且从每个相对取向簇中选择簇代表复合物用于在匹配程序步骤内的进一步加工,即匹配程序的另一个循环,例如可以包括其他最优操作的有利匹配程序。根据一些实施方案,对于所选择的复杂结构实现有利的匹配程序(步骤(x)的另一个循环),导致粗略匹配程序(图2中的框16a)。根据本发明的一些实施方案,有利的匹配程序可以包括与其他最优操作组合来实现的完整表示对接操作,所述其他最优操作例如当前复合物的重建结构中的氨基酸的改变,其在本文中称为“序列设计”。序列设计通常由规则例如PSSM值,例如对于整个重建结构重新指定的PSSM值来指示。根据这些实施方案,根据本发明的一些实施方案,对存在的重建结构氨基酸残基进行序列设计:在重建结构和目标分子表面之间的界面;在落入围绕界面的壳内的重建结构的区域处;在落入围绕区段的壳内的重建结构的区域处;和/或整个重建结构或其任何部分。根据一些实施方案,壳的特征在于2Å至20Å的半径,或可替代地2Å、3Å、4Å、5Å、6Å、7Å、8Å、9Å、10Å、11Å、12Å、15Å或20Å的半径。根据一些实施方案,壳半径为6Å。根据一些实施方案,匹配程序进一步包括匹配评价程序,其对于每个最优复合物产生一个或多个匹配得分,导致匹配程序的当前循环。术语“匹配得分”包含各种复杂属性评价,包括但不限于包埋的表面积(包埋在结构和目标分子表面之间的界面中)、结构和目标分子表面之间的互补形状、以及结构与目标分子表面之间的结构能或亲和力、及其任何组合。用于复杂评价的另一个标准是与重建结构有关的折叠稳定性,其在游离蛋白质未结合到目标分子表面时的背景下加以考虑。根据一些实施方案,重建结构的折叠稳定性可以通过包装质量进行测定,所述包装质量根据重建结构的计算自由能进行评价。匹配得分标准各自被指定用作选择过滤器的截断值,即最小包埋表面积、最小形状互补、最小结合自由能、最小包装质量和最小包装质量(图2中的框17a)。根据本发明的一些实施方案,可替代步骤(x)包括有利的匹配程序,其中重建结构-分子表面复合物被指定匹配得分,例如关于包埋表面的得分,并且如果其匹配得分通过预定的选择截断标准(例如,通过最小的包埋表面截断值)发现是可接受的(图2中的框17a),则合格的复合物可以经受当前复合物的重建结构中的权重拟合的重新闭合区段中的至少一个由另一个相应的权重拟合的重新闭合区段(图2中的框9)(在本文中被称为“替换区段”(图2中的框18))的任选取代。根据本发明的一些实施方案,从多个合格复合物中随机抽取合格复合物,经受步骤(xi),并且使所得到的复合物经受步骤(x)的附加循环,例如,另一个有利的匹配循环(图2中的框16b)。根据一些实施方案,可以通过用于随机选择的MoteCarlo例行程序来执行合格复合波的随机抽取。根据本发明的一些实施方案,任选步骤(xi)包括用另一个相应的权重拟合的重新闭合区段取代随机抽取的合格复合物的重建结构中的权重拟合的重新闭合区段中的至少一个,并且重复步骤(x)以便设计取代重建结构-分子表面复合物(图2中的框18)。根据本发明的一些实施方案,对于取代重建结构进行的步骤(x)的重复循环是如上所述的有利匹配程序(图2中的框16b)。在一些实施方案中,构成有利匹配程序的部分的序列设计可以对于整个替换区段以及任选对于取代重建结构的其他区域实现,所述其他区域例如落入围绕在目标分子表面和取代重建结构之间界面的壳内的区域、落入围绕整个替换区段的壳内的取代重建结构的区域、或可替代地整个取代重建结构或其任何部分。根据本发明的一些实施方案,有利的匹配程序可以包括旨在最优取代重建结构对分子实体的目标分子表面的亲和力且最优取代重建结构的折叠的稳定性的任何最优操作。此类另外的最优操作包括但不限于改变单独或以氨基酸组合的氨基酸同一性和构象,包括仅旋转异构体构象(构象异构)或离体旋转异构体(off-rotamer)构象,或限制/补充有在相同FSSP的天然存在的结构中观察到的构象,使用主链最小化、CCD、肽片段插入、动态环闭合或任何主链构象精制操作来改变替代区段的主链构象,使用源于在天然存在的抗体结构中观察到的取向的约束来改变且最优其他区段相对于彼此的相对构象等等。根据本发明的实施方案,对经历有利的匹配程序的复合物指定匹配得分,所述匹配得分包括但不限于包埋表面积得分、形状互补得分和结合能得分中的至少一个,以及折叠稳定性得分,及其任何组合。根据本发明的一些实施方案,匹配得分包括属性的任何组合,所述属性可以用于估计取代重建结构对分子实体的目标分子表面的亲和力,并且估计取代重建结构的折叠的稳定性,其包括以任何组合且不限于与结合界面表面有关的电荷互补性、极性和疏水性得分,如通过分子动力学评价的构象刚性得分,布朗动力学得分,蛋白质的正常模式分析(NMA)移动性得分,人源化得分(反映与人天然氨基酸序列的相似性),与给定表达系统的氨基酸序列相容性,避免氨基酸序列的不需要部分等等。根据本发明的一些实施方案,组合匹配得分用于估计已经历设计最优匹配程序的合格复合物,并选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列,其对应于合格复合物的重建和最优的结构。根据一些实施方案,步骤(xii)包括基于上文所述的匹配得分中的一个或多个,选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列(图2中的框17b)。属于起因于步骤(xii)的合格复合物的多个氨基酸序列构成对分子实体的目标分子表面具有所需亲和力的一组序列(图2中的框19)以及起于不同属性评分组合的其他所需属性。常见结构折叠:如上文讨论的,根据本发明的一些实施方案,计算构建具有共同结构折叠的氨基酸序列文库的方法,以及设计和选择对分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法,可以应用于对于其可获得实验获得的3D结构的结构上相似的蛋白质(FSSP)的家族中任一。几种广泛已知的FSSP的实例包括但不限于“α/β水解酶”、“β抓握”(泛素样折叠)、“希腊钥匙”、“胶冻卷”、“Keyroll”、“Plait”(铁氧还蛋白样折叠)、“罗斯曼折叠”、“Β三叶草”、“锚蛋白重复”、“犰狳重复”、“三角四肽重复”和“TIM桶”,因为这些天然普遍存在的折叠是本领域已知的。在本文中应注意,虽然本文呈现的方法主要在抗体的上下文中例证,但应理解相同的概念适合于由几种天然存在的大分子共享的其他常见蛋白质折叠、基序和结构域。下表1呈现了由SCOP2服务器[MurzinA.G.等人,“SCOP:astructuralclassificationofproteinsdatabasefortheinvestigationofsequencesandstructures”,J.Mol.Biol.,1995,247,536-540;Andreeva,A.等人,“SCOP2prototype:anewapproachtoproteinstructuremining”,NucleicAcidsRes.,2014,42(数据库发布),第D310-4页]汇编的常见蛋白质折叠的非限制性列表,其可以用于鉴定FSSP中的源结构。表1折叠名称(SCOP2)折叠描述DRBD型3螺旋束3个螺旋,束,上下逆时针拓扑,闭合或部分开放的,右旋扭曲GroES样含有桶,部分开放的,n*=4,S*=8珠蛋白样核心:6个螺旋,折叠叶,部分开放的超级三明治三明治,在2个片层中的18条链EF/AMT型β(6)桶桶,闭合的,n=6,S=10,希腊钥匙;反向平行片层,顺时针次序:143256规范罗斯曼折叠3层,α/β/α,6条链的平行β折叠,次序321456SD/FR插入结构域样折叠β(2)-α-β-α(n)-β-α(n)-β;反向平行β折叠,次序:21543,折叠成半桶,并在两个末端处用螺旋封端LDHC末端亚结构域样折叠α-β(3)-α,扭曲成半桶的曲折β折叠,次序123,具有在相对末端处包装的螺旋罗斯曼(2x2)oid3层:a/b/a,4条链的平行β折叠FMT/AAG型β(6)桶桶,开放的,n*=6,S*=10,希腊钥匙;反向平行β折叠,顺时针次序:125436二血红素肘两个螺旋CxxCH基序的迷你折叠,共价附着至两个血红素组的堆叠铁氧还蛋白样β-α-β(2)-α-β;2层,a/b;反向平行β折叠,次序:4132TPM/PMT结构域样3层,a/b/a;5条链的平行片层,次序51423罗斯曼(2x3)oid(黄素氧还蛋白样)3层,a/b/a,5条链的平行β折叠,次序21345叶酸结合半结构域样β(2)-α-β(3)-α;2层,α/β,反向平行β折叠,次序:12543谷氨酰tRNA-还原酶二聚化结构域6个螺旋,3螺旋结构域的同二聚体表1(续)电压门控钾通道寡聚跨膜α螺旋蛋白质GSRC末端结构域样SufE/NifU样折叠的延伸变体:β(5)-α(3),2层,a/b;反向平行β折叠,次序12543FAD/NAD(P)-结合结构域核心:3层,b/b/a,5条链的中心平行β折叠,次序32145,3条链的顶部反向平行β折叠甲酰转移酶型3层:a/b/a,7条链的混合β折叠,次序3214567,链6与其余部分反向平行硫氧还蛋白折叠核心:3层,a/b/a,4条链的混合β折叠,次序:2134,链3与其余部分反向平行罗斯曼(3x2)oid核心:3层,a/b/a,5条链的平行β折叠,次序32145黄酮还原酶插入结构域样β(4)-α-β;2层,a/b;反向平行β折叠,次序:51432ClpP型β-α超螺旋核心:(β-β-α)n右旋超螺旋的4个转角,在C末端处通过反向平行β链封端HI0933型β(6)桶桶,闭合的,n=6,S=12,希腊钥匙;顺时针次序:143256TrkAC末端结构域样β-X-β(2)-X-β-α;反向平行β折叠,次序:1423,折叠成半桶并且在一个末端端处通过螺旋封端;与HPr样折叠的拓扑相似性血影蛋白重复样3个螺旋,束,上下逆时针拓扑,闭合的,左旋扭曲TIMβ/α桶(β-α)8;平行β折叠桶,闭合的,n=8,S=8,链次序12345678(逆时针);前七个超家族具有相似的磷酸结合位点规范FwdE/GAPDH结构域样折叠α-β-α-β(3),2层,α/β三明治,混合片层,链2与链1平行,链2和3之间的psi环S13样H2TH结构域具有两个‘亲吻’循环的3-5个螺旋的阵列FAD/NAD(P)结合结构域,圆形排列的变体1核心:3层,b/b/a,5条链的中心平行β折叠,次序15234,3条链的顶部反向平行β折叠GDI类异戊二烯结合结构域样5个螺旋,阵列,两个接吻环长α发夹2个螺旋,反向平行左旋卷曲螺旋II类aaRS/BPL结构域样核心:β-X-β(2)-α-β(4);混合的,大多数反向平行β折叠,次序1237654,链1和2彼此平行含有核苷三磷酸水解酶的P环3层:a/b/a,可变大小的平行或混合β折叠PRT酶样3层,a/b/a,6条链的混合β折叠,次序321456,链3与其余部分反向平行TNF样三明治,2个片层中的10条链,胶冻卷GyrI-型α(2),β(6)桶β-α-β(2)基序的重复:反向平行β折叠形式桶(n=6,S=12),链次序132564(逆时针)FadR效应结构域样核心:6个螺旋:闭合束,希腊钥匙,内部伪二重对称,DEATH结构域样折叠的镜像拓扑DtxR二聚结构域样3个螺旋,排列成碗状结构;在单个疏水核周围形成紧密同二聚体解离酶样核心:3层:a/b/a,5条链的混合β折叠,次序21345,链5与其余部分反向平行蛋白激酶样(PK样)由夹住ATP结合位点的两个α+β亚结构域组成Profilin样核心:β(2)-α(n)-β(3);反向平行β折叠,次序:21543,2层:a/b;额外的N末端和/或C末端螺旋形成第三层表1(续)双链β螺旋螺旋的一个转角通过与短转角连接的两对反向平行链制备,具有不同体系结构和胶冻卷拓扑的三明治外观GATA锌指样含有在两对锌离子配体之间的β发夹以及在C末端处的一个或多个螺旋转角DEATH结构域样6个螺旋:闭合束,希腊钥匙,内部伪二重对称;FadR效应结构域样折叠的镜像拓扑免疫球蛋白样β三明治三明治,在2个片层中的7条链,希腊钥匙,折叠的一些成员具有另外的链分支酸裂解酶样α(2)-β(3)基序的重复,反向平行β折叠,次序123654细胞色素b5样β-α-β(2)-α(1,2)-(β)-α(2)-β,3层:a/b/a,反向平行β折叠,次序:1532(4)尿刊酸酶催化结构域样α(2)-β(3)-X-β-α(2)-β-α-β-α-β(2)-α,X是罗斯曼样结构域的插入;3层,a/b/a;混合β折叠,次序:21378645,链2、4和8与其余部分反向平行FpgN末端结构域样伪桶,在两个末端上通过α螺旋封端NDP糖基转移酶样3层:a/b/a,7条链的混合β折叠,次序3214657,次序3214657,链6与其余部分反向平行NosL/MerB样异常折叠,包含β(2)-α-β基序的两个结构重复,形成分开的β折叠,可能的重复FPGS型核糖激酶样折叠核心:3层:a/b/a,8链的混合β折叠,次序21345678,次序21345678,链7与其余部分反向平行SBP2HA样假设2型溶质结合蛋白的单结构域祖先折叠;3层,a/b/a;5条链的混合β折叠,次序21354,链5与其余部分反向平行单链右旋β螺旋超螺旋转角由平行的β链和(短)转角制成四环素阻遏物C末端结构域样多螺旋配体结合和二聚化结构域半乳糖结合结构域样三明治,2个片层中的9条链,胶冻卷核小体重塑ATP酶ISWI的HAND结构域4个螺旋,不规则阵列GHKL结构域样2层:α/β;8链混合β折叠,次序:87126345,链1和7彼此平行核糖核酸酶H样基序2层:a/b,5条链的混合β折叠,次序32145,链2与其余部分反向平行;通常随后为在β折叠的另一侧上形成第三层或在不同结构域之间交换的一个或多个螺旋DCoH样β(2)-α-β(2)-α,2层,α/β;反向平行β折叠,次序1243ATP抓握平台样β(3)-α-β(2);2层,a/b;反向平行β折叠,次序:32145Pili亚基样α-β(4);2层,a/b,螺旋沿反向平行β折叠的链包装,次序1234,曲折的Nudixβ(3)-α-β(3)-α,3层:α/β/α,混合β折叠,次序:6(2,3)154,链1和5彼此平行;含有分叉:链2和3两者均与链1H-键合;与β抓握基序的拓扑相似性核糖体蛋白质S5结构域2样核心:β(3)-α-β-α,2层:α/β,混合β折叠,次序:1243,链2与其余部分反向平行;链2和3之间的psi环;在链3和4之间的左手交叉表1(续)RIFT型β(6)桶桶,闭合的,n=6,S=10,希腊钥匙;反向平行β折叠,次序125436(逆时针)α/β水解酶核心:3层,a/b/a,8条链的混合β折叠,次序12435678,链2与其余部分反向平行限制性核酸内切酶样α-β(3)-α-β;3层,a/b/a,混合β折叠,次序:1234,链2与其余部分反向平行FPGSC末端结构域样3层:a/b/a,6条链的混合β折叠,次序126345,链1与其余部分反向平行蚯蚓血红蛋白型上下4螺旋束4个螺旋,束,上下逆时针拓扑,闭合或部分开放的,左旋扭曲II型GAT结构域样β-α-β(2)-α-β(7)-α-β(3);4层:α/β/β/α,两个反向平行β折叠中的13条链;S1次序:3241A9B;S2次序:5678CDMoeB样3层:a/b/a,8条链的混合β折叠,次序32145678,链6和8与其余部分反向平行胰蛋白酶型β(6)桶桶,闭合的,n=6,S=8,希腊钥匙;反向平行β折叠,次序:123654(顺时针)Sec63N末端结构域样6-7个螺旋,短和长螺旋的不规则阵列,一个中心螺旋α-α超螺旋多螺旋,2(弯曲)层:α/α,右手超螺旋POU结构域样核心:4个螺旋,折叠叶,闭合的甲基转移酶样核心:3层,a/b/a,7条链的混合β折叠,次序3214576,链7与其余部分反向平行伴刀豆球蛋白样三明治,在2个片层中的12-14条链,复杂拓扑SH3样桶桶,部分开放的,n*=4,S*=8,曲折最后一条链被3-10个螺旋的转角中断硫解酶样由通过伪二联体(pseudodyad)相关的两个相似的子结构域组成;5层:a/b/a/b/a,各5条链的两个相似的混合β折叠,次序:32451,链5与其余部分反向平行CBS结构域对重复:类似序列的两个β-X-β-α-β(2)-α基序的串联重复,4层:a/b/b/aSTAT型4螺旋束4个螺旋,束,左旋扭曲,右旋超螺旋MgtE膜结构域样5个跨膜螺旋,束,右旋扭曲HPr样β-α-β(2)-α-β-α,2层:a/b,反向平行片层β抓握核心:β(2)-α-β(2),2层,a/b;混合β折叠,次序:2143,次序:2143,链1和4彼此平行肌酸酶/氨基肽酶催化域样复制:由通过伪二重轴相关的两个非常相似的α(2)-β(3)单元组成;两个β折叠以“错误”的正角度异常配对以及在边缘链之间的少数H键OB折叠β(5)桶,闭合或部分开放的,n=5,S=10或S=8,希腊钥匙;混合β折叠,次序12354(逆时针),链3和5彼此平行Mor二聚结构域样单体:α(2)-β;二聚体:4螺旋束,右旋扭曲,在一个端部处通过β带封端二聚互锁连同形成4螺旋束一起互锁的α-αV形单元的二聚体PROX1亚结构域型4螺旋束4个螺旋,束:上下逆时针拓扑,右旋扭曲,菱形横截面,螺旋1和3之间的对角接触;伪二重对称圆形排列的叶酸结合半结构域样β-α-β(3)-α-β;2层,α/β,反向平行β折叠,次序:51432表1(续)四吡咯甲基化酶C末端叶状3层,a/b/a;5条链的混合片层,次序12534,链4&5与其余部分反向平行烯醇酶N末端结构域样β(3)-α(3);2层,a/b;曲折反向平行β折叠,次序:123,针对3螺旋束包装YejL样互锁6个螺旋,三螺旋单位的缠结二聚体,束FUR二聚结构域样β(2)-α-β单元的二聚体;混合β折叠,次序213,链2与其余部分反向平行;在二聚体中形成单个β折叠,具有链3和3'的反向平行H-键合红氧还蛋白样包含两个β发夹的金属离子结合折叠;每个发夹在其尖端处含有两个金属离子配位残基,通常为半胱氨酸核苷酸基转移酶样核心:α-β-转角-β-X-β-(α);混合β折叠,次序:123,链1与其余部分反向平行组蛋白折叠核心:3个螺旋;长中间螺旋在每个端部处侧面为较短的螺旋GltS中心结构域样3层,a/b/a;平行β折叠,次序15432;可能的原始形式的β/α(8)桶,起因于N末端一半的大部分的缺失SAM结构域样4-5个螺旋;两个正交包装的α发夹的束;DNA和蛋白相互作用结构域PH结构域样β(6)桶桶,部分开放的;n*=6,S*==12;曲折;通过C末端α螺旋封端Frataxin样α-β(5)-α;2层:α/β;曲折反向平行片层,次序12345EF手样对核心:4个螺旋;2个α发夹的开放阵列SMAD/FHA结构域样三明治;2个片层中的11条链;希腊钥匙E2结合域样(泛素样拓扑的圆形排列变体)β-α-β(2)-X-β(2),2层,a/b;反向平行β折叠15423;C末端链占据泛素样蛋白质的N末端链的位置,但在相反方向运行RAP型三重桶两个相关亚基的异二聚体;形成两个相似的桶,各自n=8,S=10,其融合在一起,形成第三桶,n=6,S=8RRF结构域样α-β(2)-α-β(2);2层,a/b;反向平行β折叠,次序:1243Mu转座酶型β(6)桶桶;n=6,S=8,希腊钥匙;反向平行β折叠,顺时针次序:145632CycX/PrpD结构域样6个螺旋,上下束,右旋扭曲;偶数螺旋被包装在由奇数螺旋包围的伪三重对称轴周围MurF/HprK结构域样3层,a/b/a;5条链的混合β折叠,次序15432,链1与其余部分反向平行;部分类似于回旋结构域折叠SCP样α-β(3)-X-β-α(2)-β-α;2层:a/b,交叉环X构成第三层;5条链的反向平行β折叠;次序:32145‘热狗’折叠核心:β-α-β(4);2层,a/b;反向平行β折叠,次序13452λcro蛋白样β-α(3)-β(2);2层:a/b;反向平行β折叠,次序123;三个螺旋保持与p22cro蛋白样折叠的前三个螺旋相似的排列,包括HTH基序Mad2样核心:α(2)-β(2)-α-β;混合片层,次序:213β发夹-α发夹重复β(2)-α(2)基序的多个重复表1(续)伪“翼状螺旋”β-α(2)-β(2)-α;体系结构类似于“翼状螺旋”折叠,但拓扑学不同β夹双链带在两个地方急剧弯曲;带末端形成不完整的桶;胶冻卷ZU5结构域样核心:β三明治,2个片层中的8条链;折叠的曲折IL8样β(3)-α;2层,a/b;曲折反向平行β折叠,次序:123,螺旋跨越β链包装SWIRM/ISPC型4螺旋束4个螺旋,束:上下顺时针拓扑,右旋扭曲,菱形横截面,螺旋2和4之间的对角接触;伪二重对称RpiR/Int型5螺旋阵列5个螺旋,阵列;螺旋1和5封端DRBD型3螺旋束的相同开放端部规范WHD(翼状螺旋结构域)折叠α(3)-β(2);3螺旋DRBD型束,在一个端部处用β发夹封端;可以在环中含有额外的β链,连接螺旋1和2螺旋延伸的WHD折叠α(3)-β(2)-α;3螺旋DRBD型束,在一个端部处用β-发夹和螺旋4封端片层延伸的WHD折叠SDR型延伸的罗斯曼折叠α-β(2)-α(2)-β(2);3螺旋DRBD型束,在一个端部处用4链β折叠封端OCD型延伸的罗斯曼折叠3层α/β/α,7条链的平行β折叠,次序3214567,链6和7之间的左手交叉连接CoA结合结构域型罗斯曼折叠3层α/β/α,8条链的平行β折叠,次序321458676PGDH型延伸的罗斯曼折叠3层α/β/α,7条链的混合、大多数平行的β折叠,次序3421567,链3与其余部分反向平行DAO型FAD/NAD(P)结合结构域3层α/β/α,8条链的混合β折叠,次序32145678,链7和8彼此平行并与其余部分反向平行“反转”铁氧还蛋白样折叠FAD/NAD(P)结合结构域的变体,其中α螺旋替换顶部曲折β折叠:3层,a/b/a,5条链的平行β折叠,次序32145UBA型3螺旋束β-α-β(2)-α-β;2层,a/b;反向平行β折叠,次序:1423YrdC/RibB样3个螺旋,束,上下顺时针拓扑,闭合或部分开放的,右旋扭曲HypF锌指样核心:α-β(2)-α-β-α(2)-β(2)-α-β-α-β;3层,a/b/a;7条链的混合扭曲片层,次序:7126354;链7和1彼此平行SBP2HA样圆形排列变体1具有两个CxxC基序的坐标锌离子,每个基序位于螺旋转角的N末端处Sua5结构域样3层,a/b/a;5条链的混合β折叠,次序51423,链1与其余部分反向平行MptD样3层,a/b/a;5条链的平行β折叠,次序51423规范亲环素型β(8)桶α(2)-β(4);2层:a/b;反向平行β折叠,次序:1234,曲折亲环素38型双重β桶桶,闭合的;n=8,S=10;反向平行β折叠,顺时针次序:12756438,具有彼此交叉的两个上侧连接的复杂拓扑抗密码子结合域样折叠成两个相连桶的9条链的分叉β折叠;桶1:闭合的;n=8,S=10;反向平行β折叠,逆时针次序:12659348;桶2:闭合的,n=6,S=10;混合β折叠,顺时针次序:126578,链7和8彼此平行表1(续)FAD结合/转运蛋白相关的结构域样3层:a/b/a,五条链的混合β折叠,次序21345,链4与其余部分反向平行免疫球蛋白/白蛋白结合结构域样由两个α+β亚结构域组成罗斯曼(3x4)oid3个螺旋,束,上下顺时针拓扑,闭合的,左旋扭曲;血影样折叠的镜像拓扑根据本发明的一些实施方案,上文呈现的FSSP的小子集(表1)包括但不限于免疫球蛋白、“α/β水解酶”、“β抓握”(泛素样折叠)、“希腊钥匙”、“胶冻卷”、“Keyroll”、“Plait”(铁氧还蛋白样折叠)、“罗斯曼折叠”、“Β三叶草”、“锚蛋白重复”、“犰狳重复”、“三角四肽重复”和“TIMβ/α桶”的共同折叠。根据本发明的一些实施方案,共同结构折叠是抗体的。注意到在本发明的时间,超过1900个PDB条目可用于抗体。如本文使用的,术语“抗体”包括能够结合例如巨噬细胞的完整分子及其功能片段,例如Fab、F(ab')2、scFv、scFab和Fv。这些功能性抗体片段包括但不限于:(1)Fab,含有抗体分子的单价抗原结合片段的片段,可以通过用蛋白水解酶(例如木瓜蛋白酶)消化完整抗体来产生,以获得完整的轻链和一条重链的一部分;(2)Fab',可以通过用蛋白水解酶(例如胃蛋白酶)处理完整抗体,随后还原而获得的抗体分子的片段,以得到完整的轻链和重链的一部分;每个抗体分子获得两个Fab'片段;(3)(Fab')2,可以通过用蛋白水解酶(例如胃蛋白酶)处理完整抗体不伴随后续还原而获得的抗体的片段;F(ab')2是通过两个二硫键保持在一起的两个Fab'片段的二聚体;(4)Fv,定义为含有作为两条链表达的轻链可变区和重链可变区的基因工程片段;和(5)单链抗体(“SCA”),含有通过合适的多肽接头连接的轻链可变区和重链可变区的基因工程分子,作为遗传融合的单链分子。根据本发明的一些实施方案,共同结构折叠是抗体的Fv,定义为含有作为两条链表达的轻链可变区和重链可变区的基因工程片段。关于TIMβ/α桶折叠的示例性FSSP,注意到在本发明的时候,超过2000个PDB条目可用于具有TIMβ/α桶折叠或至少含有TIMβ/α桶结构域的蛋白质。该示例性FSSP包括但不限于磷酸丙糖异构酶(TIM)、核酮糖磷酸结合桶、硫胺素磷酸合酶、吡哆醇5'-磷酸合酶、FMN连接的氧化还原酶、肌苷一磷酸脱氢酶(IMPDH)、PLP结合桶、NAD(P)连接的氧化还原酶、(反式)糖苷酶、金属依赖性水解酶、醛缩酶、烯醇酶C末端结构域样、磷酸烯醇丙酮酸/丙酮酸结构域、苹果酸合酶G、RuBisCoC末端结构域、木糖异构酶和木糖异构酶样、细菌荧光素酶和细菌荧光素酶样、烟酸/喹啉酸盐PRT酶C末端结构域样、PLC样磷酸二酯酶、钴胺素(维生素B12)依赖性酶、tRNA-鸟嘌呤糖基转移酶、二氢蝶呤合成酶和二氢蝶呤合成酶样、FAD-连接的氧化还原酶、一甲基胺甲基转移酶MtmB、同型半胱氨酸S-甲基转移酶、(2r)-磷酸-3-硫代乳酸合酶ComA、自由基SAM酶、GlpP样、CutC样、ThiG样、TM1631样和EAL结构域样蛋白。关于α/β水解酶折叠的示例性FSSP,注意到在本发明时,超过1600个PDB条目可用于具有α/β水解酶折叠或至少含有α/β水解酶结构域的蛋白质,其在下述(但不限于其)中观察到:乙酰胆碱酯酶、羧酸酯酶、分枝杆菌抗原、假定蛋白TT1662、PepX催化结构域、脯氨酰寡肽酶C末端结构域、DPP6催化结构域、丝氨酸羧肽酶、胃脂肪酶、脯氨酸亚氨基肽酶、乙酰木聚糖酯酶、卤代烷烃脱卤酶、烯醇内酯水解酶、碳-碳键水解酶、生物素生物合成蛋白质BioH、阿克拉霉素甲酯酶RdmC、羧酸酯酶/脂肪酶、环氧化物水解酶、卤素过氧化物酶、硫酯酶、羧酸酯酶/硫酯酶1、Ccg1/TafII250相互作用因子B(Cib)、细菌酯酶、真菌脂肪酶、细菌脂肪酶、胰脂肪酶的N末端结构域、羟腈裂解酶、多肽的硫酯酶结构域、聚酮化合物和脂肪酸合酶、角质酶、YdeN、推定丝氨酸水解酶Ydr428c、酰基氨基酸释放酶的C末端结构域、假定酯酶YJL068C、Atu1826、PHB解聚酶、IroE样、TTHA1544、O-乙酰基转移酶和2,6-二氢假氧基尼古丁水解酶。关于罗斯曼折叠的示例性FSSP,注意到在本发明的时候,超过1350个PDB条目可用于具有罗斯曼折叠或至少包含罗斯曼结构域的蛋白质,其在下述(但不限于其)中观察到:醇脱氢酶、酪氨酸依赖性氧化还原酶、甘油醛-3-磷酸脱氢酶、甲酸/甘油酸脱氢酶、LDHN末端结构域、6-磷酸葡糖醛酸脱氢酶、氨基酸脱氢酶、钾通道NAD-结合结构域、转录阻遏物Rex、CoA-结合结构域和鸟氨酸环化脱氨酶。关于蛋白质的三维结构中的FSSP以及保守折叠和结构域的鉴定和分类的讨论在例如Holm,L.等人[ProteinScience,1992,1,p.1691-1698;Marchler-BauerA.等人,NucleicAcidsRes.,2007,35(数据库发布),D237-40;和Marchler-BauerA.等人,NucleicAcidsRes.,2013,41(数据库发布),D348-52中提供,所述参考文献整体引入本文作为参考,如同在本文中完全阐述一样。对于FSSP的全面讨论、分类和鉴定,本领域普通技术人员可以使用例如公共可访问的服务,例如SCOP[AndreevaA.等人,NucleicAcidsRes.,2008,36(数据库发布),第D419-25页]、SCOP2[Andreeva,A.等人,“SCOP2prototype:anewapproachtoproteinstructuremining”,NucleicAcidsRes.,2014,42(数据库发布),第D310-4页]等等。生产对目标分子表面具有所需亲和力的蛋白质的氨基酸序列的方法:根据本发明的一些实施方案,本文呈现的方法的产物是一组氨基酸序列(图2中的框19),其被推荐用于使用实验性体外和/或体内程序的表达和进一步最优。因此,根据本发明的一些实施方案的另一个方面,提供了产生对目标分子表面具有所需亲和力的氨基酸序列的方法,其通过下述步骤进行:根据设计和选择对本文呈现的分子实体的目标分子表面具有所需亲和力的氨基酸序列的方法的实施方案,设计和选择对目标分子表面具有所需亲和力的氨基酸序列;和在表达系统中表达氨基酸序列,从而产生对目标分子表面具有所需亲和力的氨基酸序列。最一般地,设计的蛋白质或蛋白质片段可以被反向翻译并逆转录成编码蛋白质或片段的DNA区段,本文称为遗传模板。随后可以使用公开和商购可得的已建立的方法合成该遗传模板。允许限制性连接反应或同源重组到常用的pET或其他蛋白质表达质粒中的5'和3'片段通过标准PCR延伸加入遗传模板中。随后可以使用相容的限制性酶将遗传模板限制在表达质粒内或通过同源重组掺入表达质粒内。用相容的编码基因的质粒转化标准表达生物(细菌、酵母、噬菌体、昆虫或哺乳动物细胞),并诱导表达。考虑到蛋白质的大小和复杂性,化学合成通常不是用于表达通过本文呈现的方法中的任何一种提供的氨基酸序列的可行选项。相反,活细胞及其细胞机器可以作为生物表达系统来利用,以基于相应的遗传模板来构造和构建所设计的蛋白质。与蛋白质不同,目的蛋白质的遗传模板(DNA)相对简单地使用良好建立的重组DNA技术合成或体外构建。因此,具有或不具有附加的报道分子或亲和标签序列,通过本文呈现的方法中的任何一种提供的特定氨基酸序列的DNA模板,可以构建为用于所设计的重组蛋白质表达的模板。用于重组蛋白质表达的策略是本领域众所周知的,并且通常涉及用含有目的遗传模板的DNA载体转染细胞,并且随后培养细胞,使得它们转录和翻译所设计的蛋白。通常,随后裂解细胞以提取所表达的蛋白质用于后续纯化。原核和真核体内蛋白质表达系统两者均被广泛使用。系统的选择取决于蛋白质的类型、功能活性的要求和所需得率。细菌蛋白表达系统是最广泛使用的,因为细菌容易培养、快速生长并产生高得率的所设计的重组蛋白质。然而,在细菌中表达的多结构域真核蛋白质通常是无功能的,因为细胞未装备来完成所需翻译后修饰或分子折叠。另外,许多蛋白质变得不可溶如包涵体,所述包涵体非常难以在不含苛刻变性剂以及后续繁琐的蛋白质重折叠程序的情况下回收。哺乳动物体内表达系统通常产生具有一些显著限制的功能蛋白质。无细胞蛋白质表达是使用全细胞的翻译相容性提取物的蛋白质的体外合成。原则上,全细胞提取物含有转录、翻译和甚至翻译后修饰所需的所有大分子组分。这些组分包括RNA聚合酶、调节蛋白质因子、转录因子、核糖体和tRNA。当补充有辅因子、核苷酸和特定基因模板时,这些提取物可以相对容易地合成目的蛋白质。虽然对于大规模生产通常不可持续,但无细胞蛋白质表达系统具有超过传统体内系统的几个优点。无细胞系统允许用经修饰的氨基酸标记蛋白质,以及经历通过细胞内蛋白酶的快速蛋白水解降解的设计蛋白质的表达。另外,使用无细胞方法,更容易同时表达许多不同的蛋白质(例如,通过在小规模上从许多不同的重组DNA模板表达来测试设计蛋白质)。在本发明的一些实施方案中,共同结构折叠是抗体的折叠。产生多克隆和单克隆抗体及其片段的方法是本领域众所周知的(参见例如引入本文作为参考的Harlow和Lane,Antibodies:ALaboratoryManual,ColdSpringHarborLaboratory,NewYork,1988)。在本发明的一些实施方案中,共同结构折叠是抗体片段的折叠。根据本发明的一些实施方案的抗体片段可以通过抗体的蛋白酶解水解或者通过在大肠杆菌(E.coli)或哺乳动物细胞(例如,中国仓鼠卵巢细胞培养物或其他蛋白质表达系统)中表达编码该片段的DNA来制备。可以使用蛋白水解酶(例如胃蛋白酶或木瓜蛋白酶)通过常规方法消化完整抗体来获得抗体片段。例如,可以通过用胃蛋白酶酶促切割抗体以提供指示为F(ab')2的5S片段来产生抗体片段。该片段可以使用硫醇还原剂和任选的用于巯基(其起因于二硫键的切割)的阻断基进一步切割,以产生3.5SFab'单价片段。可替代地,使用胃蛋白酶的酶切割直接产生两个单价Fab'片段和Fc片段。这些方法在例如Goldenberg,美国专利号4,036,945和4,331,647及其中包含的参考文献中描述,所述专利在此整体引入作为参考。还参见Porter,R.R.[Biochem.J.73:119-126(1959)]。也可以使用切割抗体的其他方法,例如分离重链以形成单价轻-重链片段,进一步切割片段或其他酶促、化学或遗传技术,只要片段结合被完整抗体识别的抗原。Fv片段包含VH和VL链的结合。这种结合可以是非共价的,如Inbar等人[Proc.Nat'lAcad.Sci.USA69:2659-62]中描述的。可替代地,可变链可以通过分子间二硫键连接或通过化学品如戊二醛交联。优选地,Fv片段包含通过肽接头连接的VH和VL链。这些单链抗原结合蛋白(scFv)通过构建结构基因来制备,所述结构基因包含通过寡核苷酸连接的编码VH和VL结构域的DNA序列。将结构基因插入表达载体内,随后将所述表达载体引入宿主细胞如大肠杆菌内。重组宿主细胞合成具有桥接两个V结构域的接头肽的单个多肽链。用于产生scFv的方法例如由[Whitlow和Filpula,Methods2:97-105(1991);Bird等人,Science242:423-426(1988);Pack等人,Bio/Technology11:1271-77(1993);和美国专利号4,946,778描述,所述参考文献在此整体引入作为参考。根据本发明的一些实施方案,本文呈现的方法,包括其实施方案中的任何一个,及其任何组合,可以用于人源化蛋白质例如抗体的结构。在本发明的这些实施方案的上下文中,根据本发明的实施方案设计蛋白质的结合结构域,例如抗体的Fv片段的氨基酸序列,并且随后通过找到与其最相容的人构架将整个蛋白质人源化。此类方法遵循如本文所述的用于蛋白质人源化的一些方法的基本原理。用于人源化非人抗体的方法是本领域已知的。一般地,人源化抗体具有从非人来源引入其内的一个或多个氨基酸残基。这些非人氨基酸残基通常被称为输入残基,其在本发明的一些实施方案的上下文中是通过本文呈现的方法设计的产物。人源化可以基本上遵循Winter及同事的方法[Jones等人,Nature,321:522-525(1986);Riechmann等人,Nature332:323-327(1988);Verhoeyen等人,Science,239:1534-1536(1988)],通过用非人序列取代相应的设计氨基酸序列来执行。非人(例如鼠)抗体的人源化形式是免疫球蛋白、免疫球蛋白链或其片段(例如Fv、Fab、Fab'、F(ab')2或抗体的其他抗原结合子序列)的嵌合分子,其含有衍生自非人免疫球蛋白的最小序列。人源化抗体包括其中氨基酸序列形成互补决定区(CDR)的人免疫球蛋白(受体抗体),或者也包括受体的构架部分的较大结构域(例如,Fv结构域)替换为根据本文呈现的方法设计的氨基酸序列,其具有所需特异性、亲和力、稳定性和能力。在一些情况下,人免疫球蛋白的Fv构架氨基酸序列替换为通过本文呈现的方法提供的相应非人氨基酸序列。人源化抗体还可以包含既未在受体抗体中也未在输入CDR或构架序列中发现的残基。一般而言,人源化抗体将包含至少一个、且通常为两个可变结构域的基本上全部,其中CDR区的全部或基本上全部对应于通过本文呈现的方法提供的那些,并且FR区的大部分或基本上全部是人免疫球蛋白共有序列的那些。人源化抗体最佳地还包含免疫球蛋白恒定区(Fc)的至少一部分,通常为人免疫球蛋白的那种[Jones等人,Nature,321:522-525(1986);Riechmann等人,Nature,332:323-329(1988);和Presta,Curr.Op.Struct.Biol.,2:593-596(1992)]。完全或部分设计的抗体也可以使用本领域已知的不同技术产生,包括噬菌体展示文库[Hoogenboom和Winter,J.Mol.Biol.,227:381(1991);Marks等人,J.Mol.Biol.,222:581(1991)]。Cole等人和Boerner等人的技术也可用于制备人单克隆抗体(Cole等人,MonoclonalAntibodiesandCancerTherapy,AlanR.Liss,第77页(1985)和Boerner等人,J.Immunol.,147(1):86-95(1991)]类似地,完全或部分设计的抗体可以通过将人免疫球蛋白基因座引入转基因动物(例如其中内源免疫球蛋白基因已部分或完全失活的小鼠)内来制备。在攻击后,观察到完全或部分设计的抗体产生,其在所有方面非常类似于在人中可见的,包括基因重排、装配和抗体所有组成成分。这种方法例如在美国专利号5,545,807;5,545,806;5,569,825;5,625,126;5,633,425;5,661,016以及下述科学出版物中描述:Marks等人,Bio/Technology10,:779-783(1992);Lonberg等人,Nature368:856-859(1994);Morrison,Nature368812-13(1994);Fishwild等人,NatureBiotechnology14,845-51(1996);Neuberger,NatureBiotechnology14:826(1996);以及Lonberg和Huszar,Intern.Rev.Immunol.13,65-93(1995)。分子实体的目标分子表面:根据本发明的一些实施方案,通过提供构成分子实体的部分的目的可限定分子表面的原子的原子坐标,可以进行设计和选择对分子实体(本文称为“设计蛋白质”)的目标分子表面具有所需亲和力的氨基酸序列的方法。具有可限定分子表面意指设计蛋白质意欲与其相互作用的分子实体或至少目标分子表面可以通过三维的原子坐标来描述。如本文使用的,短语“分子实体”描述可由三维空间中的一组原子坐标表示的分子、化合物、复合物、加合物和/或复合物。根据本发明的一些实施方案,原子坐标表示靶的所有原子或至少非氢原子的相对位置。根据本发明的一些实施方案,足以提供构成分子实体的部分的目标分子表面的原子坐标,基本上是因为它是当两个实体形成复合物(彼此结合)时与设计蛋白质相互作用的分子表面。换言之,根据本发明的一些实施方案,分子实体可以通过原子坐标来限定,所述原子坐标限定至少设计蛋白质设计为与之相互作用并结合的分子表面。根据本发明的一些实施方案,目标分子表面被这样限定,以便包含比其估计与所设计的蛋白质结合更广的区域,以便允许该方法探索且鉴定大于预期的识别和结合区域。至少分子实体的目标分子表面的原子坐标的原点可以通过实验程序来获得,所述实验程序例如通过计算程序或其组合获得,对有形的天然存在的或合成物质的样品进行的X射线衍射或NMR分析。目标分子表面可以包括与其相关的原子,并且可以在主要部分的框架中被指定原子坐标,但不一定共价结合。例如,根据本发明的一些实施方案,目标分子表面由属于分子实体的原子的原子坐标以及溶剂(例如水)分子的原子坐标表示,所述溶剂分子通过氢键与分子实体结合。同样地,与分子实体相关的离子的原子坐标也可以构成目标分子表面的结构表示的部分。根据本发明的一些实施方案,目标分子表面的原子坐标可以表示在给定条件下的热力学稳定,或可替代地,代表不稳定构象的三维结构,例如在靶分子的过渡态的情况下,所述过渡态处于相同分子实体的两个更稳定构象之间。目标分子表面也可以表示单个分子实体的几种构象异构体之一,如在具有超过一种状态,因此具有超过一种可识别分子表面的细胞受体蛋白质的情况下;对于其中的每一种,使用本文公开的方法可以设计所设计的蛋白质。通常,但非排他地,分子实体是可以发挥一种或多种生物和/或药物活性的分子实体。根据本发明的一些实施方案,分子实体可以与术语“生物活性剂”、“药物活性剂”、“药物活性材料”、“治疗活性剂”、“生物学活性剂”、“治疗剂”、“药物”和其他相关术语包括例如遗传治疗剂、非遗传治疗剂、小分子和细胞中任一互换使用。可以对于其设计所设计的蛋白质用于使用本文所述方法的分子实体的代表性实例包括但不限于基于氨基酸和肽和蛋白质的物质,例如细胞因子、趋化因子、化学引诱剂、化学驱避剂、激动剂、拮抗剂、抗体、抗原、酶、辅因子、生长因子、半抗原、激素和毒素;基于核苷酸的物质,例如DNA、RNA、寡核苷酸、标记的寡核苷酸、核酸构建体和反义物;糖、多糖、磷脂、糖脂、病毒和细胞、以及亲水性或两亲性放射性同位素、放射性药物、受体、类固醇、维生素、血管生成促进剂、药物、抗组胺剂、抗生素、抗抑郁药、抗高血压药、消炎药、抗氧化剂、抗增殖剂、抗病毒剂、化学治疗剂、辅因子、胆固醇、脂肪酸、胆汁酸、皂苷、激素、金属离子、合成或天然表面、抑制剂和配体及其任何组合。本文所述的分子实体各自可以是大分子生物分子或小的有机分子。如本文使用的,术语“生物大分子”指在活生物中天然存在的聚合生物化学物质或生物聚合物。聚合生物大分子主要是有机化合物,即它们主要由碳和氢组成,任选且通常连同氮、氧、磷和/或硫一起,而其他元素可以引入其中,但通常为较低的发生率。氨基酸和核苷酸是聚合生物大分子的最重要构件块中的一些,因此生物大分子通常由聚合的氨基酸(例如肽和蛋白质)的一条或多条链、聚合的核苷酸(例如核酸)、聚合的糖、聚合的脂质及其组合组成。大分子可以包含可以彼此共价或非共价附着的几个大分子亚基的复合物。核糖体、细胞器和完整病毒在本文中也包括在术语“生物大分子”下。如本文使用的,生物大分子具有高于1000道尔顿(Da)的分子量,并且可以高于3000Da、高于5000Da、高于10kDa、且甚至高于50KDa。可以对于其设计抗体用于使用本文所述方法的生物大分子的代表性实例包括但不限于肽、多肽、蛋白质、酶、抗体、寡核苷酸和标记的寡核苷酸、核酸构建体、DNA、RNA、反义物、多糖、受体、病毒及其任何组合、以及细胞,包括完整细胞或其他亚细胞组分和细胞碎片。如本文使用的,短语“小有机分子”或“小有机化合物”指主要由碳和氢连同氮、氧、磷和硫以及以较低发生率的其他元素组成的小化合物。有机分子构成整个生命世界和所有合成制备的有机化合物,因此它们包括所有天然代谢产物和人造药物。在本发明的上下文中,关于化合物、试剂或分子的术语“小”指低于约1000克/摩尔的分子量。因此,小的有机分子具有低于1000Da、低于500Da、低于300Da或低于100Da的分子量。可以对于其设计抗体用于使用本文所述方法的的小有机分子的代表性实例包括但不限于血管生成促进剂、细胞因子、趋化因子、化学引诱剂、化学驱避剂、药物、过渡态类似物、激动剂、氨基酸、拮抗剂、抗组胺剂、抗生素、抗原、抗抑郁药、抗高血压药、消炎药、抗氧化剂、抗增殖剂、抗病毒剂、化学治疗剂、辅因子、脂肪酸、生长因子、半抗原、激素、抑制剂、配体、糖、放射性同位素、放射性药物、类固醇、毒素、维生素及其任何组合。从头设计的蛋白质:如上文呈现的,本发明的实施方案提供了用于设计氨基酸序列的方法,所述氨基酸序列能够折叠成对目标分子表面显示出所需亲和力的稳定3D结构。本文公开的方法可以应用于设计结合蛋白,其可以以高亲和力和选择性结合具有可限定分子表面的任何分子实体,同时维持可行和稳定的总体结构。“可限定的”意指分子表面可以由其每个原子或其至少一些原子的一组原子坐标表示。根据本发明一些实施方案的另一个方面,提供了对目标分子表面具有所需亲和力的氨基酸序列。该氨基酸序列可以用于产生用于在体外表达系统中使用的相应遗传模板,如下文例示的。在本发明的一些实施方案中,对目标分子表面具有所需亲和力的氨基酸序列是抗体片段,例如作为单链构建体(scFv)的抗体可变结构域(Fv)或融合至天然存在的恒定结构域作为抗体片段(Fab)、二硫键连接的或融合成单链(scFab)、或作为抗体的全长IgG。存在表达抗体和就结合测试抗体的许多可替代途径。这些包括噬菌体,核糖体和酵母展示,细菌表达和从包涵体再折叠,由细菌、酵母、哺乳动物或昆虫细胞分泌抗体。作为这些途径各自的第一步,将所设计的目的抗体克隆到表达质粒内,对于可变轻和可变重结构域分开克隆或在scFv构建体的情况下融合为一个基因区段。简言之,每种设计的抗体由DNA寡核苷酸合成或从定制DNA合成服务订购,并克隆到pCTCON2质粒内用于酵母细胞表面展示。酵母转化、表达和结合测定是本领域已知的,并且标准实验室流式细胞仪或荧光激活流式细胞仪(FACS)用于使用荧光标记的抗-cmyc抗体和结合来监测设计抗体的表达水平。设计蛋白质的示例性用途:本文呈现的方法可以用于下述但不限于下述:基于现有天然结合蛋白设计改善和精制的结合蛋白,其特征在于与天然蛋白相比较具有更高的稳定性、亲和力或特异性;通过找到与其最相容的人构架来人源化已知的非人结合蛋白,例如抗体;设计用于任何分子实体并用于任何用途的从头结合蛋白,所述用途包括药物、分析或诊断用途,作为用于合成和纯化任务的工具等等;和基于其已知配体的结构或仅基于分离蛋白质的序列,预测对于其无法获得实验性3D结构的结合蛋白的结构。因为目标分子表面可以通过原子坐标来限定,即使该表面是构象动态的,即在给定条件下改变构象的分子实体的表面,所以本文呈现的方法可以设计蛋白质,例如抗体,其可以结合分子实体的分子表面,即使该表面表现为过渡态或中间体。本文呈现的方法可以通过使用过渡态的分子表面用于设计酶和其他催化蛋白,例如催化抗体。例如,可以将蛋白质设计为在对应于底物和产物之间的过渡态的构象(confirmation)下结合分子的分子表面。蛋白质例如结合分子同时对分子赋予或促进过渡态构象,从而催化从底物到产物的过渡(反应)。本文呈现的方法还可以通过使用受体构象状态中的任何一种或其他任何分子开关作为分子表面用于设计信号传导蛋白。例如,可以将蛋白质设计为在受体构象时结合受体的分子表面,所述受体构象对应于“开”、“关”、“开放”、“闭合”或具有某些生物表达的任何其他状态。蛋白质例如结合生物系统中的相应受体,同时赋予或促进受体的“开”、“关”、“开放”、“闭合”或任何其他构象,从而充当信号传导蛋白。新型酶促功能的设计:根据本发明的一些实施方案,本文呈现的方法可以用于提供在任何给定的结合/活性位点中显示出对任何配体或目的底物结构的亲和力的结构。根据一些实施方案,该方法可以通过将来自现有蛋白质结构的活性/结合位点的催化残基的官能团与所结合的目的配体/底物或其类似物叠加到设计结构上的相应基团上来实施。如果现有的结合结构不可用,则可以计算生成模型,其中官能团围绕过渡态几何取向,使得其预测的自由能降低,提供用于催化的基础。随后可以选择关于其他支架位置的最佳残基鉴定,以相对与配体/底物形状互补的活性/结合位点,同时还使催化残基稳定在其预定构象中。还可以引入主链构象取样以最优催化残基的形状互补性和位置。基于氨基酸序列的蛋白质结构预测:结构设计方法的延伸是具有已知序列的蛋白质和已知结构的数据库的结构预测方法,所述已知结构的数据库可以被分段成如在已知序列的区段中具有精确数目的氨基酸的区段。该方法在本文中使用抗体进行例示。结构预测方法基于在蛋白质数据库(PDB)中观察到的V1/Vh刚体取向的取样,并且通过预计算每个抗体PDB条目中的轻链和重链就彼此而言的刚体取向,并将该信息存储在数据库中开始。尽管序列设计方法仅对主链和侧链构象取样,但允许氨基酸序列中的改变包括氨基酸的插入和缺失,所述结构预测方法包括从预计算数据库中取样刚体位移,同时保持靶蛋白的氨基酸固定。该方法通过组合生成具有与包含靶抗体的区段相同长度的主链区段的多个组合而继续。取样包括五类移动:建模片段(VL、VH、L3和H3)之一由来自预计算的主链文库的具有相同长度的随机构象的替换,以及刚体取向由选自预计算的刚体文库的随机选择的取向的替换。根据一些实施方案,刚体取向也被组合取样。在每次此类移动之后,使用全原子Rosetta能量函数来包装和最小化围绕建模区段的6Å壳内的氨基酸侧链构象,并且将所得到的能量与先前接受的能量比较;根据Metropolis标准接受新近生成的结构,并且使用模拟退火方案重复该过程的几个步骤(100-500),在温度0时结束。与用于抗体结构预测的其他方法形成对比,该方法依赖于用于建模所有自由度(区段和轻链/重链刚体取向)的实验结构。根据本发明的一些实施方案,发现本文提供的结构预测方法在环构象的准确性和立体化学质量两方面均优于基准中的最佳方法(数据未显示)。不允许针对易错力场的无限构象结构最优,而是更多地依赖于实验结构,预期本文提供的结构预测方法可以更准确地捕获靶结构的构象能量景观。预期在从本申请成熟的专利的寿命期间,将开发基于在天然存在的抗体中发现的序列和构象信息用于设计从头抗体结构的许多相关方法,并且术语基于在天然存在的抗体中发现的序列和构象信息用于设计从头抗体的方法的范围预期先验包括所有此类新技术。如本文使用的,术语“约”指±10%。术语“包含(comprises)”、“包含(comprising)”、“包括(includes)”、“包括(including)”、“具有”及其缀合物意指“包括但不限于”。术语“由……组成”意指“包括且不限于”。术语“基本上由……组成”意指组合物、方法或结构可以包括另外的成分、步骤和/或部分,但仅当另外的成分、步骤和/或部分实质上不改变请求保护的组合物、方法或结构的基本和新颖特征时。如本文使用的,单数形式“一个”、“一种”和“该/所述”包括复数指示物,除非上下文另有明确说明。例如,术语“支架”或“至少一个支架”可以包括多个支架,包括其混合物。在本申请自始至终,本发明的不同实施方案可以以范围形式呈现。应当理解以范围形式的描述仅为了方便和简洁起见,并且不应被解释为对本发明的范围的硬性限制。相应地,范围的描述应当被视为已具体公开了所有可能的子范围以及该范围内的各个数值。例如,例如1至6的范围的描述应当被视为已具体公开了子范围,例如1至3、1至4、1至5、2至4、2至6、3至6等,以及该范围内的各个数目,例如1、2、3、4、5和6。不管范围的宽度如何,这都适用。每当本文指示数目范围时,它意欲包括在所示范围内的任何引用数字(分数或整数)。短语“范围为第一指示数目至第二指示数目/第一指示数目和第二指示数目之间的范围”和“范围为第一指示数目到第二指示数目/第一指示数目至第二指示数目之间的范围”在本文中可互换使用,并且意欲包括第一和第二指示数目以及它们之间的所有分数和整数。如本文使用的,术语“方法”指用于完成给定任务的方式、手段、技术和程序,包括但不限于已知的方式、手段、技术和程序,或通过化学、药理学、生物学、生物化学和医学领域的从业者由已知的方式、手段、技术和过程容易开发的那些方式、手段、技术和程序。如本文使用的,术语“治疗”包括消除、基本上抑制、减慢或逆转状况的进展,基本上改善状况的临床或美学症状,或基本上预防状况的临床或美学症状的出现。当提及特定的序列表时,此类提及应理解为还包括基本上对应于其互补序列的序列,如包括起因于例如测序错误,克隆错误或者导致碱基取代、碱基缺失或碱基添加的其他改变的微小序列变异,条件是此类变异的频率小于50个核苷酸中的1个,可替代地,小于100个核苷酸中的1个,可替代地,小于200个核苷酸中的1个,可替代地,小于500个核苷酸中的1个,可替代地,小于1000个核苷酸中的1个,可替代地,小于5000个核苷酸中的1个,可替代地,小于10,000个核苷酸中的1个。应了解为了清楚起见,在分开实施方案的上下文中描述的本发明的某些特点也可以以单个实施方案的组合提供。相反,为了简洁起见,在单个实施方案的上下文中描述的本发明的不同特点也可以分开地或以任何合适的子组合或者如本发明的任何其他描述的实施方案中适当地提供。在不同实施方案的上下文中描述的某些特点不被视为那些实施方案的必要特点,除非在不含那些元件的情况下实施方案无效。如上文所述和如下文权利要求部分中请求保护的本发明的不同实施方案和方面在下述实施例中找到实验支持。实施例现在参考下述实施例,其连同上文说明书一起以非限制性方式举例说明本发明的一些实施方案。方法源代码可用性:该方法的一些部分在Rosetta大分子建模软件套件[Das&Baker,AnnuRevBiochem2008,77:363–382]内实施,并且可通过RosettaCommons协议获得。方法中的一些已通过RosettaScripts[Fleishman等人,PLoSOne2011,6:e20161]实施。该方法的一些部分使用Monte-Carlo方法[Hazewinkel,Michiel,编辑(2001),"Monte-Carlomethod",EncyclopediaofMathematics,Springer,ISBN978-1-55608-010-4]实施。结合模式标准:决定哪些设计概括了天然结合模式基于CAPRI挑战标准[Méndez等人,Proteins2003,52:51–67]。具体地,在两个抗体结构比对后,测量设计和天然结构之间的靶界面残基(具有在抗体10Å半径内的原子的所有残基)的RMSD的I_RMS。天然抗体和设计抗体之间的界面RMSD截断设定为4Å。下述脚本以Python编写,并使用Pymol[ThePyMOLMolecularGraphicsSystem,Version1.6.1Schrödinger,LLC.]实施。为了运行此脚本,将设计的复合物数据库连同天然抗体-抗原复合物一起置于文件夹中。创建文本文件。已经创建了命名为:“pdb_file_list”的该文本文件,其含有所有设计PDB文件和作为第一条目的天然抗体-抗原复合物的列表。执行命令是Bash终端中的“pymol–clig_rms.py”。该命令从设计PDB存储于其中的相同文件夹执行。来自此脚本的输出是两个文件夹,一个文件夹含有具有大于4Å的I_RMS值的所有设计的拷贝(“more_than_4”),并且第二个文件夹含有具有小于4Å的I_RMS值的所有设计的拷贝(“less_than_4”)。CDR定义:本工作中使用的CDR定义一般与本领域已知的先前定义一致。为了清楚起见,本文呈现的CDR定义遵循Chothia位置编号方案。在下述实施例中,使用两种不同的CDR定义。第一,紧密匹配的V(D)J基因区段,将CDR1和2作为一个单元处理。该定义在PSSM的构建期间和在主链取样期间使用,如下文所述。第二个定义类似于常规CDR定义,并将每个CDR(CDR1、CDR2和CDR3)作为分开的单元处理,以测定序列约束阈值的水平,如下文所述。上表2提供了上述CDR定义之间的比较。形状互补性:形状互补性使用Singer等人[J.Immunol.,1993,150,p.2844-2857]中所述的算法进行计算,并且并在Rosetta软件套件中实施。下述脚本“sc.xml”用于执行形状互补性最优。这个脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:抗体支架与靶表位的对接:如下所述构建的4,500个抗体支架各自最初与复合结构中的天然抗体构架比对。随后将靶坐标加入抗体支架结构中。随后使用简化表示对接(质心模式),使用下述dockLowRes.xmlscript,用RosettaDock[Gray等人,JMolBiol2003,331:281–299]扰乱结合模式。这个脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:结合能计算:结合能定义为结合态和非结合态的总系统能量之间的差异。在每个状态下,允许界面残基重新包装。对于数目稳定性,结合能计算重复三次并取平均值。下述ddg.xml脚本用于执行此计算。这个脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:抗体稳定性计算:稳定性能量定义为系统的抗体单体的自由能。为了评估抗体的稳定性能量,去除靶并计算抗体总能量得分(得分12)。下述AB_stability.xml脚本用于执行此计算。这个脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:包装质量评价:使用下述Packstat.xml脚本,使用在Rosetta软件套件中实施的“RosettaHoles”(Packstat)[Sheffler&Baker,ProteinSci2009,18:229–239]计算在抗体核心和抗体-靶界面处的蛋白质包装质量。这个脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:界面侧链的玻尔兹曼构象概率:玻尔兹曼构象概率如由Pantazes和Maranas[Protein.Eng.Des.Sel.,2010,23,849–858]描述的进行计算。对于每个复合物,该方法首先分离配偶体,并且对于对结合作出可观贡献的每个残基(当突变为丙氨酸时结合能增加超过1R.e.u.),它重复如在主链依赖性旋转异构体Dunbrack文库中定义的所有其旋转异构体状态,排除预测与蛋白质主链或Cβ原子形成空间碰撞的旋转异构体。对于每个旋转异构体放置,6Å壳内的所有残基被重新包装并最小化。随后使用Rosetta全原子能量函数(得分12)[Kortemme&Baker,PNAS2002,99:14116–14121]估计每个此类状态的能量E。随后假设玻尔兹曼分布来计算残基i的构象的概率Pi:(方程2)其中s是旋转异构体状态,kB是玻尔兹曼常数,并且T是绝对温度。kBT在所有模拟中设定为0.8R.e.u.。Ei是未结合状态的能量。下述脚本以“RosettaScripts”编写,并使用Rosetta建模套件实施。执行实例:主链区段聚簇:将下文描述的数据库中的抗体结构分别与抗体4m5.3,(PDBID1X9Q)[Lippow等人,NatBiotechnol2007,25:1171–1176]的可变重链和可变轻链结构域比对。随后提取根据VL、L3、VH和H3定义的CDR的坐标,并根据长度聚簇。对于L3和H3,使用Rosetta聚簇应用执行另外的构象聚簇。将主链构象聚簇成如在Cα原子之间测量的2.0ÅRMSD的bin内。就共同序列基序手动检查所得到的簇。含有具有相应主链构象差异的多重序列基序的簇通过减少聚簇bin大小而分开。同样地,在其中多个簇含有相同序列基序的情况下,增加聚簇bin大小以合并簇。实例命令行:。生成序列概况:对于每个主链构象簇,生成位置特异性评分矩阵(PSSM)。从每个结构中提取氨基酸序列,以首先生成多重序列比对,从其中去除了100%的序列冗余(比对中的每一个序列与比对中的所有其他序列具有至少单个氨基酸差异)。使用具有缺省参数和多重序列比对作为输入的PSI-BLAST套件[Biegert&Söding,PNAS2009,106:3770–3775]生成PSSM。下述脚本是以bash编写,并且依赖于两个程序,“muscle”[EdgarRC.doi:10.1093/nar/gkh340.PubMedPMID:15034147.]和“psiblast”。这个脚本在与聚簇主链区段相同的文件夹中执行,以产生单个PSSM。算法性能:典型的轨迹从在标准单CPU上提交到成功完成花费约7小时。协议分为两部分。首先,使设计的抗体支架(算法,部分d)和靶分子之间形成的复合物经受对接、设计和最优/最小化(算法,e部分)。绝大多数时间用于下游精制步骤(算法,f部分)。为了有效利用计算资源,在进行到精制之前应用能量和结构过滤;平均起来,所有轨迹中仅4%通过了这个过滤。取决于计算资源的可用性和设计问题的量级,可以调整在该步骤处的过滤器。检查点:实施检查点策略,其确保如果设计轨迹提前终止,则它可以从最后一个备份点恢复。检查点策略从主链最优程序的开始实施。每当取样主链改善目标函数(算法,部分g)时,含有复合物的坐标信息的PDB格式化文件连同精确的设计阶段、复合物稳定性和结合能一起被保存到磁盘上。当程序启动时,它自动检查检查点文件的存在。如果找到检查点文件,则计算将从上次停止的相同点继续。代码流和模块化:设计协议使用RosettaScripts[Fleishman等人,PLoSOne2011,6:e20161]实施,其为所有主要的Rosetta功能性提供方便的用户界面。这种实施形式允许没有先前编码知识的非专家用户完全控制设计协议的所有方面。该协议是有意模块化的,因此当潜在用户认为合适时,他们可以添加、更改或删除协议中的不同要素。实施例1设计的抗体-示例性程序模板抗体:如上文所述,根据本发明的一些实施方案,该方法中的两个预计算步骤涉及创建构象数据库。数据库只能与它对于其创建的任意模板抗体一起使用。对于本文呈现的结果,使用抗荧光素抗体4m5.3(PDBID1X9Q);然而,应注意可以使用任何任意抗体模板。生成PSSM和主链构象数据库:根据本发明的一些实施方案,该程序的第一步是将源抗体解析成区段并根据长度将区段分组。788条可变轻κ链和785个可变重链源抗体结构构成了在下文呈现的实施例中使用的所有源抗体的集合。如上文讨论的,源抗体的Fv片段的分段遵循分成四个区段:L1-L2(称为“VL”)和H1-H2(称为“VH”),各自侧面为轻和重可变结构域L3和H3的两个结构上保守的半胱氨酸,各自在第二个半胱氨酸后的第一个氨基酸处起始,并且在可变轻κ结构域的位置100和可变重结构域的位置103处结束(参见下表2)。下表2呈现了在基于位置的评分步骤中使用的结构同源区段型定义,以及一些广泛接受的位置编号方案之间的比较。在每个区段簇中存在不同长度的序列,因此每个簇进一步被分成长度组。在每个长度组内,进行了进一步构象聚类以根据它们编码的3D构象区分序列。对于每个此类簇,进行序列比对过程,并且使用例如PSI-BLAST软件包,对每个结构同源区段指定位置特异性评分矩阵(PSSM)。下表3列出了它们的长度分组方案中的区段,并且指示相应源抗体各自的PDB条目。这些区段各自被指定如上所述的PSSM。为了生成主链构象数据库,将所有源抗体结构叠加到模板抗体上,同时使用结构上显著的干位置(最高结构保守性位置)用于在所有抗体中在结构上非常良好地比对的构象区段,以确保插入片段可以作为可以在构建人工抗体中任意重组的模块片进行处理。对于每个结构同源区段簇(VL、L3、VH和H3)的每个长度组,从每个源抗体提取主链二面角(Φ、Ψ和Ω),并且用源的二面角替换模板抗体中相应区段中的那些,在建模区段中随机选择的位置引入主链切割位点。换言之,通过叠加具有最高结构保守性的相应位置,将来自源结构的结构同源区段在位于模板结构上的任意位置,通常是远离区段末端的几个位置处切割,并且随后基于结构约束,使区段的两个一半的二面角经受权重拟合,同时根据PSSM值改变区段的氨基酸序列,如上文所述。下述示例性splice_out.xmlRosettaScriptsxml协议用于从每个源抗体中提取每个主链区段即L1-L2(VL)、L3、H1-H2(VH)和H3的二面角,并且将其强加在模板抗体上。命令行选项(可以包装在“flag”文件中):执行实例:注意,最后三个标志被移出标志文件到命令行。另外,对于协议的这部分,输入PDB文件(在命令行中由–sflag指示)应当与模板pdb相同。下述参数可以对于不同区段进行更改:在执行“splice_out.xml”之前,当提取H1_H2、H3区段时,源抗体PDB可以与可变重结构域进行比对,或当提取模板抗体的L1_L2、L3区段时,与可变轻结构域进行比对。loop_dbase_file_name–应当给予有意义的名称,使数据库文件与正确的区段关联。From_res/to_res–应为模板抗体结构的起始和终止残基数目。Segment–将其更改为四个可用抗体区段(L1_L2,L3,H1_H2和H3)之一。生成抗体构象代表:如上文讨论的,Fv氨基酸序列文库起因于衍生自抗体的Fv源结构的四个权重拟合和重新闭合区段(将VL、L3、VH和H3指示为区段组)的组合性组合。在本实施例中,源结构的数目为约700,并且对于每个考虑四个区段,重建结构的数目将是700^4,或约10^11。如果不进行简化,则该方法将导致抗体的重建结构的过大的文库。然而,在先前研究中进行的观察强调,除H3外的每个抗体主链区段落入少数规范构象内,因此,本文呈现的方法的结合蛋白设计程序通过生成抗体主链或支架的简化文库开始,所述简化文库取样这些规范构象的空间加上一组H3主链构象。这些观察导致本发明人考虑通过下述减少重组区段的数目:通过长度分选区段,并根据RMSD对其构象取样,从而允许选择减少数目的代表性区段,其充分代表构象空间。实际上,取样程序将区段的数目减少为可管理的数目,所有的排列对于其组合使用以形成重建结构,其构成具有共同结构折叠的氨基酸序列的简化和代表性文库。一旦生成构象数据库,下一步就是创建构象代表;这些是跨越抗体构象空间的抗体结构。在预计算步骤中,使用splice_out.xml协议成功插入模板抗体中的主链区段通过构象进行聚簇。生成抗体构象代表:从每个簇中选择主链代表,其随后与其他主链簇代表性区段组合以产生构象代表。下述“splice_in.xml”脚本用于从指定抗体的数据库文件中提取二面角,并将其插入支架抗体中。随后将输出抗体用作下一个主链区段插入的输入,以此类推(例如,首先插入H3主链区段,并且随后输出结构用于插入L3主链构象等等)。该程序可以重复,直到生成所有主链构象变体。下述脚本用于将L1_L2主链构象簇代表插入模板抗体中,对于插入其他区段的必要修饰在下文说明。命令行选项(可以包装在“flag”文件中):下述参数可以对于不同区段进行更改:database_pdb_entry–插入构象表示的名称,因为它出现在应该插入模板抗体的扭曲数据库中(名称是扭曲数据库文件中的每个条目的最后一个值)。torsion_database-根据待取样的主链构象更改(例如,如果取样L1_L2主链区段,则扭曲数据库应为“L1_L2.db”)。执行实例:应注意,来自该示例性执行的输出PDB文件将是具有来自1AHW的L1_L2的主链构象插入其中的1X9Q抗体结构。这个结构随后应当用作下一区段插入的输入结构。其中插入区段的次序是无关紧要的。对于该实施例,从每个簇中选择主链代表,其随后与其他主链簇代表性区段组合以产生构象代表。表4呈现了从源抗体结构中提取的区段,经受区段减少程序,并且随后用于构建约4,500个结构支架代表的简化文库。如表4中可见的,在该示例性情况下,已经受用于区段闭合程序的权重拟合的表3中呈现的区段被减少至5(VL)x2(L3)x9(VH)x50(H3)。将这些代表性区段组合成所有排列,提供了约4,500个结构代表的简化文库。除了可变轻和可变重结构域中二硫化物键合的半胱氨酸的相对取向,并且PSSM衍生的序列约束用于指导序列设计选择之外,来自模板抗体的所有序列和构象信息在该程序中消除。根据本发明的一些实施方案,具有共同结构折叠的氨基酸序列文库包含扭曲数据库,其中每个扭曲数据库含有“n”个条目,n是在模板抗体的背景下成功权重拟合和重新闭合的区段的数目。在一些实施例中,扭曲数据库中的每个条目具有4(N+1)个字段,N是插入的主链区段的序列长度。在一些实施方案中,字段是关于插入的主链区段的每个残基的Ψ、Φ和Ω,二面角值和残基鉴定。在一些实施方案中,最后四个字段是相对于模板抗体的移植区段的起始、终止和拆分位点残基数目,以及其他鉴定标签例如源抗体的文件名。在本文呈现的方法的执行期间,每当对不同的主链构象取样时,根据当前区段重新指定整个抗体的PSSM,使序列约束与主链构象同步。为了效率,在该方法的不同阶段,使不同的残基集合经受设计。例如,几个初始设计阶段仅最优配体结合表面,而在设计方案结束时,存在所有抗体位置(遭受序列约束)的全序列最优的几个迭代。序列约束,例如PSSM,可观地减少了组合设计问题;在代表性的情况下,长超过230个氨基酸的可变片段的完全设计的最后一个步骤具有总共约10^117种不同的可能序列组合,等价于仅93个位置的完全重新设计;增加PSSM截断将进一步减少该组合空间。作为通过CDR和构架中的位置之间的关联性发挥的作用的示例性证明,主链构象簇L1.16_L2.8(参见表3)在构架位置L71处具有完全保守的苯丙氨酸,其与在CDR1上的位置L30处的亮氨酸或异亮氨酸相互作用。主链构象簇L1.10_L2.8在构架中使用不同的保守残基来稳定CDRL1-在L30处的缬氨酸或异亮氨酸,以及在位置71处的完全保守的酪氨酸。通过使用与区段构象关联的序列约束,这些保守的序列构象关联性先前被鉴定为维持CDRL1稳定性的关键,设计过程解释了这些关系。重和轻链刚体取向的取样:除主链构象和序列之外,Fv结构的第三个决定簇是轻链和重链之间的刚体取向(RBO)。认识到需要对RBO取样,编写RBOut以便基于实验κ轻链抗体结构来构建RBO的数据库。在数据库中,每种抗体的RBO表示为每个Ig结构域中的第二个半胱氨酸(大约质心)之间的转化矩阵。具体地,通过对其RBO应该去往数据库的所有抗体运行下述来构建RBOut数据库:另外,实施算法“RBIn”以允许在抗体设计方案期间从RBO数据库中的RBO取样。RBIn的使用显示于下文design_refine.xml中。代替使用来自1X9Q的RBO,实施了抗脑膜炎球菌抗体(PDBID:2BRR)的刚体取向,对于轻链对比抗体,其具有主链和c-β原子对于重链中的第二个半胱氨酸的最平均放置,与其他抗体相比较。类似于序列和CDR构象之间的协方差,可能能够根据界面残基的身份指导RBO的取样。这可以通过如其他地方所述的神经网络方法[Abhinandan,K.R.等人,ProteinEngineering,Design&Selection,2010,23(9),第689-697页]来完成。在此类方法中,在数据库中的RBO取样的概率可以与估计的RBO可能性成比例。用于接种对接和精制算法的方法:通过组合关于VL(5)、L3(3)、VH(8)的规范构象与50种多样化H3构象(参见上文,生成构象代表),来构建6000个成员的最大多样性嵌合体文库。随后使用optimize_chimera.xml脚本对这些种子进行精制,以改善包装、区段彼此的相容性,并且总体改善蛋白质的稳定性。PatchDock算法[Schneidman-DuhovnyD.等人,Nucl.Acids.Res.,2005,33,W363-367]用于对于嵌合体文库的每个成员生成约5000个候选抗原-抗体复合物。为了运行Patchdock算法,使用“perlbuildMS.pl$target”构建分子表面文件,随后在通过“submit_patchdock.shparams.txtout/$binder.pd”最终运行Patchdock之前,构架参数文件“buildParams.plbinders/$bindertarget/$targetname”。替代取样方法:用于取样和精制主链构象的策略基于坐标下降,其中每个区段依次在选择最佳前从实验主链构象的数据库中取样且最优50次。为了提供构象多样性的替代取样,在主链构象方案的取样和精制中的坐标下降替换为MonteCarloSimulatedAnnealing(MCSA)。五种不同的移动可用于MCSA取样器:VL、L3、VH、H3和RBO。每种移动包含不同的构象多样性并且具有不同的接受概率。考虑到这一点,使随机选择的移动偏向,使得H3最有可能被取样,而其他移动更不频繁地被取样。具体地,如下文呈现的design_and_refine.xml协议中所示,以0.40、0.05、0.15、0.25和0.15的概率对H3、L3、VL、VH和RBO取样。算法最优:为了最优AbDesign算法,已提出可以通过抢占对多变的轨迹花费的时间来改善效率。因此,不同结构统计学对于最终姿势质量的预测性进行评价。发现在环剪接之前的不同阶段时的包埋表面积和结合能被初始化,预测最终姿势质量。参见下文xml协议中的soft_dock_sasa_filter、2nd_hard_min_sasa_filter和ddg_final_commitmentfilter。在环剪接期间,发现主链冲突预测环是否将被成功插入。AbDesign算法:将最优的嵌合体、Patchdock文件和抗原作为输入的设计和精制算法可以如下执行:使用MinMover进行最小化造成问题,因为当姿势处于规范结构阶段时,决定最小化何种dofs在parsetime时生成的movemap。在设计期间,姿势长度改变,并且这意味着最小化将应用于不适当的dof。TaskAwareMinMover在运行时应用任务操作。仅限于重新包装的残基将仅使其侧链最小化,允许设计的残基将使侧链和主链两者最小化产生设计的抗体:该方法使用如上所述生成的结构代表作为输入结构。为了生成基准设计,将每个代表性抗体构象与复合物结构中的天然抗体进行比对,并且随后将靶蛋白坐标加入代表性抗体构象中。随后使用简化表示对接扰乱原始结合模式。最终设计并不都具有与针对相同靶蛋白的天然抗体相同的结合模式。使用python脚本执行另外的过滤,以获得在天然抗体结合模式的4Å截断内结合的结构。如上文在本发明的一些实施方案的上下文中描述的,使用低分辨率刚体表面互补性(取向)精制程序,将在先前步骤中生成的重建抗体文库的成员对接至分子实体的目标分子表面,本文统称为粗略匹配程序。一旦对接,重建抗体就经历对于结合位点的残基的氨基酸序列最优,其受到PSSM约束的修饰。粗略匹配程序随后最优重建抗体上的侧链残基,并根据自由能和结构稳定性过滤器评价复合物。其后,根据对接和稳定性得分对所得到的复合物进行排序,并通过RMSD聚类,由此该步骤提供了代表性复合物的简化集合,编号为几十万至几百万个复合物,这取决于可用的计算力和其他实际考虑。对于通过前一步骤的结构过滤器的复合物,程序用于使用Monte-Carlo选择例行程序从排序且分选的复合物中随机选择复合物。应注意,在本发明的一些实施例下,可以考虑其他选择标准和随机抽取例行程序。如上文在本发明的一些实施方案的上下文中讨论的,使所选择的复合物经受更精细的解析对接过程,其伴随在抗原-支架界面区处的氨基酸残基的基于PSSM的随机序列最优(PSSM-SSO),在本文中统称为有利匹配程序。例如,对于在结合时的最小包埋表面积(在抗体的情况下通常预定为1000Å2),达到匹配得分的预定截断值的复合物被传递到下一个有利匹配程序,而未达到这些截断的复合物被放弃。下一个有利匹配程序实施由预计算的主链构象数据库指导的快速主链取样策略(图1和图2中的框9)。简言之,根据本发明的一些实施方案,随机选择权重拟合和重新闭合的区段之一以用相同簇的另一个权重拟合和重新闭合的区段取代,允许区段选择在长度中改变0-4个氨基酸残基(图2中的框18)。在本文呈现的方法的目前示例性证明中,该程序从相关构象数据库中随机取样50个不同的权重拟合和重新闭合的区段,所述区段在关于输入区段的预定序列长度变化内。例如,如果经历设计的代表性权重拟合和重新闭合的区段具有长度x氨基酸的H3,则精制样品H3长度x±4的主链区段。允许的长度变化取决于当前待设计的段。限制区段长度取样减少了较长区段的偏差,其可能具有更有利的稳定性和结合能。例如,在下文呈现的基准重现测试中,允许的长度变化参数对于区段类型VL、VH和L3设定为±2,并且对于H3设定为±4。在扭曲数据库中将当前权重拟合和重新闭合的区段更改为任何其他权重拟合和重新闭合的区段由强加在数据库中指定的主链二面角组成,并且在标准CPU上一秒钟内完成。对于每个取样的主链,该方法使用组合侧链包装来设计受上述PSSM约束的序列。该程序随后通过高分辨率刚体取向精制同时最优复合物结合界面,以及在靶结合表面中以及使用PSSM-SSO在替换区段周围3-10Å半径内的侧链构象和氨基酸身份最优。有利匹配程序重复三次,从软排斥势开始,并以标准的全原子能量函数结束。该程序随后使用旋转异构体试验最小化方案,由此单个侧链被随机选择、包装和最小化。这种迭代程序导致抗体核心和抗体-靶界面中的侧链包装的改善。因此,如上文所述,将设计方案分成两个部分:粗略匹配程序(图3中的框14)和旨在最优抗体和靶蛋白之间的结合能的有利匹配程序(图2中的框16a和16b);以及计算密集精制步骤,其需要对所有四个抗体区段的主链取样(图2中的框18)。第一部分在命名为“start_fresh”的设计xml中的子协议下解析。第二部分在子协议“recover”下解析,其使用坐标下降对四个区段各自执行主链取样,并经受通过结合和稳定性得分约束的最优函数。命令行选项(可以包装在“flag”文件中)执行实例:蛋白质功能设计中的关键挑战是蛋白质需要在其设计的构象中是稳定的并且结合其靶分子两者。在抗体的本文示例性情况下,对于四个主链区段(VL、L3、VH和H3)各自,该程序随机取样衍生自该区段的构象数据库的50个主链构象(图2中的框18),计算重新设计的抗体的能量(EB)和稳定性(ES),并且各自根据下述S形函数进行变换:(方程1)其中E是结合能(EB)或未结合抗体(ES)的能量,o是S形中点,其中f(E)采取½的值,并且s是中点周围的S形的陡度。S形在非常低的能量时接近值一,并且在非常高的值时接近零。在对每个区段取样构象之前,方程1中的参数o被重新设定为当前设计的抗体的能量值,因此两个S形接近其在每个区段的精制开始时的中点。最优目标函数是两个S形的乘积:o=f(ES)xf(EB),导致当ES和EB两者均较低时,值接近一,并且如果能量标准之一很高,则值接近零。最优该目标函数的效果是找到其为足够稳定和高亲和力两者的主链构象。例如,使结合能改善10个Rosetta能量单位(R.e.u.)(0.99的转化S形值)和稳定性改善10R.e.u(0.97的转化值,乘积(ESxEB)等于0.963)的主链构象将优于使结合能改善1R.e.u(0.61的转化值)和稳定性改善30R.e.u(转化值0.999,乘积等于0.6)的主链构象,如图4中所示。图4呈现了目标函数、稳定性的乘积值和如由方程1定义的结合S形的3D图,其中在设计期间取样的主链构象通过由受抗体稳定性和结合亲和力两者约束的目标函数估计;如图4中可见的,结合和稳定性中的-10R.e.u变化的转化值优于稳定性中的-30R.e.u变化和结合中的-1R.e.u变化。两种转化的乘积测量了相对于基线得分(迄今为止的中期最佳得分抗体结构),所掺入的区段对抗体的稳定性和结合亲和力的作用。在区段最优之前和之后结合能和稳定性的变化的实例显示于图5中。图5A-B呈现了比较在精制之前和之后所设计的抗体的稳定性(图5A)和结合能(图5B)之间的散点图。X轴是在序列最优后和在精制前抗体-靶复合物的计算能量(R.e.u)。Y轴是主链精制阶段后设计的抗体能量(R.e.u)。如图5A-B中可见的,刚体、构象和序列精制改善了所设计的抗体的结合能和稳定性两者。平均起来,观察到在主链精制阶段后,对于结合能约5R.e.u(等价于大约2.5kcal/摩尔)和对于抗体稳定性100R.e.u.(等价于大约50kcal/摩尔)的改善。过滤与自然结合模式类似的设计在本文呈现的方法的该步骤中,使用对应于匹配得分的不同参数的四个截断参数进行设计抗体的最终过滤,即预测的结合能、包埋表面积、设计的抗体的可变轻和重结构域与结合的配体之间的包装质量、以及在抗体和结合的配体之间的形状互补。如上文讨论的,关于这些参数各自的截断衍生自属于源抗体家族的抗体复合物的实验结构数据,并且关于这些参数各自的截断衍生自下文列出的303种天然抗体-蛋白复合物集合,其使用“SabDab”数据库从蛋白质数据库(ProteinDataBank)中提取。在下文呈现的示例性基准重现测试中使用这些相同的截断值。使用蛋白质结构预测的关键评估(CriticalAssessmentofproteinStructurePrediction)(CASP)I_RMS标准[Méndez,R.等人,Proteins,2003,52(1),第51–67页]限定与天然结合模式类似的设计抗体。简言之,设计抗体在结构上与靶向相同分子的天然抗体比对。相对于天然复合物,计算设计复合物中的靶的界面残基(抗体结构的10Å距离截断内的所有靶残基)之间的均方根偏差(RMSD)。具有小于4Å的I_RMS值的设计的抗体复合物视为类似于天然结合模式。用于设定关于结合能、包埋表面积、包装质量和形状互补的截断值的天然抗体-蛋白质复合物的303个PDB条目是:PDBIDs1A14、1A2Y、1AR1、1BJ1、1BVK、1C08、1CZ8、1DQJ、1EGJ、1EO8、1FDL、1FE8、1FJ1、1FNS、1G7H、1G7I、1G7J、1G7L、1G7M、1G9M、1G9N、1GC1、1H0D、1IC4、1IC5、1IC7、1IQD、1J1O、1J1P、1J1X、1JHL、1JPS、1JRH、1K4C、1K4D、1KB5、1KIP、1KIQ、1KIR、1LK3、1MHP、1MLC、1N8Z、1NBY、1NBZ、1NCA、1NCB、1NCC、1NCD、1NDG、1NDM、1NFD、1NMB、1NMC、1NSN、1OAK、1OB1、1ORS、1OSP、1QFU、1R0A、1R3I、1R3J、1R3K、1R3L、1RJL、1RZJ、1RZK、1S5H、1TPX、1TQB、1TZH、1TZI、1UA6、1UAC、1UJ3、1V7M、1VFB、1WEJ、1XF5、1XGP、1XGQ、1XGR、1XGT、1XGU、1XIW、1YJD、1YQV、1YYL、1YYM、1ZTX、1ZWI、2ADF、2AEP、2ARJ、2ATK、2B2X、2BDN、2BOB、2CMR、2DQC、2DQD、2DQE、2DQF、2DQG、2DQH、2DQI、2DQJ、2DWD、2DWE、2EIZ、2EKS、2FD6、2FJG、2H8P、2H9G、2HFE、2HG5、2HJF、2HMI、2HVJ、2HVK、2I5Y、2I60、2IFF、2IH1、2IH3、2ITD、2J88、2JEL、2JK5、2NLJ、2NR6、2NXY、2NXZ、2NY0、2NY1、2NY2、2NY3、2NY4、2NY5、2NY6、2NY7、2NYY、2OZ4、2P7T、2Q8A、2Q8B、2QQK、2QQN、2R0L、2R56、2UZI、2VDK、2VDL、2VDM、2VDO、2VDP、2VDQ、2VDR、2VXQ、2VXT、2W0F、2W9E、2WUC、2XQY、2XRA、2XTJ、2Y5T、2YBR、2YC1、2YPV、2YSS、2ZCH、3A67、3A6B、3A6C、3B2U、3B9K、3BDY、3BE1、3BN9、3BT2、3CVH、3D85、3D9A、3DET、3DVG、3DVN、3EHB、3EOA、3FB5、3GB7、3GRW、3HB3、3HI1、3HI6、3IGA、3IU3、3JWD、3K2U、3KLH、3KR3、3L5W、3L5X、3L95、3LD8、3LDB、3LEV、3LH2、3LHP、3LIZ、3MXW、3NID、3NIF、3NIG、3O2D、3OR6、3OR7、3P0Y、3PGF、3PNW、3Q3G、3QWO、3RKD、3RVV、3RVW、3RVX、3S35、3S37、3SDY、3SE9、3SKJ、3SO3、3SOB、3SQO、3STL、3STZ、3T3M、3T3P、3U30、3U9P、3UC0、3V6O、3VG9、3VI3、3VI4、3VW3、3W9E、3WKM、3ZDX、3ZDY、3ZDZ、3ZE0、3ZE2、3ZKM、3ZKN、4AEI、4AG4、4AL8、4ALA、4CAD、4D9Q、4D9R、4DGI、4DKF、4DN4、4DTG、4DVR、4DW2、4ENE、4ETQ、4F15、4F37、4F3F、4FFV、4FFW、4FFY、4G3Y、4G6J、4G6M、4H88、4HC1、4HCR、4HLZ、4HT1、4HWB、4I2X、4I9W、4IRZ、4JPK、4JQI、4JR9、4JRE、4K2U、4K3J、4K94、4K9E、4KI5、4KJQ、4KK8、4KK9、4KKL、4L5F、4LBE、4LCU、4LEO、4LF3、4LMQ、4LOU、4LSP、4LSQ、4LSR、4M48、4MSW和4MWF。图6A-D呈现了当对于抗体的Fv结构域证明时,显示在本文呈现的方法的最后步骤中用于过滤所设计的抗体结构的能量和结构标准的概率密度图,其中基于四个参数过滤所设计的抗体:预测的结合能(图6A)、包埋表面积(图6B)、抗体结构和配体之间的形状互补性(图6C)、以及可变轻和重结构域结构域与配体之间的包装质量(图6D),而截断值由虚线表示,并且衍生自303种天然蛋白质结合抗体的集合,而通过所有过滤器的抗体设计(黑色曲线)与天然蛋白质结合抗体(灰色曲线)进行比较。算法AbPredict用于从序列不知情预测抗体结构的用途:为了验证设计抗体的稳定性,可以在optimize_chimera.xml中禁用序列设计,并且采用来自不同结构区段和刚体取向的样品的MCSA方案。对于针对实验结构的基准,已观察到天然结构与具有约8个Rosetta能量单位的能隙的可替代构象很好地分离(数据未显示)。当观察到此类能隙时,预测和设计变得可行[Fleishman,S.等人,Cell,2012,149(2),第262–273页]。实施例2设计的抗体-重现基准为了测试该方法的能力,根据本发明的一些实施方案,为了预测蛋白质结合抗体的结构和序列,使用结构抗体数据库“SAbDab”选择九种高亲和力(Kd<10nM)、高分辨率(X射线分辨率<2.5Å)蛋白质结合抗体的多样化基准(参见下表5)。天然抗体集合包含人抗体Fab40、D5中和mAb和BO2C11(分别为PDBID3K2U、2CMR和1IQD),鼠抗体E8、D1.3mAb、F10.6.6、JEL42和5E1Fab(分别为PDBID1WEJ、1VFB、1P2C、2JEL和3MXW)和人源化鼠抗体D3H44(PDBID1JPS)。靶分子包含含有螺旋(2CMR)、片层(1JPS)和环(1P2C、3K2U)二级结构元件的凸形(2JEL、1IQD)、平坦(1P2C)和凹形(3MXW)表面。基准重现测试已从4500个计算重建和设计的抗体结构集合开始。将每个结构与处于其结合构象的天然结合抗体的位置进行比对,并且生成包含设计结构和包含目的表面的分子实体的复合结构。消除来自天然抗体的所有序列和主链构象信息。使用RosettaDock应用简化表示对接(参见上文的描述),以扰乱抗体和靶之间的初始结合模式。为了测试该方法的性能,分离了对于基准集合中的每种抗体-靶复合物计算的最高结合亲和力的设计抗体,并且根据下述参数将该设计与天然抗体进行对比:序列同一性、RMSD、界面形状互补性(Sc)、包装统计、包埋表面积、结合能和主链构象聚类。表5呈现了用于重现基准的结合抗体复合物,并且概括了基准重现试验实验的结果。如表5中可见的,本文呈现的设计方法不专门产生在PDB中观察到的天然结合模式。为了分析在实验观察的结合模式中的设计构象和序列,其中靶界面大于4ÅRMSD的结合构象从天然构象中消除。如表5中进一步可见的,本文呈现的设计方法以高概率重现天然抗体构象。基准集合中的九种抗体中的五种在计算的结合能方面处于前10%的排序处。具有大的包埋表面积(大于1800Å2)的结合构象比具有较低包埋表面积的那些结构更一致地准确预测。作为成功重现的结合模式的代表性实例,考虑了靶向与人源化抗组织因子抗体D3H44(PDBID1JPS)和抗跨膜糖蛋白D5中和mAb(PDBID2CMR)相同表面的设计。包含设计抗体的所有主链构象区段属于与实验测定的1JPS(L1.11_L2.8、L3.10.1、H1.14_H2.15、H3.16.5)和2CMR(H1.14_H2.15、H3.18.7、L1.11_L2.8、L3.10.1)的结构相同的主链簇。这些设计的主链构象和结合模式显示与天然抗体一致的高水平。其中与天然抗体具有相似构象的设计具有差预测结合能排序的情况突出了设计方法中的潜在偏差。在抗溶菌酶抗体F10.6.6(PDBID1P2C)的情况下,天然抗体包埋相对小的表面积(参见表5)。靶向相同溶菌酶表位的大多数排序最靠前的设计通过使用更长的L1区段包埋更大的表面(超过1600Å2)。优选通过过滤程序选择较长的段,其包埋较大的表面并维持对于靶的高互补性;需要实验来证明此类设计是否确实对其靶具有比天然抗体更高的亲和力。抗肝细胞生长因子激活物抗体(PDBID3K2U)具有1980Å2的结合表面积,而最佳排序的相似构象设计仅包埋1700Å2(参见表5)。包埋表面积中的这种差异是由于天然和设计的抗体的轻和重可变结构域之间的包装角度中的差异;可以进行可变结构域之间的包装角度的更广泛建模,以便解决此类不准确性。序列重现率在先前描述的设计基准的范围内。然而,因为先前描述的设计工作尝试处理功能位点设计或蛋白质核心,而本文呈现的抗体设计基准处理两者,并且因为在基于实验数据约束序列变异和构象选择的同时,在本文呈现的方法中进行,所以值无法直接比较。如先前描述的基准研究中,抗体核心内的序列重现至大致60-80%同一性内,并且结合表面序列同一性为约30%,类似于蛋白质结合和酶设计基准。高度重现自然相互作用的两个突出的例子是抗组织因子设计的抗体和抗跨膜糖蛋白设计的抗体。在这两种情况下,界面序列重现高于30%(参见表5),并且在界面处的保守残基也保存侧链构象。如对于天然抗体观察到的,设计抗体集合中的大多数序列变异限于靶结合表面;抗体核心位置明显更保守。这种高序列保守性也反映在抗体核心中的侧链构象的高度重现。因为氨基酸构象可塑性具有减少结合特异性和亲和力的潜力,所以在结合表面上固定侧链的设计算法一般成功设计蛋白质抑制剂以及蛋白质和小分子结合剂。提出评价侧链刚度的计算度量,其计算当结合剂与其靶解离时所有侧链构象的整体中结合的侧链构象的玻尔兹曼重量。使用现有策略的设计结合剂通常显示比天然结合剂更低的侧链玻尔兹曼重量,以及可能更低的刚性。将侧链刚度掺入其设计方案内的先前的设计尝试,在设计期间明确地考虑其或已将其用作用于设计估计的附加过滤器。假设在主链构象文库和相关PSSM中编码的序列结构规则隐含地将设计抗体结合表面中的残基限制为更严格的选择。使用本文所述的方法比较303种天然高亲和力抗体与本文设计的抗体在结合表面处的侧链构象可塑性,显示在结合表面处设计的芳香族残基,其对预测的结合能贡献超过1R.e.u,显示出构象概率密度非常接近天然抗体。在非结合状态下不太可能处于其预期构象的极低概率侧链构象(小于5%概率)的比例小于10%,并且所有设计抗体的界面残基的超过一半显示出概率高于15%的侧链构象。实施例3从头设计抗体的表达通过使用本文所述的方法,获得数千个蛋白质序列,其中19个被选择用于实验表达和活性测试。将氨基酸序列转录为作为单链可变片段的DNA,其中GS接头将VH链的C末端连接到VL链的N末端,对于在面包酵母酿酒酵母(S.cerevisiae)中表达进行密码子最优。在外部定制合成后,通过PCR扩增具有上游和下游侧翼区的DNA区段,并且通过在酿酒酵母菌株EBY100中的同源重组插入pETCON质粒内。通过测序验证所获得的质粒,并且用于在酵母表面展示中测试,其中通过设计抗体的荧光染色监测表达,同时通过生物素化配体的荧光标记监测配体识别。随后通过体外进化增强初始设计的相对低的亲和力;通过易错PCR在每个基因得到1-3个突变的条件下扩增原始设计,并且由所得到的DNA生成酵母文库(在酿酒酵母菌株EBY100中)。通过几轮FACS分选,分离具有增加亲和力的克隆,并且必要时,重复该过程。根据本发明的实施方案,获得显示出75%及以上的表达水平的设计蛋白质。这些速率以前不能通过目前已知的计算设计方法实现,并且与抗荧光素抗体4m5.3的表达水平可比较,所述抗荧光素抗体4m5.3在设计过程中充当模板以及用于作为酵母表面展示中的scFv的高表达和稳定性的黄金标准。另外,本文呈现的方法得到识别靶的抗体结构,它们被设计为以与由随机抗体结构提供的亲和力相比较明显更高的亲和力结合。图7呈现了酵母表面展示的散点图,其中细胞就表达水平和结合染色用于测试两种抗体:使用本文描述的算法设计以结合ACP(由灰点标记)的“设计#1”(SEQIDNO.1),以及不结合ACP(由黑点标记)的抗组织因子抗体“4m5.3”,其充当模板结构以及结合测试的对照,以证明ACP和设计#1(SEQIDNO.1)之间的结合是特异性的,其中表达水平的特征在于在X轴上的左移,并且结合的特征在于在Y轴上的上移。如图7中可见的,表达抗ACP设计#1(SEQIDNO.1)的细胞显示出比对照4m5.3更强的与10μMACP的结合,如可以衍生自这个群体的y位移。结合剂随后经历体外进化,其导致具有增加亲和力的单个氨基酸取代的克隆的鉴定。图8A-B呈现了在引入点突变之前和之后的抗ACP设计的滴定曲线(设计#1-5;SEQIDNOs.1-5,图8A),以及抗ACP设计(设计#1SEQIDNO.1)由其设计的底物ACP(在图8B中由圆圈标记)和阴性对照(TEM,在图8B中由正方形标记)的滴定曲线。根据使用所述算法生成的模型,将点突变引入设计#1(SEQIDNO.1)的结合界面。预测这些突变对结合是有害的。引入了两个分开的点突变:A34N(设计#2;SEQIDNO.2)和S100W(设计#3;SEQIDNO.3),以及分别引入来自不结合ACP的抗荧光素抗体(4m5.3)的环H2和H3的完整区段改变(分别为设计#4(SEQIDNO.4)和设计#5(SEQIDNO.5))。如图8中可见的,已用所呈现的算法生成用于ACP的第二结合剂,具有885nM的初始估计kD,其可以通过体外进化增加至45nM。如图8中进一步可见的,预测干扰结合的单个氨基酸和环交换的引入显示急剧减少结合。还如图8A中可见的,对设计#1(SEQIDNO.1)的结合界面的所有改变消除了与ACP的结合,从而支持设计#1(SEQIDNO.1)和ACP之间的建模的相互作用。为了证明设计#1(SEQIDNO.1)特异性结合ACP并且不是混杂(“粘性”)的,测试针对另一种底物(TEM)的结合,并且显示出无结合(图8B)。实施例4修饰磷酸三酯酶(PTE)酶的特异性本文所述的方法用于生成磷酸三酯酶(PTE)的活性位点的模型,所述磷酸三酯酶是属于TIM桶的共同蛋白质折叠的双核金属依赖性酶。PTE可以在扩散极限下水解对氧磷,广泛使用的农药。已假设PTE已从水解内酯的另一种TIM桶折叠酶进化而来[Afriat-Jurnou,L.等人,Biochemistry,2012,51,6047–6055]。虽然PTE不能水解内酯,但已知内酯酶显示出混杂的PTE活性[Hiblot,J.,Sci.Rep.,2012,2,第779页]。假设由于内酯酰基链和酶的环7之间的碰撞,PTE无法水解内酯。本文呈现的方法已用于生成具有叶片7的修饰构象的磷酸三酯酶TIM桶,同时保持结构的所有其他部分构象不变。来自缺陷假单胞菌(Pseudomonasdiminuta)的对硫磷酸水解酶(PDBID:1HZY)用作模板蛋白。关于叶片7的茎残基(最高结构保守性位置)是216和263(根据PDB结构编号)。生成一百七十七(177)种可能的构象变体,其中选择九(9)种用于在使用Pymol目视检查和手动添加突变后的实验测试,主要是使用Foldit[Khatib,F.等人,Proc.Natl.Acad.Sci.,2011,108,第18949–18953页]来增加蛋白质包装。因为所有设计的结构共享相同的N末端区域(氨基酸残基1-196),所以N末端基因和可变C末端区域分开排序,并且使用PCR将其组合。所有设计均与N末端MBP融合以帮助蛋白质的清洁和稳定性,并且无伴侣在大肠杆菌BL21细胞中表达。在九种设计的结构中,两种在野生型水平下表达,如在聚丙烯酰胺凝胶电泳中可见的(数据未显示)。对于两个良好表达的设计,设计#6(SEQIDNO.6)具有来自嗜热地芽孢杆菌(Geobacilluskaustophilus)内酯酶(PDBID:4HA0)的叶片7构象,并且设计#7(SEQIDNO.7)具有衍生自单核细胞增生性李斯特菌(Listeriamonocytogenes)内酯酶(PDBID:3PNZ)的叶片构象。两种设计均展示高于背景的关于对氧磷的PTE活性,但是没有可检测的内酯酶活性,如用硫代烷基丁内酯底物(TBBL)底物所测试的。设计的检查揭示在位置220处应该存在先前已报道对预内酯酶活性重要的精氨酸。在两种设计中,在位置220处重新引入精氨酸残基,并且两种修订的设计均展示与以前相似的表达水平。在突变后,设计#6(SEQIDNO.6)显示出内酯酶活性以及对氧磷酶活性。设计#6展示改变的底物特异性;原始模板(来自缺陷假单胞菌的对硫磷水解酶)具有约3×10−5的TBBL/对氧磷比率,而设计#6(SEQIDNO.6)具有0.15的TBBL/对氧磷比率,其为103倍的变化。而设计#7(SEQIDNO.7)未展示关于对氧磷或其他测试的内酯底物的可检测活性。实施例5修饰内酯酶的特异性为了进一步证明该方法可以使用主链设计提供对酶特异性的完全控制,该方法用于改变内酯酶的底物特异性。选择来自硫磺矿硫化叶菌(Sulfolobussolfataricus)的超热稳定性内酯酶(PDBID:2VC7)作为模板结构。模板结构具有针对对氧磷的中度Kcat/Km效率(参见上表6)。生成设计构象类似于上文对于磷酸三酯酶描述的方法,除了两个差别之外:(i)在主链设计期间改变四个叶片(叶片4-7),而叶片1、2、3和8构成二聚化界面的一部分,并且因此保持不变,以及(ii)叶片7的构象限制于衍生自已知PTE的那些。通过将与对氧磷类似物(PDBID:2R1N)结合的PTE的晶体结构与模板蛋白的晶体结构比对,并且将配体坐标拷贝到模板蛋白质结构,来生成对氧磷的原始对接取向。选择具有不同叶片7构象和序列设计选择的五种设计结构用于实验测试(SEQIDNO.8-12)。图9呈现了与充当设计模板的原始酶(来自硫磺矿硫化叶菌的内酯酶;PDBID:2VC7;SEQIDNO.13)的序列相比较,具有设计的改变底物特异性的指定为设计#8-12(分别为SEQIDNO.8-12)的五种TIM桶折叠设计的序列比对。如图9中可见,将十四个氨基酸插入模板蛋白中,并且在插入的区段(用粗线标记)周围引入另外的序列修饰以容纳插入。引入来自PTE家族中的其他成员的插入的先前尝试已导致无功能蛋白质[Afriat-Jurnou,L.等人,Biochemistry,2012,51,6047–6055]。虽然本发明已与其具体实施例结合进行描述,但显而易见的是许多替代、修改和变化对于本领域技术人员将是显而易见的。相应地,预期包括落入所附权利要求的精神和广泛范围内的所有此类替代、修改和变化。在本说明书中提及的所有出版物、专利和专利申请通过引用整体并入本说明书内,至如同每个个别出版物、专利或专利申请特别且个别指示引入本文作为参考相同的程度。另外,本申请中的任何参考文献的引用或鉴定不应被解释为承认此类参考文献可用为本发明的现有技术。就使用章节标题的程度,它们不应被解释为必然限制。序列表<110>YedaResearchAndDevelopmentCo.Ltd.FLEISHMAN,SarelLAPIDOTH,GideonPSZOLLA,MariaGabrieleNORN,Christoffer<120>计算蛋白质设计的方法、由此设计的蛋白质及其生产方法<130>62938<150>62/021,309<151>2014-07-07<160>13<170>PatentInversion3.5<210>1<211>825<212>DNA<213>人工序列<220><223>抗ACP-AB设计1核苷酸序列<400>1ggtggaggcggtagcggaggcggagggtcggaagtgaaactggacgaaaccggtggtggt60ctggttcagccgggtggtgcgatgaaactgtcttgcgttacctctggtttcgacttcggt120gactactacatgctgtgggttcgtcagtctccggaaaaaggtctggaatgggttgcggtt180gttggtccagacaactcttacaccaactacgcggactctgttaaaggtcgtttcaccatc240tctcgtgacgactctaaatcttctgtttacctgcagatgaacaacctgcgtaccgaagac300accggtatctactactgcatgggctcttcttggtcccaggactcctcttccgaatctgtt360atgaaatacctcggtcagggtacttctgtgaccgtttctggaggtggcggtagcggaggc420ggcggttctggaggtggcgggagcaacgttgttatgacccagaccccgctgtctctgcca480gtttccctgggtgaccaggcgtctatctcttgccgttcttctcagtccctcaccgcggaa540gcgggtctgaccgttctggcttggttcctgcagaaaccgggtcagtctccaaaggtgctg600atctacaaagtttctaaccgtgtgtctggtgttccggaccgtttctctggttccggttct660ggtaccgacttcaccctgaaaatcaaccgcgttgaagctgaagacctcggtgtttacttc720tgcgcggcttggaccaactctaaatgggttttcggtggtggcaccaagctggaaattaag780ggtggcggatccgaacaaaagcttatttctgaagaggacttgtaa825<210>2<211>825<212>DNA<213>人工序列<220><223>抗ACP-AB设计2核苷酸序列<400>2ggtggaggcggtagcggaggcggagggtcggaagtgaaactggacgaaaccggtggtggt60ctggttcagccgggtggtgcgatgaaactgtcttgcgttacctctggtttcgacttcggt120gactactacatgctgtgggttcgtcagtctccggaaaaaggtctggaatgggttgcggtt180gttggtccagacaactcttacaccaactacgcggactctgttaaaggtcgtttcaccatc240tctcgtgacgactctaaatcttctgtttacctgcagatgaacaacctgcgtaccgaagac300accggtatctactactgcatgggctcttcttggtcccaggactcctcttccgaatctgtt360atgaaatacctcggtcagggtacttctgtgaccgtttctggaggtggcggtagcggaggc420ggcggttctggaggtggcgggagcaacgttgttatgacccagaccccgctgtctctgcca480gtttccctgggtgaccaggcgtctatctcttgccgttcttctcagtccctcaccgcggaa540aatggtctgaccgttctggcttggttcctgcagaaaccgggtcagtctccaaaggtgctg600atctacaaagtttctaaccgtgtgtctggtgttccggaccgtttctctggttccggttct660ggtaccgacttcaccctgaaaatcaaccgcgttgaagctgaagacctcggtgtttacttc720tgcgcggcttggaccaactctaaatgggttttcggtggtggcaccaagctggaaattaag780ggtggcggatccgaacaaaagcttatttctgaagaggacttgtaa825<210>3<211>825<212>DNA<213>人工序列<220><223>抗ACP-AB设计3核苷酸序列<400>3ggtggaggcggtagcggaggcggagggtcggaagtgaaactggacgaaaccggtggtggt60ctggttcagccgggtggtgcgatgaaactgtcttgcgttacctctggtttcgacttcggt120gactactacatgctgtgggttcgtcagtctccggaaaaaggtctggaatgggttgcggtt180gttggtccagacaactcttacaccaactacgcggactctgttaaaggtcgtttcaccatc240tctcgtgacgactctaaatcttctgtttacctgcagatgaacaacctgcgtaccgaagac300accggtatctactactgcatgggctcttcttggtcccaggactcctcttccgaatctgtt360atgaaatacctcggtcagggtacttctgtgaccgtttctggaggtggcggtagcggaggc420ggcggttctggaggtggcgggagcaacgttgttatgacccagaccccgctgtctctgcca480gtttccctgggtgaccaggcgtctatctcttgccgttcttctcagtccctcaccgcggaa540gcgggtctgaccgttctggcttggttcctgcagaaaccgggtcagtctccaaaggtgctg600atctacaaagtttctaaccgtgtgtctggtgttccggaccgtttctctggttccggttct660ggtaccgacttcaccctgaaaatcaaccgcgttgaagctgaagacctcggtgtttacttc720tgcgcggcttggaccaactggaaatgggttttcggtggtggcaccaagctggaaattaag780ggtggcggatccgaacaaaagcttatttctgaagaggacttgtaa825<210>4<211>831<212>DNA<213>人工序列<220><223>抗ACP-AB设计4核苷酸序列<400>4ggtggaggcggtagcggaggcggagggtcggaagtgaaactggacgaaaccggtggtggt60ctggttcagccgggtggtgcgatgaaactgtcttgcgttacctctggtttcgacttcggt120gactactacatgctgtgggttcgtcagtctccggaaaaaggtctggaatgggttgcgcag180ttccgtaacaaaccgtacaactacgaaacctactacgcggactctgttaaaggtcgtttc240accatctctcgtgacgactctaaatcttctgtttacctgcagatgaacaacctgcgtacc300gaagacaccggtatctactactgcatgggctcttcttggtcccaggactcctcttccgaa360tctgttatgaaatacctcggtcagggtacttctgtgaccgtttctggaggtggcggtagc420ggaggcggcggttctggaggtggcgggagcaacgttgttatgacccagaccccgctgtct480ctgccagtttccctgggtgaccaggcgtctatctcttgccgttcttctcagtccctcacc540gcggaagcgggtctgaccgttctggcttggttcctgcagaaaccgggtcagtctccaaag600gtgctgatctacaaagtttctaaccgtgtgtctggtgttccggaccgtttctctggttcc660ggttctggtaccgacttcaccctgaaaatcaaccgcgttgaagctgaagacctcggtgtt720tacttctgcgcggcttggaccaactctaaatgggttttcggtggtggcaccaagctggaa780attaagggtggcggatccgaacaaaagcttatttctgaagaggacttgtaa831<210>5<211>801<212>DNA<213>人工序列<220><223>抗ACP-AB设计5核苷酸序列<400>5ggtggaggcggtagcggaggcggagggtcggaagtgaaactggacgaaaccggtggtggt60ctggttcagccgggtggtgcgatgaaactgtcttgcgttacctctggtttcgacttcggt120gactactacatgctgtgggttcgtcagtctccggaaaaaggtctggaatgggttgcggtt180gttggtccagacaactcttacaccaactacgcggactctgttaaaggtcgtttcaccatc240tctcgtgacgactctaaatcttctgtttacctgcagatgaacaacctgcgtaccgaagac300accggtatctattactgtactggtgcgtcttacggtatggaatacctgggtcagggtact360tctgtgaccgtttctggaggtggcggtagcggaggcggcggttctggaggtggcgggagc420aacgttgttatgacccagaccccgctgtctctgccagtttccctgggtgaccaggcgtct480atctcttgccgttcttctcagtccctcaccgcggaagcgggtctgaccgttctggcttgg540ttcctgcagaaaccgggtcagtctccaaaggtgctgatctacaaagtttctaaccgtgtg600tctggtgttccggaccgtttctctggttccggttctggtaccgacttcaccctgaaaatc660aaccgcgttgaagctgaagacctcggtgtttacttctgcgcggcttggaccaactctaaa720tgggttttcggtggtggcaccaagctggaaattaagggtggcggatccgaacaaaagctt780atttctgaagaggacttgtaa801<210>6<211>320<212>PRT<213>人工序列<220><223>设计#6氨基酸编码序列<400>6AspArgIleAsnThrValArgGlyProIleThrIleSerGluAlaGly151015PheThrLeuThrHisGluHisIleCysGlySerSerAlaGlyPheLeu202530ArgAlaTrpProGluPhePheGlySerArgLysAlaLeuAlaGluLys354045AlaValArgGlyLeuArgArgAlaArgAlaAlaGlyValArgThrIle505560ValAspValSerThrPheAspIleGlyArgAspValSerLeuLeuAla65707580GluValSerArgAlaAlaAspValHisIleValAlaAlaThrGlyLeu859095TrpTyrAspProProLeuSerMetArgLeuArgSerValGluGluLeu100105110ThrGlnPhePheLeuArgGluIleGlnTyrGlyIleGluAspThrGly115120125IleArgAlaGlyIleIleLysValAlaThrThrGlyLysAlaThrPro130135140PheGlnGluLeuValLeuLysAlaAlaAlaArgAlaSerLeuAlaThr145150155160GlyValProValThrThrHisThrAlaAlaSerGlnArgAspGlyGlu165170175GlnGlnAlaAlaIlePheGluSerGluGlyLeuSerProSerArgVal180185190CysIleGlyHisSerAspAspThrAspAspIleSerTyrLeuThrAla195200205LeuAlaAlaArgGlyTyrLeuIleAlaPheAspArgPheGlyHisGln210215220GlyMetAsnGlyAlaProThrAspGluGluArgIleArgThrLeuVal225230235240AlaLeuLeuArgAspGlyTyrGluLysGlnIleLeuLeuSerAsnAsp245250255TrpLeuPheGlyTyrSerSerTyrThrThrAsnIleMetAspValMet260265270AspArgTyrAsnProAspGlyMetAlaHisIleProLeuArgValIle275280285ProHisLeuArgGluLysGlyValProGlnGluThrLeuAlaGlyIle290295300ThrValThrAsnProAlaArgPheLeuSerProThrLeuArgAlaSer305310315320<210>7<211>317<212>PRT<213>人工序列<220><223>设计#7氨基酸编码序列<400>7AspArgIleAsnThrValArgGlyProIleThrIleSerGluAlaGly151015PheThrLeuThrHisGluHisIleCysGlySerSerAlaGlyPheLeu202530ArgAlaTrpProGluPhePheGlySerArgLysAlaLeuAlaGluLys354045AlaValArgGlyLeuArgArgAlaArgAlaAlaGlyValArgThrIle505560ValAspValSerThrPheAspIleGlyArgAspValSerLeuLeuAla65707580GluValSerArgAlaAlaAspValHisIleValAlaAlaThrGlyLeu859095TrpTyrAspProProLeuSerMetArgLeuArgSerValGluGluLeu100105110ThrGlnPhePheLeuArgGluIleGlnTyrGlyIleGluAspThrGly115120125IleArgAlaGlyIleIleLysValAlaThrThrGlyLysAlaThrPro130135140PheGlnGluLeuValLeuLysAlaAlaAlaArgAlaSerLeuAlaThr145150155160GlyValProValThrThrHisThrAlaAlaSerGlnArgAspGlyGlu165170175GlnGlnAlaAlaIlePheGluSerGluGlyLeuSerProSerArgVal180185190CysIleGlyHisSerAspAspThrAspAspLeuSerTyrLeuThrAla195200205LeuAlaAlaArgGlyTyrPheValSerPheAspArgIleAlaLeuIle210215220LysTyrAlaProGluSerAlaArgIleAlaLeuIleLeuTyrLeuVal225230235240SerGluGlyPheGluAsnGlnIleLeuValSerGlyAspTrpLeuPhe245250255GlyPheSerSerTyrThrThrAsnIleMetAspAsnMetAspArgVal260265270AsnProAspGlyMetAlaPheIleProLeuArgValIleProTyrLeu275280285ArgGluLysGlyValProGlnGluThrLeuAlaGlyIleThrValThr290295300AsnProAlaArgPheLeuSerProThrLeuArgAlaSer305310315<210>8<211>328<212>PRT<213>人工序列<220><223>TIM桶折叠设计#8氨基酸编码序列<400>8MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TrpIlePheValAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyPheValLysIleAlaAlaAspGlnProGly130135140IleThrLysAspValGluLysValIleArgAlaAlaAlaArgAlaSer145150155160LysGluThrGlyCysProIleIleSerHisSerAsnAlaHisAsnAsn165170175AspGlyGluAlaGlnGlnGluIleLeuAlaCysGluGlyValAspPro180185190CysLysIleLeuIleGlyHisLeuGlyAspThrAspAsnLeuAspTyr195200205IleArgLysIleAlaGlnArgGlySerPheIleGlyIleAspArgIle210215220ProHisSerGlyIleGlyAlaGluGlyAsnAlaSerAlaSerAlaLeu225230235240PheGlyAsnArgSerTrpGlnGluArgAlaSerValIleLysAlaMet245250255IleAspAspGlyTyrAlaAspLysIleLeuMetSerHisAspTyrCys260265270CysThrPheAspValGlyAlaAlaLysProGluTyrLysProSerMet275280285AlaProArgTrpSerIleThrValIlePheGluAspThrIleProPhe290295300LeuLysArgAsnGlyValAsnGluGluValLeuAlaThrIlePheLys305310315320GluAsnProLysLysPhePheSer325<210>9<211>327<212>PRT<213>人工序列<220><223>TIM桶折叠设计#9氨基酸编码序列<400>9MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TrpIlePheValAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyPheIleLysLeuAlaSerSerLysGlyArg130135140IleThrProTyrGluGluLysValLeuArgAlaAlaAlaArgAlaGln145150155160LysGluThrGlyAlaProIleIleSerHisThrGlnGluGlyGlnGln165170175GlyProGlnGlnAlaGluLeuLeuLysGlnGluGlyAlaAspProGlu180185190LysIleLeuIleGlyHisSerAspAspThrAspAspLeuAspTrpIle195200205ArgLysMetAlaAlaLeuGlySerPheIleGlyPheAspArgIlePro210215220HisSerGlyIleGlyAlaGluAspAsnAlaSerAlaThrAlaLeuPhe225230235240GlyAsnArgSerAspGlnGluArgAlaArgIleIleLysAlaMetIle245250255AspGluGlyPheAlaAsnLysValIleMetSerHisAspTyrCysCys260265270ThrPheAspValGlyThrAlaLysProGluTyrLysProSerAlaAla275280285ProArgTrpSerIleThrLeuMetPheGluAspThrIleProPheLeu290295300LysArgAsnGlyValAsnGluGluValIleAlaThrIlePheLysGlu305310315320AsnProLysLysPhePheSer325<210>10<211>327<212>PRT<213>人工序列<220><223>TIM桶折叠设计#10氨基酸编码序列<400>10MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TrpIlePheValAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyPheIleLysValAlaThrThrGlyLysAla130135140ThrProAspGluGluLysValIleArgAlaAlaAlaArgAlaSerLys145150155160GluThrGlyCysProIleIleThrHisThrAlaAlaSerGlnArgAsp165170175GlyGluGluGlnAlaGluIleGluGluCysGluGlyGlyProProCys180185190ArgIleMetIleGlyHisSerAspAspThrAspAspLeuAspTrpIle195200205ArgLysLeuAlaGlnLysGlyTyrPheIleGlyPheAspArgMetPro210215220HisSerGlyIleGlyAlaGluAspAsnAlaSerAlaThrAlaLeuPhe225230235240GlyThrArgSerAspGlnThrArgAsnGluAlaIleLysArgIleIle245250255AspAspGlyTyrAlaGluLysIleLeuMetSerHisAspTyrCysCys260265270ThrIleAspValGlyAlaAlaLysProGluHisLysProSerAlaAla275280285ProArgTrpSerIleThrLeuIlePheGluAspThrIleProPheLeu290295300LysArgAsnGlyValAsnGluGluValIleAlaThrIlePheLysGlu305310315320AsnProLysLysPhePheSer325<210>11<211>329<212>PRT<213>人工序列<220><223>TIM桶折叠设计#11氨基酸编码序列<400>11MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TrpIlePheValAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyMetIleAlaGluIleGlyThrSerGluGly130135140GluMetAlaProGluGluGluLysLeuLeuArgAlaAlaAlaGlnAla145150155160AlaAsnGluThrGlyArgProIleThrThrHisThrAlaAlaSerGln165170175ArgValGlyAspGluValAlaArgValLeuGluGluGlyGlyValPro180185190ProCysLysValCysIleGlyHisSerAspAspThrAspAspLeuAsp195200205TrpIleArgLysLeuAlaAlaArgGlyTyrPheIleGlyPheAspArg210215220IleGlyHisSerGlyIleGlyAlaGluAspAsnAlaSerAlaSerAla225230235240LeuAlaGlyAsnArgSerAspGlnThrArgAlaGluCysValLysArg245250255MetAlaAspGluGlyPheAlaGluLysIleMetValSerHisAspTyr260265270CysCysThrIleAspValGlyThrAlaLysProGluTyrLysProSer275280285AlaAlaProArgTrpSerIleThrLeuIlePheGluAspThrIlePro290295300PheLeuLysArgAsnGlyValAsnGluGluValIleAlaThrIlePhe305310315320LysGluAsnProLysLysPhePheSer325<210>12<211>329<212>PRT<213>人工序列<220><223>TIM桶折叠设计#12氨基酸编码序列<400>12MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TrpIlePheValAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyMetIleAlaGluIleGlyThrSerGluGly130135140GluCysAlaProGluGlnGluLysAlaLeuArgAlaAlaAlaGlnAla145150155160AlaAsnGluThrGlyArgProIleThrThrHisThrAlaAlaSerGln165170175ArgThrGlyGluGluGlnAlaArgValLeuGluCysGlyGlyValPro180185190GlyCysLysIleCysIleGlyHisMetCysGlyAsnThrAspLeuGlu195200205GlnHisArgLysLeuAlaAspArgGlyTyrPheLeuGlyPheAspArg210215220IleGlyHisSerGlyIleGlyLeuGluAspAsnAlaSerAlaSerAla225230235240LeuMetGlyAsnArgSerTrpGlnGluArgAlaGluLeuIleLysAla245250255IleIleAspGluGlyTyrAlaAspLysIleMetValSerHisAspTyr260265270CysCysThrIleAspValGlyAlaAlaLysProGluTyrLysProLys275280285MetAlaProArgTrpSerIleThrLeuIlePheGluAspThrIlePro290295300PheLeuLysArgAsnGlyValAsnGluGluValIleAlaThrIlePhe305310315320LysGluAsnProLysLysPhePheSer325<210>13<211>314<212>PRT<213>硫磺矿硫化叶菌<400>13MetArgIleProLeuValGlyLysAspSerIleGluSerLysAspIle151015GlyPheThrLeuIleHisGluHisLeuArgValPheSerGluAlaVal202530ArgGlnGlnTrpProHisLeuTyrAsnGluAspGluGluPheArgAsn354045AlaValAsnGluValLysArgAlaMetGlnPheGlyValLysThrIle505560ValAspProThrValMetGlyLeuGlyArgAspIleArgPheMetGlu65707580LysValValLysAlaThrGlyIleAsnLeuValAlaGlyThrGlyIle859095TyrIleTyrIleAspLeuProPheTyrPheLeuAsnArgSerIleAsp100105110GluIleAlaAspLeuPheIleHisAspIleLysGluGlyIleGlnGly115120125ThrLeuAsnLysAlaGlyPheValLysIleAlaAlaAspGluProGly130135140IleThrLysAspValGluLysValIleArgAlaAlaAlaIleAlaAsn145150155160LysGluThrLysValProIleIleThrHisSerAsnAlaHisAsnAsn165170175ThrGlyLeuGluGlnGlnArgIleLeuThrGluGluGlyValAspPro180185190GlyLysIleLeuIleGlyHisLeuGlyAspThrAspAsnIleAspTyr195200205IleLysLysIleAlaAspLysGlySerPheIleGlyLeuAspArgTyr210215220GlyLeuAspLeuPheLeuProValAspLysArgAsnGluThrThrLeu225230235240ArgLeuIleLysAspGlyTyrSerAspLysIleMetIleSerHisAsp245250255TyrCysCysThrIleAspTrpGlyThrAlaLysProGluTyrLysPro260265270LysLeuAlaProArgTrpSerIleThrLeuIlePheGluAspThrIle275280285ProPheLeuLysArgAsnGlyValAsnGluGluValIleAlaThrIle290295300PheLysGluAsnProLysLysPhePheSer305310当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1