用于治疗脑转移瘤的方法与流程
优先权的要求
本申请要求2014年9月19日提交的美国临时申请第62/052,966号的优先权,在本文中其全部内容并入本文以供参考。
资助信息
本发明在美国国立卫生研究院(nationalinstitutesofhealth)颁发的基金号为p01-ca129243和p30-ca008748,以及美国国防部(departmentofdefense)(dod)颁发的的基金号为w81xwh-12-0074的政府资助下进行的。政府对本发明享有一定的权利。
本发明涉及用于治疗脑转移瘤的间隙连接(gapjunction)抑制剂。由此,这些抑制剂可以用在治疗癌症患者的方法中。
背景技术:
成年人中在20-40%的晚期癌症中发生脑转移瘤,并且代表着最常见的颅内恶性肿瘤(gavrilovic和posner,2005;maher等人,2009)。肺癌和乳腺癌是最常见的来源。尽管在其他转移位点有治疗进展,但是目前的脑转移瘤临床管理仅施用有限的疾病控制,并且大多数患者在诊断后不到12个月内死于肿瘤进展(gavrilovic和posner,2005;stelzer,2013)。因此,必须了解该疾病进程之下的机理,从而将其成功地用在合理的治疗策略中。
脑部的独特微环境对转移性癌细胞造成了强大的屏障。最近的进展已经开始揭示负责引发脑转移瘤的复杂的细胞间和分子间相互作用。机理上停留在脑部毛细血管中的循环的癌细胞必须首先跨过构成血脑屏障(bbb)的加固的血管壁(eichler等人,2011)。已经确认了在实验模型中介导癌细胞通过bbb外渗并在临床中预测脑转移瘤的基因(bos等人,2009;li等人,2013)。一旦进入脑实质中,转移的细胞与微血管保持相关联(kienast等人,2010;lorger和felding-habermann,2010)。癌细胞中细胞粘附分子l1cam的表达介导它们与外腔毛细血管基底膜的紧密粘附,作为对于引发转移性过度增生的要求(valiente等人,2014)。wnt是支持过度增生的信号转导通路之一(nguyen等人,2009)。然而,绝大多数渗入脑部的癌细胞死亡(chambers等人,2002;heyn等人,2006;kienast等人,2010),并且它们被脑中最丰富的一类细胞(星形胶质细胞)所排斥(valiente等人,2014)。
功能多效性的星形胶质细胞在严格的代谢需求下维持bbb、协调神经与血管的偶联、维持组织的体内稳态(homeostasis)(oberheim等人,2012),并针对类似于损伤或者浸润性细胞的紊乱进行精准的反应(pekny和nilsson,2005)。反应性星形胶质细胞生成纤溶酶(plasmin),其调动促凋亡因子fasl以杀灭浸润性癌细胞(valiente等人,2014)。此外,纤溶酶剪切癌细胞中细胞表面的l1cam以抑制其拉拢血管系统(vasculature)的能力(valiente等人,2014)。为了逃避星形胶质细胞的攻击,来自乳腺癌和肺癌的脑转移瘤细胞表达出纤溶酶原激活子(pa)的丝氨酸蛋白酶抑制剂(valiente等人,2014)。尽管这些观察表明星形胶质细胞保护脑部免受转移侵袭,也有证据表明星形胶质细胞在转移中的作用可能并非一致地拮抗性。在体外,星形胶质细胞共培养物保护黑色素瘤细胞系免受化疗药影响(kim等人,2011),并且在体内,星形胶质细胞能够活化癌细胞中的notch信号转导(xing等人,2013)。
技术实现要素:
本发明涉及通过抑制间隙连接功能性来治疗脑转移瘤的方法。其至少部分地基于以下发现:表达原钙粘附蛋白7(protocadherin7)和连接蛋白43(connexin43)的癌细胞与星形胶质细胞形成间隙连接促进了脑转移瘤的生长,并且抑制原钙粘附蛋白7和/或连接蛋白43在癌细胞中的表达减少了脑转移瘤的进展。其还基于以下发现:借助间隙连接抑制剂托那博沙(tonabersat)和甲氯芬那酸盐(meclofenamate)的治疗抑制了脑转移瘤病变的进展,并增强了常规化疗剂卡铂的抗肿瘤活性。
某些非限制性的实施方式提供一种用于治疗患有癌症的受试者的方法,其包括向受试者施用一定量的抑制癌症在脑中的转移进展的间隙连接抑制剂。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂(carboplatin)。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种用于抑制受试者脑中转移癌细胞的生长和/或存活的方法,其包括用治疗有效量的间隙连接抑制剂治疗受试者。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种用于治疗患有癌症的受试者中的脑转移瘤的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种在患有癌症的受试者中预防癌症转移至脑部的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种在患有癌症的受试者中降低癌症向脑部的可检测转移的风险的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用能在脑中保持治疗水平的治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种在患有癌症的受试者中降低癌症向脑部的可检测转移的风险的方法,其包括向受试者施用治疗有效量的原钙粘附蛋白7抑制剂。在具体的非限制性实例中,原钙粘附蛋白7抑制剂是干扰rna。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种用于延长患有癌症的受试者的存活时间的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,例如,其中施用间隙连接抑制剂抑制癌症在脑中的转移进展。在具体的非限制性实例中,间隙连接抑制剂是连接蛋白43抑制剂或原钙粘附蛋白7抑制剂或其组合。在具体的非限制性实例中,抑制剂是托那博沙或甲氯芬那酸盐或其组合。在具体的非限制性实例中,癌症是乳腺癌或肺癌,和/或受试者的癌细胞表达连接蛋白43和/或原钙粘附蛋白7。在具体的非限制性实例中,该方法还包括向受试者施用治疗有效量的抗癌剂,比如,但不限于卡铂。当采用发明的方法时,受试者在治疗前可以已知患有一种或多种脑转移瘤,或者可选地,不知道患有脑转移瘤。
某些非限制性的实施方式提供一种通过测量cgamp的水平来评价间隙连接活性(例如评估抑制性)的试验,其中cgamp的降低与间隙连接抑制性相关联。具体的非限制性实施方式提供一种用于抑制受试者脑中转移性癌细胞的生长和/或存活的方法,其包括用治疗有效量的间隙连接抑制剂治疗受试者,相对于不存在该量的间隙连接抑制剂下的cgamp水平,该治疗有效量的抑制剂产生cgamp水平的降低。另一个非限制性实施方式提供一种确定受试者中性脑肿瘤或者转移性脑肿瘤是否将从借助间隙连接抑制剂的治疗中获得治疗益处的方法,其包括确定将来自所述肿瘤的样品暴露于间隙连接抑制剂是否导致cgamp的降低,其中cgamp的降低表示治疗益处。
附图说明
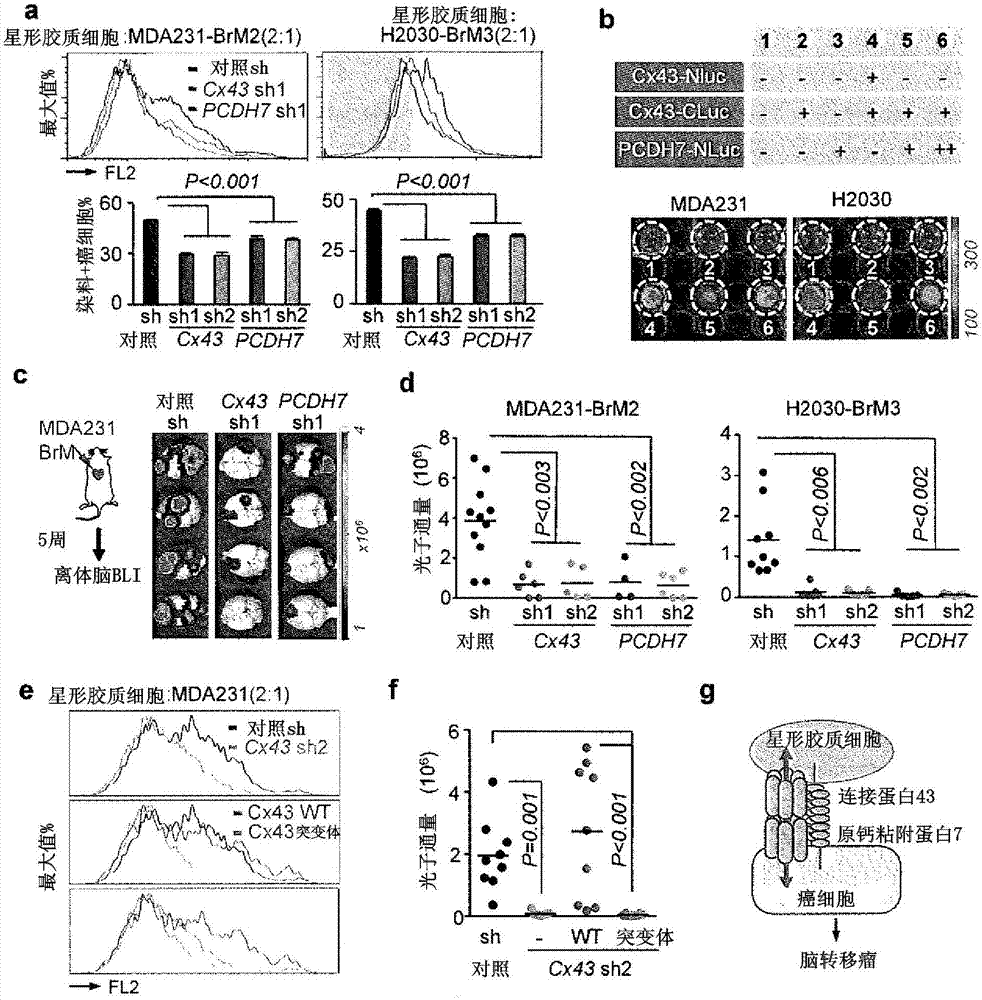
图1a-k.与脑转移瘤相关的cx43和pcdh7。(a)在小鼠心内接种后早期(7天)和后期(21天)的时间点处,在脑实质中gfp+h2030-brm3细胞(绿色)被gfap+激活的星形胶质细胞(红色)所包围。蓝色,容器中的胶原蛋白iv(coliv)染色。比例尺:10μm。(b)在gfp+h2030-brm3(绿色)和gfap+星形胶质细胞(蓝色)的界面处的cx43染色(箭头)。比例尺:10μm。(c)来自三阴性乳腺癌和非小细胞肺癌的人类脑转移瘤样本中的cx43染色的代表性图像。在原发性(1ry)肿瘤、脑转移瘤和正常肺部组织中定量cx43阳性样品的比例。比例尺:100μm。(d)在来自非小细胞肺癌患者的匹配的原发瘤和脑转移瘤样本中的cx43免疫染色的代表性图像以及定量。比例尺:100μm。(e)染料转移试验的示意性说明。(f)染料从星形胶质细胞向癌细胞的转移的定量。柱状图显示了亲本(par)和brm细胞中的红色荧光信号。所有值均为平均值±s.e.m.(n=3个生物重复)。n=3次独立实验。(g-i)在所示的亲本和脑转移瘤衍生物中((g)n=3次独立实验)、在与脑细胞类型相比的脑转移瘤细胞中((h)n=2次独立试验),以及在转移至脑、肺(lm)或骨(bom)的mda231衍生物中((i)n=2次独立实验)的cx43和pcdh7蛋白质免疫印迹分析。(j-k)基于在原发性肿瘤中的cx43/pcdh7表达,在189例三阴性乳腺癌(j),以及129例(mskcc组2)和58例(gse3141)肺腺癌(adenocarcinoma)(k)中的累积无脑转移瘤存活的kaplan-meier曲线。
图2a-g.cx43/pcdh7癌-星形胶质细胞间隙连接介导脑转移瘤。(a)染料从星形胶质细胞到对照以及cx43缺失或pcdh7缺失的脑转移瘤细胞的柱状图(顶部)和定量(底部)。所有值均为平均值±s.e.m(n=3个生物重复)。n=3次独立实验。(b)检测cx43-pcdh相互作用的荧光素酶互补试验。nluc和cluc:n末端和c末端半个萤火虫荧光素酶。表(顶部)以数字确定试验中使用的细胞系组合和(底部)代表性板的生物发光成像(bli)。由对照、cx43缺失或pcdh7缺失的脑转移细胞形成的脑转移性病变的bli(c)和定量(d)。n=3次独立实验。(e、f)野生型(wt)或t154a突变体(mut)cx43在cx43缺失的mda231-brm2细胞(cx43sh2)中重新表达。将细胞进行星形胶质细胞染料转移分析((e)n=3次独立实验)或进行脑转移瘤测定和bli定量((f)n=2次独立实验)。(g)脑转移瘤中cx43和pcdh7介导的癌细胞和星形胶质细胞之间相互作用的概述。
图3a-i.间隙连接激活癌细胞中的stat1和nf-rb通路。(a)与星形胶质细胞共培养后,来自mda231-brm2细胞的trap-seq数据的信号转导通路分析。在多核糖体免疫沉淀和mrna测序之前,将对照(ctrl)或表达l10a-gfp核糖体蛋白融合物的cx43-缺失的mda231-brm2细胞与星形胶质细胞共培养24小时。热力图描绘了蓝色(下调)和红色(上调)通路。n=2个生物重复。(b、c)在借助来自星形胶质细胞共培养物的条件培养基(cm)孵育2小时后在mda231-brm2细胞中stat1和nf-κbp65的磷酸化。在星形胶质细胞与对照或者cx43缺失的mda231-brm2细胞共培养24小时之后收集cm(b),或者从用野生型cx43(wt)或cx43(t154a)突变体(mut)转导的cx43缺失的mda231-brm2细胞收集(c)。n≥3次独立实验。(d)来自星型胶质细胞与指定的mda231-brm2细胞共培养物的cm中的ifnα和tnfα的elisa。所有值均为平均值±s.e.m.(n=4次技术重复)。n≥2次独立实验。(e)在与mda231-brm2癌细胞共培养后重新分离的星形胶质细胞中ifna和tnfa的相对mrna水平。所有值均为平均值±s.e.m.(n=3次生物重复)。n=2次独立实验。(f)在存在或不存在10单位/ml(39单位/ng)的ifnαa或10pg/ml的tnfα的情况下,用各种浓度的卡铂(carbo)治疗的mda231-brm2细胞中剪切的半胱天冬酶3的相对水平。所有值均为平均值±s.e.m.(n=5次技术重复)。n=3次独立实验。(g)在对照和stat1敲减的mda231-brm2细胞中的stat1水平。(h)表达对照pbabe或sr-iκbα载体的mda231-brm细胞中的nf-κb海肾荧光素酶报告子测定。所有值均为平均值±s.e.m.(n=3次技术重复)。(i)由对照、stat1敲减、和sr-irbαmda231-brm2细胞形成的脑转移瘤中的bli信号定量。n=2次独立实验。
图4a-h.间隙连接介导星形胶质细胞中的细胞溶质dsdna应答。(a)将表达对照shrna(ctrlsh)或靶向cx43的shrna的mda231-brm2细胞在有或者没有星形胶质细胞的情况下培养18小时,并进行磷酸化的tbk1和irf3的免疫印迹分析(n=3次独立实验)。(b)为了样品制备和通过lc-ms/ms的cgamp分析收集单独的mda231-brm2、单独的星形胶质细胞或18小时共同培养物。直方图(右)对应于(左)中的标准化的cgamp峰,并且代表5个生物重复。n=3次独立实验。还参见图16。(c)irf3和gfp的双重免疫荧光染色的代表性图像。dapi:细胞核染色。在共培养物中:白色箭头:irf3在星形胶质细胞中的积累;绿色箭头:均匀分布在gfp+mda231-brm2细胞中的irf3。比例尺:20μm。n=2次独立实验。(d)来自2×107个细胞的指定细胞级分中的dsdna的定量。值是平均值±s.e.m.(n=3次生物重复)。n=2次独立实验。(e)mda231-brm2细胞中dsdna、gfp和coxiv(线粒体标志物)的免疫荧光染色的代表性图像。dapi:细胞核染色。比例尺:10μm。n=2次独立实验。(f、g)将edu标记的mda231-brm2细胞与星形胶质细胞共培养6小时。使用共聚焦显微镜将edu标记的dna从癌细胞到星形胶质细胞的转移可视化(f),或通过流式细胞仪定量(g)。癌细胞和星形胶质细胞分别由绿色和白色虚线描绘。比例尺:10μm。值是平均值±s.e.m.(n=3次生物重复,n=2次独立实验)。(h)间隙连接介导的抗dsdna应答、星形胶质细胞中ifnα和tnfα的产生、以及随后癌细胞中stat1和nf-κb通路的激活以支持脑转移瘤的示意性概括。
图5a-i.间隙连接活性的抑制控制脑转移瘤过度增生。(a)在指定浓度的托那博沙或甲氯芬那酸盐存在下,从星形胶质细胞向mda231-brm2细胞的染料转移。n≥3次独立实验。(b)在具有指定浓度的托那博沙(tona)或甲氯芬那酸盐(meclo)存在下,在来自mda231-brm2细胞和星形胶质细胞共培养的条件培养基中的ifnα和tnfα的elisa。所有图表均为平均值±s.e.m.(n=4次技术重复)。n=2次独立实验。(c)在小鼠接种癌细胞后1天,开始每天施用托那博沙或甲氯芬那酸盐。基于bli定量脑转移瘤病变。n=2次独立实验。(d)14天脑转移瘤病变的gfp染色。代表性的图像显示出大的进行性病变。dapi:细胞核染色。比例尺:40μm。n=10只实验小鼠。(e)在用可诱导型对照、cx43或pcdh7shrna转导的mda231-brm2细胞接种后14天,用多西环素和卡铂治疗小鼠,如方案所示。基于bli定量脑转移瘤病变。(f、g)与离体脑bli和红色荧光成像相匹配的代表性图像。n=2次独立实验。(h)接种mda231-brm2细胞14天后,用托那博沙、甲氯芬那酸盐和卡铂治疗小鼠。按照指定的方案,基于bli定量脑转移瘤病变。n=2次独立实验(i)。
图6a-d.癌细胞与星形胶质细胞相互作用。(a)本研究中使用的癌细胞。(b)星形胶质细胞共培养保护癌细胞。如方案(左)所示,在sfasl治疗或化疗治疗后定量剪切的半胱天冬酶3+/gfp+凋亡的brm细胞。n=3次独立实验。(c、d)星形胶质细胞和brm细胞之间的间隙连接通讯。染料从mda231-brm2细胞转移到星形胶质细胞的延时观察(time-lapse)图像(c)。比例尺:100μm。通过流式细胞仪随着时间的推移定量染料从星形胶质细胞到mda231-brm2细胞的转移(d)。n=3次独立实验。
图7.cx43和pcdh7在脑转移瘤的癌细胞和星形胶质细胞中的表达升高。(a)亲本(par)和brm细胞中的cx43和pcdh7mrna。值是平均值±s.e.m.(n=3次技术重复)。n=3次独立实验。(b)erbb2亲本和脑细胞以及kras/p53细胞系中的cx43和pcdh7蛋白质印迹分析。n=3次独立实验。(c)与脑细胞相比,brm细胞中的cx43和pcdh7mrna。n=3次独立实验。(d)mda231亲本(par)和脑(brm2)、肺(lm)和骨(bom)的转移性衍生物中的cx26和cx30mrna。(e)kaplan-meier曲线显示基于cx43/pcdh7在原发性肿瘤中的表达,63例肺腺癌(gse8893)中累积的无转移存活概率。(f、g)如通过rt-pcr(f)和蛋白质印迹分析(g)评估的cx43和pcdh7用短发夹rna(shrna)的敲减。ctrl:对照。值是平均值±s.e.m.(n=3次技术重复)。n=3次独立实验。
图8a-h.pcdh7促进间隙连接通讯。(a、b)从星形胶质细胞到对照和cx43缺失的或pcdh7缺失的kras/p53-393n1细胞的染料转移的柱状图和定量(a),以及与生胃酮(carbenoxolone)(50μm)治疗相比,从星形胶质细胞到对照或cx43缺失的mda231-brm2细胞的染料转移的柱状图和定量(b)。(c、d)星形胶质细胞中的pcdh7促进间隙连接。在对照或pcdh7缺失的星形胶质细胞中的pcdh7蛋白质印迹分析(c)。从mda231-brm2细胞到pcdh7缺失的星形胶质细胞的染料转移的定量(d)。(e)从人脑微血管内皮细胞(hbmec)到对照、cx43缺失的或pcdh7缺失的mda231-brm2细胞的染料转移的定量。(f)从mda231-brm2细胞向星形胶质细胞与hbmec的混合群体的染料转移。(g)从对照或cx43缺失的mda231-brm2细胞到人小神经胶质细胞的染料转移的定量。(h)如方案所示,小神经胶质细胞、星形胶质细胞或癌细胞24小时共培养后,检测通过transwell分离的mda231-brm2细胞(左)或星形胶质细胞(右)中的cx43mrna。对于染料转移试验,值是平均值±s.e.m.(n=3次生物重复)。n≥2次独立实验。在h中,值是平均值±s.e.m.(n=4次生物重复)。
图9a-d.cx43与pcdh7直接相互作用,但不与e钙粘附蛋白或n钙粘附蛋白相互作用。(a)在过表达融合蛋白的癌细胞中cx43和pcdh7的蛋白质免疫印迹分析。(b)cx43-cluc/pcdh7-nluc(+)癌细胞和星形胶质细胞共培养15分钟后bli的定量。c-e,检测cx43-e钙粘附蛋白或cx43-n钙粘附蛋白相互作用的萤光素酶分裂试验。nluc和cluc:n末端和c末端半个萤火虫荧光素酶。表(c)以数字确定试验中使用的细胞系组合,蛋白质免疫印迹分析(d)表明过表达融合蛋白的癌细胞中的e或n钙粘附蛋白表达,以及代表性板的生物发光成像(bli)(e)。n≥2次独立实验。
图10a-e.间隙连接活性的抑制阻止脑转移瘤过度增生。(a)由对照(ctrl)、cx43缺失的或pcdh7缺失的kras/p53-393n1细胞形成的脑转移瘤病变的生物发光成像(bli)定量。n=2次独立实验。(b)由对照、cx43缺失的或pcdh7缺失的mda231-brm2细胞形成的gfp+脑转移瘤病变的代表性图像。脑部分或脑转移瘤病变分别用白色虚线或红色虚线表示。比例尺:1000μm。(c)由mda231-brm2细胞形成的肺转移性病变的bli(图像)和定量(柱状图)。值是平均值±s.e.m.(每组n=5只小鼠)。n=2次独立实验。(d、e)间隙连接介导的脑转移瘤需要cx43的通道功能。在cx43缺失的mda231-brm2细胞(cx43sh2)中重新表达野生型(wt)或t154a突变体(mut)cx43。通过蛋白质印迹分析检测cx43表达(d),并用bli定量由这些细胞形成的脑转移瘤(e)。n=2次独立实验。
图11a-d.cx43和pcdh7在脑转移瘤中的作用。(a)cx43和pcdh7不介导转-bbb迁移。在7天脑病变中量化对照(ctrl)、cx43缺失的或pcdh7缺失的mda231-brm2细胞。值是平均值±s.e.m.(每组n=5个脑)。(b)在14天脑病变中cx43和pcdh7介导癌细胞定植。代表性的图像是gfp(绿色)和ki67(红色)染色。dapi:细胞核染色。比例尺:20μm。柱状图是ki67+癌细胞的比例。值是平均值±s.e.m.(每组n=5个脑)。(c)cx43和pcdh7介导癌细胞的存活。脑切片试验:代表性的图像是gfp(绿色)和剪切的半胱天冬酶3(casp3)(红色)染色。比例尺:30μm。直方图是半胱天冬酶3+凋亡癌细胞的比例。值是平均值±s.e.m.(每组n=5个脑切片)。比例尺:30μm。(d)在14天脑病变中cx43和pcdh7不影响癌细胞的血管凝集。代表性的图像是gfp(绿色)染色和用tritc葡聚糖填充的血管结构(红色)。比例尺:20μm。n=2次独立实验。
图12a-d.翻译核糖体亲和纯化(trap)和细胞因子阵列。(a)在3个条件(#1、#2、#3)下,建立trap实验以从mda231-brm2细胞将翻译mrna分离的示意性说明。(b)trapmrna测序的主要组分(pc)分析。(c)由brm细胞和星形胶质细胞之间的间隙连接通讯和星形胶质细胞调节的log2倍数变化的散点图。(d)在来自星形胶质细胞共培养物的条件培养基(cm)中孵育2小时后h2030-brm3细胞中的stat1和nf-κbp65磷酸化。在将星形胶质细胞与对照或cx43缺失的h2030-brm3细胞共培养24小时后收集cm。n=3次独立实验。
图13a-f.间隙连接产生的信号转导激活癌细胞中的ifn和nf-κb通路。(a)在星形胶质细胞与对照或cx43缺失的mda231-brm2细胞共培养24小时后收集的条件培养基的细胞因子阵列分析。绘制log2倍数变化。(b)来自星形胶质细胞与指示的h2030-brm3细胞的共培养物的cm中的ifnα和tnfα的elisa。所有值均为平均值±s.e.m.(n=4次技术重复)。n=2次独立实验。(c)在存在或不存在10单位/ml(39单位/ng)的重组ifnαa或10pg/ml的重组tnfα的情况下,用各种浓度的紫杉醇治疗的h2030-brm3细胞中剪切的半胱天冬酶3的相对水平。所有值均为平均值±s.e.m.(n=5次技术重复)。n=3次独立实验。(d、e)在对照和stat1敲减的h2030-brm3细胞中的stat1水平。(f)来自由对照、stat1敲减的细胞形成的脑转移瘤的bli信号的定量。n=2次独立实验。
图14a-g.间隙连接引发星形胶质细胞中的细胞溶质dna应答。(a)对照或cx43缺失的h2030-brm3细胞与或不与星形胶质细胞共培养18小时,并进行磷酸化的tbk1和irf3的免疫印迹分析(n=3次独立实验)。(b)cgamp鉴定。4.47分钟处的峰包含cgamp特有的所有3个srm转换(transition)。rt:保留时间,aa:自动整合峰面积。(c)在来自2×107个h2030-brm3细胞的指定细胞级分中的dsdna的定量。值是平均值±s.e.m.(n=3次生物重复)。n=2次独立实验。(d)在指定的癌细胞和非肿瘤细胞中细胞溶质dsdna和细胞核dsdna的比例。(e)h2030-brm3细胞中dsdna、gfp、coxiv(线粒体标志物)免疫荧光染色的代表性图像。dapi:细胞核染色。比例尺:10μm。(f)星形胶质细胞中dsdna、coxiv(线粒体标志物)免疫荧光染色的代表性图像。dapi:细胞核染色。鬼笔环肽:细胞骨架染色。比例尺:10μm。(g)将edu标记的h2030-brm3细胞与星形胶质细胞共培养6小时。使用共聚焦显微镜使edu标记的dna从癌细胞到星形胶质细胞转移可见化。癌细胞或星形胶质细胞分别通过绿色或白色虚线描绘。比例尺:10μm。n=2次独立实验。
图15.间隙连接活性的抑制防止脑转移瘤过度增生。(a-d)用托那博沙(tona)或甲氯芬那酸盐(meclo)治疗(a)后,脑转移瘤(b)、乳腺脂肪垫中的原发性肿瘤生长(c)或肺转移瘤(d)通过bli定量。值是平均值±s.e.m.(每组n=5只小鼠)。n=2次独立实验。(e、f)在体外多西环素治疗后,如通过rt-pcr(e)和蛋白质免疫印迹分析(f)所评估的,用tet-on可诱导型短发夹rna(shrna)敲减mda231-brm2细胞中的cx43和pcdh7。n=2次独立实验。(g)接种mda231-brm2细胞14天后的离体脑部生物发光成像(bli)。
图16.cgamp识别的确认。通过lc-ms/ms分析图4b中所示的来自所有实验条件的合并样品。只有4.47分钟处的峰包含cgamp特有的所有3个srm转换。通过向合并样品中加入5μl的40nm环[g(2’,5’)pa(3’,5’)p](cgamp),4.47分钟处的峰增加。作为内部和阴性对照,在4.97分钟峰处,c-双-gmp包含所有2个srm转换,并且通过添加标准cgamp不改变该峰。rt:保留时间,aa:自动整合的峰面积。
具体实施方式
为了清楚且并非通过限制性的方式,将发明的详细描述分为以下部分:
(i)间隙连接抑制剂;
(a)连接蛋白43抑制剂;和
(b)原钙粘附蛋白7抑制剂;
(c)用于间隙连接活性/抑制的试验;
(ii)癌症靶标;
(iii)药物配方;和
(iv)治疗方法。
1间隙连接抑制剂
本发明提供用于公开的方法中的间隙连接抑制剂(例如,间隙连接拮抗剂)。在某些实施方式中,间隙连接抑制剂可以包括抑制和/或减少间隙连接组分的表达和/或活性、或者抑制和/或减少间隙连接的形成、开放、信号转导和/或活性的化合物、小分子、化学制品、多肽、核酸和蛋白质。
在某些非限制性实施方式中,小分子间隙连接抑制剂包括生胃酮(carbenoxolone)、甘草次酸(glycyrrhetinicacid)、奎宁(quinine)、奎尼丁(quinidine)、甲氟喹(mefloquine)、庚醇、辛醇、花生四烯酸乙醇胺(anandamide)、灭酸酯类(fenamates)、2-氨基乙氧基-二苯基-硼酸酯(2-apb)、视黄酸(retinoicacid)、油酰胺(oleamide)、精胺(spermine)、氨基磺酸盐类、丙酸钠、托那博沙(tonabersat)和甲氯芬那酸盐(meclofenamate)(甲氯芬那酸)。间隙连接抑制剂另外的非限制性实例在美国专利第5,843,989、6,211,211、7,632,866、6,251,931、7,704,946号和pct专利申请号wo1999/026584中公开。
在某些实施方式中,间隙连接抑制剂包括具有以下结构的式i化合物:
在某些实施方式中,间隙连接抑制剂包括具有以下结构的式ii化合物:
在某些实施方式中,间隙连接抑制剂包括具有以下结构的式iii化合物:
在某些非限制性实施方式中,间隙连接抑制剂可以为式i-iii化合物的盐、立体异构体、类似物或衍生物的形式。例如,但并非通过限制的方式,间隙连接抑制剂可以包括式ii的钠盐形式。
在某些非限制性实施方式中,间隙连接抑制剂可以为能够部分或全部地阻断间隙连接的形成和/或细胞之间的间隙连接的开放、间隙连接的信号转导和/或活性的抗体或者抗体片段。参见,例如emestooviedo-orta等人,thefasebjournal,vol.15:768-774(2001)。在某些非限制性实施方式中,间隙连接抑制剂可以是抗-连接蛋白化合物和/或连接蛋白模拟肽。参见,例如evans和boitano,biochem.soc.trans.,vol.29(4):606-612(2001);dahl,biophys.j.,vol.67(5):1816-1822(1994);欧洲专利申请第ep2510939和ep2252320号,以及美国专利申请第2009/0142295号。
间隙连接抑制剂的另一个非限制性实例包括特异性地抑制和/或减少间隙连接组分的表达或活性的核酶、反义寡核苷酸、短发夹rna(shrna)分子和sirna分子。“核酶”是指能够剪切特异性核酸序列的核酸。在某些非限制性实施方式中,核酶是指包含用于特异性识别的反义序列以及rna剪切酶活性的rna分子,参见,例如美国专利第6,770,633号。相反地,“反义寡核苷酸”通常为与基因的一部分互补以影响该基因表达的小的寡核苷酸。能够通过将寡核苷酸杂交至其特异性基因或信使rna(mrna)而抑制基因表达。使用反向技术以特异性地抑制已知序列的基因的基因表达的方法是本领域熟知的(例如,参见美国专利第6,566,135、6,566,131、6,365,354、6,410,323、6,107,091、6,046,321和5,981,732号)。“小干扰rna”或“短干扰rna”或“sirna”或“短发夹rna”或“shrna”是rna干扰(rnai)的形式。干扰rna可以是与靶标核酸序列互补的双链rna或部分双链的rna分子。微小rna(mirna)也可以落入此分类中。对本发明的寡核苷酸的各种修饰,例如,反义基因(antisense)、shrna或sirna分子能够作为提高细胞内稳定性和半衰期的手段被引入。此类修饰的非限制性实例包括向分子的5’和/或3’末端加入核糖核酸或脱氧核糖核酸的侧翼序列,或者在寡核苷酸主链中使用非典型的或者非天然存在的残基,比如硫代磷酸酯或2’-o-甲基,而不是磷酸二酯酶(phosphodiesterase)键合。
发明的rna分子可以由载体表达或者化学地或合成地制备。对已知序列的基因而言,选择适当的dsrna或dsrna-编码载体的方法是本领域熟知的(例如,参见tuschl,t.等人(1999);elbashir,s.m.等人(2001);hannon,gj.(2002);mcmanus,mt.等人(2002);brummelkamp,tr.等人(2002);美国专利申请第6,573,099和6,506,559号;以及pct专利申请第wo2001/036646、wo1999/032619和wo2001/068836号)。
1.1连接蛋白43抑制剂
在某些非限制性实施方式中,间隙连接抑制剂可以是对间隙连接组分特异性的。例如,并且并非通过限制性方式,间隙连接组分包括蛋白质的连接蛋白家族。连接蛋白的非限制性实例为连接蛋白43(cx43),其由基因间隙连接蛋白质α1(gja1)所编码。cx43核酸或蛋白质可以是具有在ncbi数据库登录号第nm_000165、ng_008308或m65188号中所示的序列的人cx43核酸,或者编码具有ncbi数据库登录号第np_000156号中所示的氨基酸的人cx43蛋白质分子的核酸。根据本发明,此类cx43核酸和/或蛋白质的表达和/或功能的抑制剂可以被用作间隙连接抑制剂。例如,并且并非通过限制性方式,间隙连接抑制剂可以包括cx43抑制剂,比如,但不限于,碘苯腈或碘苯腈辛酸酯。在某些实施方式中,cx43抑制剂可以包括cx43抗体、抗体片段或模拟肽(参见danesh-meyer等人,brain,135:506-520(2012))。
间隙连接抑制剂的一个非限制性实例包括与以上公开的cx43核酸序列至少一部分同源的反义基因、shrna或sirna核酸序列,其中该部分与cx43序列的同源性至少为约75%,或者至少为约80%,或者至少为约85%,或者至少为约90%,或者至少为约95%,或者至少为约98%,其中同源性百分比可以通过例如blast或fasta软件进行确定。在某些非限制性实施方式中,互补部分可以由至少10个核苷酸、或者至少15个核苷酸、或者至少20个核苷酸、或者至少25个核苷酸、或者至少30个核苷酸组成,并且反义核酸、shrna或sirna分子在长度上可以为至多15个、或者至多20个、或者至多25个、或者至多30个、或者至多35个、或者至多40个、或者至多45个、或者至多50个、或者至多75个、或者至多100个核苷酸。抑制cx43的shrna的非限制性实例在以下实施例中示出。在非限制性实施方式中,作为核酸的cx43抑制剂可以在表达cx43的癌细胞中通过载体(例如,通过慢病毒)提供,其可以选择性地靶向所述癌细胞,并且/或者其中在肿瘤细胞中有选择性活性的启动子引导cx43抑制剂核酸的表达。
1.2原钙粘附蛋白7抑制剂
本发明提供用于所公开的方法中的原钙粘附蛋白7(pcdh7)抑制剂。pcdh7抑制剂的非限制性实例包括抑制和/或减少pcdh7的表达和/或活性的化合物、分子、化学制品、多肽、蛋白质。pcdh7核酸或蛋白质可以为具有ncbi数据库登录号第nm_001173523、nm_032457、nm_032456或nm_002589号所示的序列的人pcdh7核酸,或者编码具有ncbi数据库登录号第np_001166994、np_115832、np_115833或np_002580号所示的氨基酸的人pcdh7蛋白质分子的核酸。
在某些非限制性实施方式中,pcdh7抑制剂可以包括特异性抑制和/或减少pcdh7的表达或活性的核酶、反义寡核苷酸、shrna分子和sirna分子。pcdh7抑制剂的一个非限制性实例包括与pcdh7核酸序列的至少一部分同源的反义基因、shrna或sirna核酸序列,其中该部分相对于pcdh7序列的同源性为至少约75%、或者至少约80%、或者至少约85%、或者至少约90、或者至少约95%、或者至少约98%,其中同源性百分比可以通过例如blast或fasta软件进行确定。在某些非限制性实施方式中,互补部分可以由至少10个核苷酸、或者至少15个核苷酸、或者至少20个核苷酸、或者至少25个核苷酸、或者至少30个核苷酸组成,并且反义核酸、shrna或sirna分子在长度上可以为至多15个、或者至多20个、或者至多25个、或者至多30个、或者至多35个、或者至多40个、或者至多45个、或者至多50个、或者至多75个、或者至多100个核苷酸。在某些实施方式中,本发明的反义基因、shrna或sirna分子可以包括如上所公开的dna或者非典型或非天然存在的残基,例如,但并非限制性的,硫代磷酸酯残基。抑制pcdh7的shrna的非限制性实例在以下实施例中示出。在非限制性实施方式中,作为核酸的pcdh7抑制剂可以在表达pcdh7的癌细胞中通过载体(例如,通过慢病毒)提供,其可以选择性地靶向所述癌细胞,并且/或者其中在肿瘤细胞中有选择性活性的启动子引导pcdh7抑制剂核酸的表达。
在非限制性实施方式中,pcdh7抑制剂可以为与pcdh7特异性结合的抗体或者抗体片段或者单链抗体。此类抗体的非限制性实例包括ab555066(abcamlnc.)和hpa011866(sigma-aldrich)。在某些非限制性实施方式中,抗-pcdh7抗体或抗体片段可被用于制备对根据本发明所用的pcdh7有特异性的人、人源化的或别的嵌合的抗体。
1.3用于间隙连接活性/抑制性的试验
发明的某些非限制性实施方式提供一种用于通过测量环一磷酸鸟苷-一磷酸腺苷(例如[g(2’,5’)pa(3’,5’)p](“cgamp”))的水平评价间隙连接活性(例如评估抑制性)的试验,其中cgamp的减少与间隙连接抑制性相关联。发明的这一方面至少部分地基于以下发现:当在星形胶质细胞和已经转移至脑部的癌细胞之间形成间隙连接时,cgamp增加,并且所述升高的cgamp随着连接蛋白43抑制而降低(参见,例如,图4b和14b)。
具体的非限制性实施方式提供一种用于抑制受试者脑中转移性癌细胞生长和/或存活的方法,其包括用治疗有效量的间隙连接抑制剂治疗受试者,相对于不存在该量的间隙连接抑制剂下的cgamp水平,治疗有效量的间隙连接抑制剂产生cgamp的降低。
具体的非限制性实施方式提供一种确定受试者中脑肿瘤或转移性脑肿瘤是否将从借助间隙连接抑制剂的治疗中获得治疗益处的方法,其包括确定将来自所述肿瘤的样品暴露于间隙连接抑制剂是否导致cgamp的降低,其中cgamp中的降低表示治疗益处。
发明的另一个非限制性实施方式提供一种抑制受试者脑中转移性癌细胞的生长和/或存活的方法,其包括:(i)确定受试者是否将从借助间隙连接抑制剂的治疗中获得治疗益处,这包括确定相对于不存在抑制剂时的cgmap水平,在暴露于间隙连接抑制剂时受试者的癌细胞(其可以获得自脑转移瘤、原发肿瘤或脑外部的转移肿瘤)是否显示出cgamp的降低,其中cgamp的降低表示治疗益处;以及(ii)如果观察到cgamp的降低,则用间隙连接抑制剂治疗受试者,或者如果未观察到cgamp的降低,则测定另一种间隙连接抑制剂在肿瘤细胞中降低cgamp的能力,或者用另一种方式(modality)治疗受试者,比如化疗、免疫疗法、放疗等。所述测定可以使用例如在以下操作实施例中所述的体外(invitro)试验、或者现有技术中已知的类似的cgamp测量体系来实施。
发明的另一个非限制性实例提供一种抑制受试者中脑肿瘤生长的方法,其包括:(i)确定受试者是否将从借助间隙连接抑制剂的治疗中获得治疗益处,这包括确定相对于不存在抑制剂时的cgmap水平,在暴露于间隙连接抑制剂时受试者的肿瘤细胞是否表现出cgamp的降低,其中cgamp的降低表示治疗益处;以及(ii)如果观察到cgamp的降低,则用间隙连接抑制剂治疗受试者,或者如果未观察到cgamp的降低,则测定另一种间隙连接抑制剂在肿瘤细胞中降低cgamp的能力,或者用另一种方式治疗受试者,比如化疗、免疫疗法、放疗等。所述测定可以使用例如在以下操作实施例中所述的体外(invitro)试验、或者现有技术中已知的类似的cgamp测量体系来实施。
cgamp可以通过现有技术已知的任何方法测量。在发明的某些非限制性实施方式中,cgamp水平是通过液相色谱-质谱/质谱(“lc-ms/ms”)测量的。可以将lc-ms/ms标准化为内部标准物(例如,以计算在纯化步骤中的任何损耗)。作为一个特定的非限制性实例,在以下操作实施例中“通过lc-ms/ms的cgamp定量”部分描述的试验并入本详细说明中以供参考。还参见图16。
在某些非限制性实施方式中,本发明提供一种用在所述试验中的试剂盒,其包括至少一种cgamp标准物,以及关于在脑肿瘤中借助间隙连接抑制性降低cgamp的信息。
在某些非限制性实施方式中,本发明提供一种用于检测样品中存在的cgamp的量的试剂盒。在某些实施方式中,试剂盒可以包括同位素标记的cgamp。例如,并且并非通过限制性方式,同位素标记的cgamp在分析化学技术中例如在质谱(ms)和液相色谱(lc)-ms/ms中可被用作内部对照。在某些实施方式中,同位素标记的cgmap可以借助低丰度的稳定同位素比如但不限于2h(氘)、13c(碳-13)、15n(氮-15)或18o(氧-18))进行富集。
在某些非限制性实施方式中,本发明的试剂盒还可以包括用于使用该试剂盒检测样本中的cgamp的量的操作说明。例如,并且并非通过限制性的方式,该操作说明可以记载在分析之前向样品加入的同位素标记的cgamp的量。在某些实施方式中,该操作说明还能记载如何从加入至样品的同位素标记的cgamp的量来计算样品中的cgamp的量。在某些非限制性实施方式中,该操作说明能记载:相对于参考对照水平,在来自对间隙连接抑制剂应答的受试者的样品中的cgamp的量或水平的减少,其表示来自使用间隙连接抑制剂的治疗益处。
2肿瘤靶标
在某些实施方式中,本发明提供一种用于治疗脑转移瘤的方法。如本文使用的“转移瘤”是指在与癌症(例如,原发性癌症)的初始部位并非身体上邻近的部位存在一种或多种癌细胞。例如,并且并非通过限制性的方式,癌症可以包括肺癌、乳腺癌、黑色素瘤、结肠癌、肾癌、肾细胞癌、间皮瘤、卵巢癌、胰腺癌、肉瘤、白血病、淋巴瘤、尿路上皮癌、头颈癌、骨肉瘤和膀胱癌。在某些实施方式中,癌症可以包括成胶质细胞瘤和星形胶质细胞瘤。
“可检测的”转移瘤是可通过磁共振成像、计算机x射线断层成像或正电子发射断层成像识别的一簇细胞。在某些非限制性实施方式中,一簇转移细胞可以包括至少约1×107个细胞。在某些实施方式中,可检测的转移瘤可以包括一簇尺寸大于约5mm或约10mm的细胞。
3药物配方
在某些非限制性实施方式中,本发明提供以上在1部分公开的间隙连接抑制剂的药物配方用于治疗用途。在某些实施方式中,药物配方包括间隙连接抑制剂和药学上可接受的载体。
如本文所用,“药学上可接受的”包括不干扰活性成分(例如,抑制剂)的生物活性效用的任何载体,并且其对施用的患者而言是无毒的。合适的药物载体的非限制性实例包括磷酸盐缓冲溶液、水、乳液(比如油/水乳液)、各种类型的湿润剂和无菌溶液。药学上可接受的载体另外的非限制性实例可以包括凝胶、生物可吸收基材、包括抑制剂和/或任何其他合适赋形物的植入成分、递送或分散手段或材料。此类载体可以通过常规方法进行配制并施用于受试者。
在某些非限制性实施方式中,本发明的药物配方可以用本领域所熟知的、适合于口服给药的药学上可接受的载体进行配制。此类载体允许将药物组合物配制成片剂、丸剂、胶囊、液体、凝胶、糖浆、软膏、混悬剂等,用于使待治疗的患者口腔或鼻腔摄入。在某些实施方式中,药物配方可以是固体剂型。在某些实施方式中,片剂可以是速释片剂。可选地或者另外地,片剂可以是延迟或控制释放的片剂。在某些实施方式中,固体剂型可以既包括速释部分也包括延迟或控制释放部分。在某些实施方式中,本发明的药物配方可以用本领域熟知的适合于肠胃外给药的药学上可接受的载体进行配制。
在某些实施方式中,适用于本发明的药物配方可以包括以下配方,其中以治疗有效量包括活性成分(例如,间隙连接抑制剂)。“治疗有效量”是指能够达到一种或多种抗癌效果、延长存活时间和/或延长直到复发的时间的量。活性成分的治疗有效量可以根据活性成分(例如,间隙连接抑制剂)、采用的配方、癌症及其严重程度、以及待治疗的受试者的年龄、体重等而变化。在某些实施方式中,患者可以以一种或多种配方的单次或多次给药来接受治疗有效量的间隙连接抑制剂,这可以取决于患者所需的和可承受的剂型和频次。
如本文所用的“抗癌效果”和“治疗益处”是指癌细胞团聚集体(aggregatecancercellmass)的减少、癌细胞生长速率的降低、癌细胞增殖的降低、肿瘤质量的降低、肿瘤体积的降低、肿瘤细胞增殖的降低、肿瘤生长速率的降低和/或肿瘤转移的减少中的一种或多种。在某些实施方式中,抗癌效果可以指在诊断有癌症的患者中的完全应答、部分应答、病情稳定(没有进展或复发)和/或具有更晚复发或无进展存活的应答。在某些实施方式中,抗癌效果可以指预防和/或减少受试者中原发性癌症的转移,例如,预防和/或减少受试者中癌症向脑部的转移。
在某些非限制性实施方式中,以上公开的间隙连接抑制剂可单独使用或者与一种或多种抗癌剂联合使用。本文所用的“抗癌剂”可以是具有抗癌效果的任何分子、化合物、化学制品或组合物。抗癌剂包括,但不限于化疗剂、放疗剂、细胞因子类、抗血管生成剂、凋亡诱导剂、抗癌抗体、抗细胞周期蛋白依赖性激酶抑制剂和/或促进免疫系统活性的药剂,包括但不限于,细胞因子类,比如但不限于白细胞介素2、干扰素、抗-ctla4抗体和/或抗-pd-1抗体。抗癌剂的非限制性实例包括紫杉醇(paclitaxel)、替莫唑胺(temozolomide)、长春瑞滨(vinorelbine)、甲基苄肼(procarbazine)、洛莫司汀(lomustine)、长春新碱(vincristine)、sfasl和卡铂。例如,但并非通过限制性的方式,间隙连接抑制剂(例如甲氯芬那酸盐和/或托那博沙)可与卡铂联合使用。如本文所用的“联合”意味着将间隙连接抑制剂和一种或多种抗癌剂作为治疗方案或计划的一部分施用于受试者。在某些实施方式中,联合使用并不需要在给药前将抑制剂与一种或多种抗癌剂在物理上结合或者在相同的时间框内施用。
在某些实施方式中,如果抑制剂与抗癌剂联合使用,在某些情况下每种的量可以小于单独采用该药剂时的治疗有效量,但当共同使用时达到了治疗效果。
4治疗方法
本发明涉及一种通过抑制间隙连接功能性治疗脑转移瘤的方法。如在以下实施例部分详述的,在本申请中呈现的研究表明癌细胞与星形胶质细胞之间的间隙连接的信号转导和/或形成的抑制可以被用于治疗脑转移瘤。这至少部分地基于以下发现:表达原钙粘附蛋白7和连接蛋白43的癌细胞与星形胶质细胞形成间隙连接,其促进了脑转移瘤生长,并且抑制癌细胞中原钙粘附蛋白7和连接蛋白43的表达降低了脑转移瘤的进展。还基于以下发现,借助间隙连接抑制剂托那博沙和甲氯芬那酸盐的治疗抑制脑转移瘤病变的进展,并增强了常规化疗剂卡铂的抗癌活性。
因此,如上所述,本发明提供通过施用间隙连接抑制剂通过抑制间隙连接的信号转导和/或形成来治疗脑转移瘤的方法。间隙连接抑制剂的非限制性实例及其药物配方如在以上1和3部分所述。可以用本发明的方法治疗的癌症在2部分有述。由此,本发明涉及用来抑制间隙连接功能性以在受试者中产生抗癌效果的方法。
在本文中可替换使用的“受试者”或“患者”是指人或非人类受试者。非人类受试者的非限制性实例包括非人类的灵长目动物、狗、猫、小鼠、大鼠、豚鼠、兔、猪、家禽、马、奶牛、山羊和绵羊。
在某些非限制性实施方式中,本发明提供一种治疗患有癌症的受试者的方法,其包括向受试者施用一定量的抑制癌症在脑中的转移进展的间隙连接抑制剂。在某些实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症可以是乳腺癌。在某些实施方式中,癌症可以是肺癌。在某些非限制性实施方式中,受试者的癌症中一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在某些实施方式中,受试者在治疗之前已知患有一种或多种脑转移瘤。在本发明某些非限制性实施方式中,受试者在治疗之前不知道患有脑转移瘤。
在某些实施方式中,治疗患有癌症的受试者的方法包括:向受试者施用一定量的托那博沙,以抑制癌症在脑中的转移进展。
在某些实施方式中,治疗患有癌症的受试者的方法包括:向受试者施用一定量的甲氯芬那酸盐,以抑制癌症在脑中的转移进展。
在某些实施方式中,治疗患有癌症的受试者的方法包括:向受试者施用一定量的cx43抑制剂,以抑制癌症在脑中的转移进展。
在某些实施方式中,治疗患有癌症的受试者的方法包括:向受试者施用一定量的pcdh7抑制剂,以抑制癌症在脑中的转移进展。
在某些非限制性实施方式中,本发明还提供一种用于抑制受试者脑中的转移性癌细胞的生长和/或存活的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症是肺癌和/或乳腺癌。在某些非限制性实施方式中,受试者的癌症的一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在某些实施方式中,受试者在治疗之前已知患有一种或多种脑转移瘤。在本发明某些非限制性实施方式中,受试者在治疗之前不知道患有脑转移瘤。
在某些实施方式中,用于抑制受试者脑中的转移性癌细胞的生长和/或存活的方法包括:向受试者施用治疗有效量的托那博沙。
在某些实施方式中,用于抑制受试者脑中的转移性癌细胞的生长和/或存活的方法包括:向受试者施用治疗有效量的甲氯芬那酸盐。
在某些实施方式中,用于抑制受试者脑中的转移性癌细胞的生长和/或存活的方法包括:向受试者施用治疗有效量的cx43抑制剂。
在某些实施方式中,用于抑制受试者脑中的转移性癌细胞的生长和/或存活的方法包括:向受试者施用治疗有效量的pcdh7抑制剂。
在某些非限制性实施方式中,本发明提供一种治疗受试者中脑转移瘤的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些非限制性实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症是肺癌和/或乳腺癌。在某些非限制性实施方式中,受试者的癌症中一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在某些实施方式中,脑转移瘤是可检测的转移瘤。
在某些实施方式中,治疗受试者中脑转移瘤的方法包括:向受试者施用治疗有效量的托那博沙。
在某些实施方式中,治疗受试者中脑转移瘤的方法包括:向受试者施用治疗有效量的甲氯芬那酸盐。
在某些实施方式中,治疗受试者中脑转移瘤的方法包括:向受试者施用治疗有效量的cx43抑制剂。
在某些实施方式中,治疗受试者中脑转移瘤的方法包括:向受试者施用治疗有效量的pcdh7。
在某些非限制性实施方式中,本发明提供一种预防受试者中癌症向脑部转移的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症是肺癌和/或乳腺癌。在某些非限制性实施方式中,受试者的癌症中一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在本发明某些非限制性实施方式中,受试者在治疗之前不知道患有脑转移瘤。
在某些实施方式中,预防受试者中癌症转移至脑部的方法包括:向受试者施用治疗有效量的托那博沙。
在某些实施方式中,预防受试者中癌症转移至脑部的方法包括:向受试者施用治疗有效量的甲氯芬那酸盐。
在某些实施方式中,预防受试者中癌症转移至脑部的方法包括:向受试者施用治疗有效量的cx43抑制剂。
在某些实施方式中,预防受试者中癌症转移至脑部的方法包括:向受试者施用治疗有效量的pcdh7抑制剂。
在某些非限制性实施方式中,本发明提供一种在患有癌症的受试者中降低癌症向脑部的可检测转移风险的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症是肺癌和/或乳腺癌。在某些非限制性实施方式中,受试者的癌症中一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在某些实施方式中,受试者在治疗之前已知具有脑转移瘤。在本发明的某些非限制性实施方式中,受试者在治疗之前不知道患有脑转移瘤。
在某些实施方式中,在患有癌症的受试者中降低癌症向脑部的可检测转移风险的方法包括:向受试者施用治疗有效量的托那博沙。
在某些实施方式中,在患有癌症的受试者中降低癌症向脑部的可探测转移风险的方法包括:向受试者施用治疗有效量的甲氯芬那酸盐。
在某些实施方式中,在患有癌症的受试者中降低癌症向脑部的可探测转移风险的方法包括:向受试者施用治疗有效量的cx43抑制剂。
在某些实施方式中,在患有癌症的受试者中降低癌症向脑部的可探测转移风险的方法包括:向受试者施用治疗有效量的pcdh7抑制剂。
在某些实施方式中,本发明提供一种用于延长患有癌症的受试者的存活时间的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些实施方式中,间隙连接抑制剂可以是甲氯芬那酸盐、托那博沙、cx43抑制剂和/或pcdh7抑制剂。在某些实施方式中,癌症是肺癌和/或乳腺癌。在某些非限制性实施方式中,受试者的癌症中一个或多个细胞表达连接蛋白43和/或原钙粘附蛋白7。在某些实施方式中,受试者在治疗之前已知具有脑转移瘤。在本发明的某些非限制性实施方式中,受试者在治疗之前不知道患有脑转移瘤。
在某些实施方式中,用于延长患有癌症的受试者的存活时间的方法包括:向受试者施用治疗有效量的托那博沙。
在某些实施方式中,用于延长患有癌症的受试者的存活时间的方法包括:向受试者施用治疗有效量的甲氯芬那酸盐。
在某些实施方式中,用于延长患有癌症的受试者的存活时间的方法包括:向受试者施用治疗有效量的cx43抑制剂。
在某些实施方式中,用于延长患有癌症的受试者的存活时间的方法包括:向受试者施用治疗有效量的pcdh7抑制剂。
在某些实施方式中,本发明的方法可以使患有癌症的受试者的存活时间延长约1个月、约2个月、约3个月、约4个月、约6个月、约8个月、约10个月、约12个月、约14个月、约18个月、约20个月、约2年、约3年、约4年、约5年、约6年或更长。
在某些实施方式中,用于在需要此类治疗的受试者中治疗癌细胞转移的方法包括:向受试者施用治疗有效量的间隙连接抑制剂,如上所述,以抑制癌细胞-星形胶质细胞间隙连接功能性。
在某些实施方式中,本发明提供一种在患有癌症的受试者中产生抗癌效果的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述。
在某些实施方式中,本发明提供在患有癌症的受试者中产生抗癌效果的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂,如上所述,以抑制癌细胞-星形胶质细胞间隙连接功能性。
在某些实施方式中,本发明提供在患有癌症的受试者中产生抗癌效果的方法,其包括向受试者施用治疗有效量的间隙连接抑制剂以抑制间隙连接功能性。
在某些实施方式中,本发明提供用于治疗患有癌症的受试者、用于抑制癌细胞的生长和/或存活、用于预防和/或延迟癌症的复发、用于抑制癌细胞的浸润以及用于延长患有癌症的受试者的存活时间的方法,其包括:向受试者施用治疗有效量的间隙连接抑制剂,如上所述。在某些实施方式中,癌症是成胶质细胞瘤和/或星形胶质细胞瘤。
在某些实施方式中,本发明的方法还包括向受试者施用抗癌剂,如上所述。例如,并且并非通过限制性方式,本发明的方法包括向受试者施用治疗有效量的间隙连接抑制剂以及能够穿透血脑屏障以实现治疗水平的治疗有效量的抗癌剂,比如,而不限于acnu、bcnu、ccnu、羟基脲、拓扑替康(topotecan)、替莫唑胺、氮烯咪胺(dacarbazine)、甲氨蝶呤(methotrexate)、ara-c、卡培他滨(capecitabine)、顺铂(cisplatin)、长春瑞滨、卡铂或其组合。
在某些实施方式中,本发明的方法包括向受试者施用治疗有效量的甲氯芬那酸盐和治疗有效量的卡铂。
在某些实施方式中,本发明的方法包括向受试者施用治疗有效量的托那博沙和治疗有效量的卡铂。
在某些实施方式中,本发明的方法包括向受试者施用治疗有效量的cx43抑制剂和和治疗有效量的卡铂。
在某些实施方式中,本发明的方法包括向受试者施用治疗有效量的pcdh7抑制剂和和治疗有效量的卡铂。
在特定的非限制性的实施方式中,可以以约1mg/kg至约30mg/kg的量施用间隙连接抑制剂。例如,并且并非通过限制性的方式,可以以约1mg/kg至约25mg/kg、约1mg/kg至约20mg/kg、约1mg/kg至约15mg/kg、约1mg/kg至约10mg/kg、约1mg/kg至约5mg/kg、约5mg/kg至约30mg/kg、约10mg/kg至约30mg/kg、约15mg/kg至约30mg/kg、约20mg/kg至约30mg/kg或者约25mg/kg至约30mg/kg的量施用间隙连接抑制剂。在某些非限制性实施方式中,可以以约0.08mg/kg至约3.6mg/kg的量施用间隙连接抑制剂(参见reagan-shaw等人,thefasebj.,vol22:659-661(2008))。在某些非限制性实施方式中,可以以约0.15mg/kg至约18mg/kg的量施用间隙连接抑制剂。
在某些非限制性实施方式中,可以以约1mg至约200mg的量施用间隙连接抑制剂。例如,并且并非通过限制性的方式,可以以约1mg至约200mg、约10mg至约200mg、约20mg至约200mg、约30mg至约200mg、约40mg至约200mg、约50mg至约200mg、约60mg至约200mg、约70mg至约200mg、约80mg至约200mg、约90mg至约200mg、约100mg至约200mg、约110mg至约200mg、约120mg至约200mg、约130mg至约200mg、约140mg至约200mg、约150mg至约200mg、约160mg至约200mg、约170mg至约200mg、约180mg至约200mg、约190mg至约200mg、约1mg至约190mg、约1mg至约180mg、约1mg至约170mg、约1mg至约160mg、约1mg至约150mg、约1mg至约140mg、约1mg至约130mg、约1mg至约120mg、约1mg至约110mg、约1mg至约100mg、约1mg至约90mg、约1mg至约80mg、约1mg至约70mg、约1mg至约60mg、约1mg至约50mg、约1mg至约40mg、约1mg至约30mg、约1mg至约20mg、约1mg至约10mg或者约1mg至约5mg的量施用间隙连接抑制剂。
在某些实施方式中,可以以约10mg/kg的量施用间隙连接抑制剂托那博沙。在某些实施方式中,可以以约0.8mg/kg至约1.2mg/kg的量施用间隙连接抑制剂托那博沙。在某些实施方式中,可以以约0.01mg/kg至约9mg/kg的量施用间隙连接抑制剂托那博沙。在某些实施方式中,可以以约20mg/kg的量施用间隙连接抑制剂甲氯芬那酸盐。在某些实施方式中,可以以约1.6mg/kg至约2.4mg/kg的量施用间隙连接抑制剂甲氯芬那酸盐。在某些实施方式中,可以以约0.1mg/kg至约19mg/kg的量施用间隙连接抑制剂甲氯芬那酸盐。在某些实施方式中,可以以每天约100mg至约400mg之间的量施用间隙连接抑制剂甲氯芬那酸盐。在某些实施方式中,可以以每天两次约100mg的量施用间隙连接抑制剂甲氯芬那酸盐。在某些实施方式中,同时用质子泵抑制剂和甲氯芬那酸盐来治疗受试者。在某些实施方式中,可以以每天两次约100mg的量施用间隙连接抑制剂甲氯芬那酸盐,可以同时用质子泵抑制剂和甲氯芬那酸盐来治疗受试者,并且治疗时间可以为至少约2个月、至少约4个月、或者至少约6个月。
在特定的非限制性实施方式中,可以以约1nm至约1μm、或者约10mg/kg至约100mg/kg的量施用抗癌剂。在特定的非限制性实施方式中,可以以约0.8mg/kg至约8mg/kg的量施用抗癌剂。在特定的非限制性实施方式中,可以以约1.2mg/kg至约60mg/kg的量施用抗癌剂。例如,并且并非通过限制性的方式,可以以约500nm或约50mg/kg的量施用抗癌剂卡铂。在某些实施方式中,可以以约4mg/kg至约6mg/kg的量施用抗癌剂卡铂。在某些实施方式中,可以以约25nm的量施用抗癌剂紫杉醇。
在某些实施方式中,可以每周一次、两次、三次、四次、五次或六次或者每天地施用本发明的间隙连接抑制剂。在某些实施方式中,可以每周一次、两次、三次、四次、五次或六次或者每天地施用本发明的抗癌剂。在某些实施方式中,可以每天一次或多次地施用本公开主题的抑制剂和/或抗癌剂。例如,并且并非通过限制性方式,可以每天一次、两次、三次、四次、五次或更多次地施用本发明的间隙连接抑制剂和/或抗癌剂。
本文公开的抑制剂和/或抗癌剂可以使用标准给药的方法施用于受试者。在某些实施方式中,抑制剂可以以口服或者肠道外给药施用于受试者。例如,并且并非通过限制性方式,给药途径可以为静脉注射、动脉内给药、鞘内注射、腹腔内给药、肌肉注射、皮下注射、局部给药(topical)、皮内注射、局部给药(locally)或其组合。在某些实施方式中,可以由患者中植入的来源将抑制剂施用于患者。在某些实施方式中,抑制剂的施用可以通过在选定时间段内的连续输注来发生。
以下实施例仅用作说明本公开的发明,而不应当将其理解为以任何方式的限制。
实施例1:原钙粘附蛋白7和连接蛋白43介导的恶性肿瘤-星形胶质细胞间隙连接和脑转移瘤
1材料和方法
细胞培养.在具有10%胎牛血清(fbs)和2mml-谷氨酰胺的dmem中培养人mda-mb-231(mda231)、鼠mmtv-neu、其转移衍生物以及鼠373n1、393n1、482n1、2691n1细胞系。在补充有10%的fbs和2mml-谷氨酰胺的rpmi1640培养基中培养人h2030细胞和转移衍生物。为了制备慢病毒,在补充有10%胎牛血清和2mml-谷氨酰胺的dme培养基中培养293t细胞。在供应商(sciencell)指定的培养基中用2-6之间的通道培养人原代星形胶质细胞、脑微血管内皮细胞(hbmec)、成人真皮成纤维细胞和小神经胶质细胞。就支原体(micoplasma)而言所有细胞均测试为阴性。
动物研究.所有使用动物的实验均是根据mskcc机构动物护理及使用委员会批准的方案完成的。使用5-6周龄的无胸腺ncrnu/nu小鼠(nci-frederick)、cr:nihbg-nu-xid小鼠(nci-frederick)和b6129sf1/j小鼠(jackson实验室)。对于长期脑转移瘤试验,我们遵循了在先所述的步骤(bos,nguyen等人2010)。简言之,将悬浮在100μlpbs中的104个mda231-brm2细胞、5x104个h2030-brm3细胞或105个393n1细胞注入左心室中。在实验的终点,向被麻醉的小鼠(氯胺酮(ketamine)100mg/kg、赛拉嗪(xylazine)10mg/kg)眼球后注射d-虫荧光素(150mg/kg),并通过离体生物发光成像(bli)对脑定植进行定量。对于短期(7天和14天)脑转移瘤实验,我们注射5x105个细胞。将tritc葡聚糖(70kd)(lifetechnologies)静脉注射至染色血管结构。对于可诱导的敲减(knockdown)实验,在注射癌细胞之后给予小鼠含有盐酸多西环素(sigma-aldrich)的饮用水(2mg/ml)和食物(harlan)14天。对于肺定植试验,将100μlpbs中的2x105个mda231-brm2细胞注射入侧尾静脉。对于原位肿瘤移植,将在50μl1∶1的pbs/生长因子减少的基质胶(bdbiosciences)中的5x103个细胞注射入雌性小鼠的第四右侧乳腺脂肪垫中。对于药物治疗实验,向小鼠腹膜内注射卡铂(hospira)(50mg/kg/5天)、托那博沙(medchemexpress)(10mg/kg/天)或甲氯芬那酸钠盐(sigma-aldrich)(20mg/kg/天)。在对照小鼠中使用载体(聚乙二醇400中10%的dmso)。用ivisspectrumxenogen仪器(caliperlifesciences)实施bli并用活体成像(livingimage)软件v.2.50进行分析。对于脑转移瘤试验,每组中使用8-10只小鼠。对于药物治疗实验,用癌细胞对小鼠进行接种并随机分配至治疗组中。在mskcc抗肿瘤核心评价(antitumourassessmentcore)中将间隙连接调节剂和化疗剂进行盲法施用。
敲减与过表达构建体.对于cx43和pcdh7的稳定敲减,我们使用慢病毒载体中的shrna。对于可诱导的敲减,使用tripz慢病毒载体中的shrna。加入1μg/ml的盐酸多西环素(sigma-aldrich)以诱导shrna的表达。shrna的靶向序列在下表1中列出。对于sr-iκb的稳定表达,使用pbabe-puro-ikbα-突变体(addgene)。对于野生型cx43(origene)或cx43(t154a)突变体(acc至gcc)的表达,我们使用plvx载体。
mrna和蛋白质检测.使用prepeaserna旋转试剂盒(usb)提取总rna。为了制备cdna,使用转录子第一链cdna(transcriptorfirststrandcdna)合成试剂盒(roche)来处理1μg的总rna。通过taqman基因表达分析引物:(cx43:hs00748445_s1,mm00439105_ml;cx30:hs00922742_s1,mm00433661_s1;cx26:hs00269615_s1,mm00433643_s1;appliedbiosystems)对cx43、cx30和cx26表达进行定量。相对于β2-微球蛋白(hs99999907_ml、mm00437762_ml)对相对的基因表达进行标准化。将pcdh7引物对设计为检测所有pcdh7异构体:5’-agttcaacgtggtcatcgtg-3’(正义)、5’-acaatcagggagttgttgctc-3’(反义)。使用sybrgreenimaster混合器(appliedbiosystems)进行反应。使用abiprism7900ht序列检测系统(appliedbiosystems)对定量表达数据进行分析。对于蛋白质免疫印迹分析,用ripa缓冲液裂解细胞颗粒,并且通过bca蛋白分析试剂盒(pierce)测定蛋白质浓度。从sciencell购买原代人星形胶质细胞、神经元细胞、小神经胶质细胞和hbmec的蛋白质裂解物。通过sds-page对蛋白质进行分离并将其转移至硝酸纤维素膜(biorad)。用于蛋白质印迹分析的抗体在下表2中列出。
染料转移和edu转移试验.用2.5ug/ml钙黄绿素红-橙am染料(lifetechnologies)在37℃下对单层癌细胞或星形胶质细胞标记30分钟。将单细胞悬浮液以已标记的:未标记的细胞为2∶1的比率混合6小时。某些实验采用mda231-brm2(gfp+)、hbmec(用ebioscience的细胞增殖染料fluor@670预标记)和未标记的星形胶质细胞的三种细胞群的混合。染料转移通过zeisslsm5live共聚焦显微镜(20-分钟延时观察)而可视化,或者通过facscalibur流式细胞仪(bdbiosciences)在不同的时间点进行定量。对于dna转移试验,将癌细胞用edu(10μm,molecularprobes)进行过夜标记并保持额外3天的培养。将标记的癌细胞与星形胶质细胞的单细胞悬浮液以比率为2∶1混合6小时。edu转移使用zeisslsm5live共聚焦显微镜而可视化,并遵循制造商的说明(molecularprobes)通过facscalibur流式细胞仪(bdbiosciences)进行定量。
癌细胞和星形胶质细胞共培养实验.将星形胶质细胞与癌细胞以1∶1的比率混合。对于细胞凋亡试验,用500ng/ml无血清培养基中的sfasl(peprotech)、500nm卡铂(sigma-aldrich)或者25nm紫杉醇(sigma-aldrich)对过夜的共培养物处理24小时。用apc-缀合的剪切的半胱天冬酶3抗体(cellsignaling)对单细胞悬浮液进行染色,通过流式细胞仪检测调亡的gfp+癌细胞。对于翻译核糖体亲和纯化(trap),将表达egfp-l10a的癌细胞与星形胶质细胞共培养24小时。遵循在先公开的方案(heiman,schaefer等人2008,zhang,jin等人2013)),借助truseqrna试样制备试剂盒(illumina)遵循制造商的说明,将从癌细胞纯化的mrna用作基因库构建。使用truseqsbs试剂盒v3(illumina)将样品编码(barcoded)并在hiseq2000平台以50bp/50bp的配对末端运行下运行。每个样品产生平均50,000,000个成对的读数。对于条件培养基分析,在24小时之后收集培养基,并用人细胞因子阵列(humancytokinearray)(r&dsystems)鉴定条件培养基中的细胞因子或者通过ifnα或tnfαelisa试剂盒(r&dsystems)对其进行测量。为了检测收集的条件培养基中的ifnα或tnfα活性,用收集的条件培养基对癌细胞处理2小时并通过蛋白质印迹分析测定stat1或nf-κbp65的磷酸化状态。对于cgamp和tbi-irf3活性实验,将癌细胞与星形胶质细胞共培养18小时。通过蛋白质免疫印迹分析测定tbk1、irf3的磷酸化状态。借助zeisslsm5live共聚焦显微镜通过免疫荧光染色来测定irf3的核转运。通过lc-ms/ms测定cgamp水平。
细胞因子处理和通路报告子试验.用10单位/ml(39u/ng)的重组ifnαa(r&dsystems)或者10pg/ml的重组tnfα(r&dsystems)与卡铂或紫杉醇(sigma-aldrich)联合对癌细胞处理24小时。通过caspase-glo3/7试验(promega)对细胞凋亡进行定量。对于nfκb报告子试验,将来自phagenfkb-ta-luc-ubc-dtomato-w构建体(addgene)(wilson,kwok等人2013)的nfκb应答序列克隆至pgl4.82海肾荧光素酶报告子(promega)中。用该载体和lego-c2mcherry载体(addgene)对癌细胞进行共转染。用renillagloluciferase系统(promega)测定海肾荧光素酶的活性。红色荧光信号用于使转染效率标准化。
免疫组化染色.用4%的多聚甲醛固定小鼠大脑,通过振动切片机(leica)或低温恒温器(leica)切片,并遵循已建立的方案(valiente,obenauf等人2014)进行染色。对于大脑切片试验(valiente,obenauf等人2014),借助振动切片机(leica)制备250μm厚的成年小鼠大脑切片并放置在脑切片培养介质(dmem,完全的hbss,5%fbs,1mml-谷氨酰胺,100iu/ml青霉素,100μg/ml链霉素)中的0.8μm孔膜(millipore)顶部。将3x105个癌细胞放置在切片的表面。在48小时的孵育之后,用4%的多聚甲醛固定大脑切片并染色。对于腔室玻片培养物中的免疫染色,用4%的多聚甲醛对细胞进行固定并染色。用于免疫组化染色的抗体在表2中列出。借助zeissaxio成像仪、z1显微镜或leicasp5直立共聚焦显微镜获得图像,并用imagej、imaris和metamorph软件进行分析。用于免疫染色的抗体在表2中列出。
分裂荧光素酶试验.通过缺失人cx43(origene)、pcdh7(origene)、e-粘附蛋白(addgene)或n-粘附蛋白(addgene)cdna中的终止密码子并将萤火虫荧光素酶(addgene)的n-末端或c-末端片段接合,产生融合cdna(luker,smith等人2004)。将构建体克隆入plvx慢病毒表达载体中并转导入非-gfp-荧光素酶-标记的亲本mda-mb-231或h2030细胞中。为检测荧光素酶的活性,将7.5mg/ml的d-虫荧光素钾盐加入培养介质中。通过ivisspectrumxenogen仪器、采用活体成像软件v.2.50来进行bli。
细胞溶质dsdna检测.对于dsdna的可视化,将细胞用抗-dsdna抗体进行免疫染色。抗-gfp染色用于勾画癌细胞主体,dapi用于区分细胞核,且抗-coxiv抗体(线粒体标示物)用于区分线粒体。鬼笔环肽染色(molecularprobe)用于勾画星形胶质细胞主体。对于dsdna的定量,使用线粒体分离试剂盒(thermoscientific)来制备细胞核、细胞溶质和线粒体部分。通过qiaampdna微型试剂盒(qiagen)对来自所有亚细胞部分的dna进行纯化,并通过quantofluordsdna系统(promega)进行定量。
生物信息学和统计学分析.除非另有规定,在r(ver.3.1.2)中进行生物信息学分析。使用tophat2-htseq-deseq2管线对数据进行分析(anders,mccarthy等人2013,kim,pertea等人2013,love,huber等人2014)。将差异化的基因表达与cookscutoff和independentfiltering关闭进行比较。散点图显示,使用ggplot2包产生成倍的改变。使用prcomp进行主成分分析(pca)。通过z-评分法(zhang,jin等人2013)的总和分析并对通路基因应答特征进行评分,如在先所述(nguyen,chiang等人2009,gatza,lucas等人2010)。使用benjamini&hochberg错误发现率方法校正多重假设检验。使用graphpad软件(prism)和student氏t检验(双尾)进行统计学分析。认为p值<0.05是统计学上显著的。值为平均值±均值标准误差(s.e.m.)。
临床样本分析.在原发性乳腺癌(emc-msk)和腺癌数据集(mskcc组2、gse3141和gse8893)的微阵列数据中分析cx43和pcdh7转录水平。通过借助跨越样本的最大方差来选择探针,将定位于相同基因的多重探针相合并。基于数据集的临床注释或者基于esr1和erbb2的转录水平,鉴定三阴性乳腺癌亚型。基于cox比例风险模型计算cx43和pcdh7值的风险比例,如通过r中的“coxph”指令所执行的。p值由cox比例风险模型来计算,以cx43和pcdh7表达作为连续变量处理。对于cx43免疫组化,从usbiomax购买正常肺组织阵列(75例)、原发性三阴性乳腺癌组织阵列(98例)和原发性非小细胞肺癌组织阵列(138例)。从符合mskcc机构审查委员会(institutionalreviewboard)的mskcc病理部门(departmentofpathology)获得来自脑转移瘤(117例三阴性乳腺癌、91例非小细胞肺癌)的石蜡包埋的组织微阵列。从所有受试者获得知情许可。对于cx43的免疫组化染色通过mskcc病理学核心机构(pathologycorefacility)使用标准化、自动化的方案来进行。对于匹配的原发性脑转移瘤病变,通过阳性染色区域对cx43染色图像进行定量(metamorph软件)。
通过lc-ms/ms定量cgamp.将细胞(单独的2,400,000的mda231-brm2或人星形胶质细胞、2,400,000的人星形胶质细胞+2,400,000的mda231-brm2共培养物)接种在10cm的皿中。在18小时后将培养介质吸出并用包含4nmc-双-gmp内部标准物的2ml80∶20的甲醇∶水进行置换。将皿在-80℃下孵育过夜以促进蛋白质沉淀,将其刮除并转移至2ml的离心管中。使样品经过2次涡旋、在液氮中冷冻/解冻循环,在冰水浴中全功率超声粉碎5分钟,并4℃下通过以21,000xg离心20分钟使之澄清。使用台式蒸发器(genevac)将提取物干燥并在100μl水中的0.1%甲酸中进行重组。使用shimadzuhplc、accelaopen自动进样器(thermo)和cortecsc18+柱(waters,150mmx2.1mm,2.7μm)进行液相色谱分离。将样本保持在4℃下,且注射体积为15μl。含水流动相(a)为水中0.1%的甲酸,且有机流动相(b)为乙腈中0.1%的甲酸。初始条件为0%b,梯度程序:1.0分钟:0%b;7分钟:20%b;7.1分钟:90%b;9.0分钟:90%b和5分钟的再平衡时间。流速为400μl/分钟,用无死体积三通(zero-deadvolumetee)在120μl/分钟下将90∶10的乙腈∶dmso柱后溶剂加入至lc流以促进检测灵敏度。使用在srm和正离子化模式下操作的tsqvantage质谱仪(thermo)检测环状核苷酸。源参数为:喷雾器电压:4000v;蒸馏器温度:200℃;鞘气压力:70psi;辅助气压力:20psi;毛细管温度:400℃。化合物特异性s-透镜值为:164v(cgamp)和190v(c-双-gmp)。监测到的独立反应和碰撞能为:cgampm/z675.1→m/z512.1(ce:19v),m/z312.0(ce:40v),m/z136.0(ce:39v)*和c-双-gmpm/z675.1→m/z540.1(ce:19v),m/z248.0v(ce:27v),m/z152.0(ce:31v)*,*表示用于定量每种环状核苷酸的初级转换。相对于环状[g(2’,5’)pa(3’,5’)p]和c-双-gmp代谢物标准物(biolog)确认保留时间和转换。使用xcalibur软件(thermo)和prism(graphpad)进行数据分析。
2结果
脑转移瘤链接到cx43间隙连接形成.肺癌和乳腺癌是最常见的脑转移瘤的来源(gavrilovic和posner2005)。我们采用了衍生自人或鼠源的乳腺(mda231-brm2、erbb2-brm)或肺腺癌(h2030-brm3、kras/p53-brm)的四种脑转移瘤模型(图6a)(bos,zhang等人2009,nguyen,chiang等人2009,winslow,dayton等人2011,valiente,obenauf等人2014)。当作为原位肿瘤植入或者接种进入小鼠的动脉循环时,包括标记的星形胶质细胞在内的这些细胞形成复制脑转移瘤的关键组织病理学特征的病变(图1a)(bos,zhang等人2009,nguyen,chiang等人2009,valiente,obenauf等人2014)。在所有这些模型中,脑转移瘤细胞产生了抗-pa丝氨酸蛋白酶抑制蛋白(serpin)以通过活性的星形胶质细胞来防止致死性血纤维蛋白溶酶的生成(valiente,obenauf等人2014)。然而,与星形胶质细胞的共培养保护癌细胞免于化疗和促细胞凋亡因子fasl的影响(图6b),这与在先的体外发现(kim,kim等人2011)是一致的。这些结果表明了星形胶质细胞在脑转移瘤中可能的双重作用。
星形胶质细胞在巨大的间隙连接网络中相互作用(theis和giaume2012,haydon和nedergaard2015)。连接蛋白43(cx43)是星形胶质细胞中的一种主要的间隙连接蛋白。在我们的脑转移瘤小鼠模型中,我们观察到在癌细胞和周围的星形胶质细胞的界面处的cx43表达(图1b)。cx43能够介导癌细胞与内皮细胞之间的相互作用(cai,jiang等人1998),并且星形胶质细胞(zhang,iwakuma等人2009)提供为促转移性(pollmann,shao等人2005)或抗转移性(sharma,abraham等人2010)。为确定cx43与脑转移瘤的临床联系,我们测定了患者的组织样本。在三阴性乳腺癌和非小细胞肺癌(nsclc)中,我们发现,相比原发性肿瘤或正常组织,脑转移瘤中的cx43染色水平更高(图1c-d)。
间隙连接通过六聚体连接蛋白半通道来形成。相邻细胞上的半通道之间的成对相互作用形成了用于细胞溶质的分子通行的孔(bennett和goodenough1978,oshima2014)。并非所有间隙连接均形成功能性孔(stoletov,stmadel等人2013)、(sharma,abraham等人2010)。然而,我们观察到钙黄绿素从脑转移瘤细胞到星形胶质细胞的时间依赖性转移,如通过延时观察荧光显微镜所示(图le;图6c),以及从星形胶质细胞到转移性细胞的时间依赖性转移,如通过流式细胞仪所示(图6d)。
脑转移瘤上调原钙粘附蛋白7.星形胶质细胞钙黄绿素与脑转移瘤细胞的转移比与其亲本对应物(counterpart)更容易发生(图1f)。该表型无法完全通过脑转移瘤衍生物中更高的cx43表达进行解释(图1g,图7a、b)。而且,转移性细胞中的cx43表达低于或类似于其在星形胶质细胞、神经元细胞或者脑微血管内皮细胞中的表达(图1h,图7c)。在脑转移瘤细胞中其他星形胶质细胞连接蛋白(cx26、cx30)的表达水平与亲本细胞的表达类似(图7d)。这些观察提出以下问题,为何转移性细胞能够与固有的星形胶质细胞竞争间隙连接的形成。
推论癌细胞必须使用除cx43之外的另一种组分以与星形胶质细胞紧密连接,我们研究了原钙粘附蛋白7(pcdh7),其是在来自乳腺和肺部肿瘤的脑转移瘤细胞中上调的一小组基因之一(bos,zhang等人2009,nguyen,chiang等人2009,valiente,obenauf等人2014)。原钙粘附蛋白是具有七个钙粘附蛋白重复单元的整合的膜蛋白,其通过同类亲合性相互作用引导细胞-细胞接触。pcdh7(还被称为钙粘附蛋白相关的神经元受体)是在脑部主要表达的唯一的原钙粘附蛋白(yoshida,yoshitomo-nakagawa等人1998,kim,chung等人2007);其功能是未知的。在脑转移瘤衍生物中的pcdh7水平高于在亲本细胞系中(图1g,图7a、b)的水平,或者高于在高度转移至骨骼或者肺部而不是脑部的匹配衍生物中的水平(图1i;参考图6a)。在脑转移瘤细胞中的pcdh7水平高于在星形胶质细胞、神经元细胞、小神经胶质细胞或内皮细胞中的水平(图1h,图7c)。
在具有复发标注位点的三阴性乳腺癌临床队列中,在原发肿瘤中的pcdh7和cx43的联合表达与脑转移瘤相关,而不于骨转移瘤或肺转移瘤相关(图1j)。尽管大多数nsclc数据集并没有以位点特异性转移信息进行标注,但这些患者中大比率的复发(高达70%)包括脑转移瘤(gaspar,chansky等人2005)。由于与脑转移瘤相关的严重的发病率和死亡率(gaspar,scott等人2000),这些不成比例地促进了无转移存活。事实上,cx43和pcdh7表达与三个队列中nsclc患者降低的无转移存活有关(图1k,图7e)。这些结果均支持了pcdh7和cx43在脑转移瘤相关的假设。
pcdh7引导恶性肿瘤-星形胶质细胞的间隙连接.与对照相比(图2a,图8a),通过短发夹rna(shrna)的手段缺失pcdh7或cx43的脑转移瘤细胞(图7f、g)显示出染料转移至星形胶质细胞的能力降低。在cx43缺失之后染料转移抑制的程度与用全-连接蛋白抑制剂(pan-connexininhibitor)carbenoloxone获得的相当(图8b)。考虑到钙粘附蛋白在相邻细胞上的分子间建立同类亲合性结合的能力(yagi和takeichi2000),我们假设星形胶质细胞pcdh7可能参与在与癌细胞的间隙连接的形成中。事实上,星形胶质细胞中的pcdh7缺失(图8c)也抑制了染料从mda231-brm2细胞的转移(图8d)。
人脑微血管内皮细胞(hbmec)表达的cx43水平大大低于星形胶质细胞,并且没有可检测的pcdh7表达(图1h,图7c)。在癌细胞与hbmec之间发生低水平的非pcdh7-依赖性间隙连接通讯(图8e)。在竞争性实验中,癌细胞与星形胶质细胞之间的染料转移比癌细胞与内皮细胞之间的染料转移更受青睐(图8f)。原代小神经胶质细胞表达极低水平的cx43和pcdh7,并且不接受来自癌细胞的钙黄绿素(图8g)。在与小神经胶质细胞共培养之后,星形胶质细胞和癌细胞中的cx43水平保持恒定(图8h)。因此,pcdh7引导癌细胞以有倾向性地形成与星形胶质细胞的cx43间隙连接。
我们采用了分裂荧光素酶互补分析(luker,smith等人2004)以检测肝细胞中pcdh7与cx43的相互作用。编码与n-末端(nluc)和c-末端(cluc)半个的萤火虫荧光素酶融合的pcdh7和cx43的构建体在非-gfp-荧光素酶标记的亲本细胞中以有关组合进行表达(图2b)。当nluc和cluc进入接近时,荧光素酶活性被重新组建。因为cx43自组装进入细胞膜中的六聚体半通道中,将用cx43-nluc和cx43-cluc载体转导细胞作为阳性对照(图2b)。我们在既表达cx43-cluc也表达pcdh7-nluc的细胞中检测到特异性的荧光素酶活性(图2b)。pcdh7和cx43的表达水平高于在亲本细胞中的内源水平,但是比在脑转移瘤细胞中的水平低或与之相当(图9a)。而且,与星形胶质细胞的共培养增加了癌细胞中的荧光素酶信号(图9b),这表明星形胶质细胞cx43和pcdh7诱导癌细胞cx43-cluc和pcdh7-nluc的进一步的聚集。当n-钙粘附蛋白或e-钙粘附蛋白与nluc融合并与cx43-cluc共表达时,没有检测到活性(图9c-e)。
cx43和pcdh7介导脑转移瘤定植.shrna介导的cx43或pcdh7的缺失抑制了在异种移植(图2c-d)和免疫活性模型(图10a)中由乳腺癌和肺癌形成脑转移瘤。在脑切片中对gfp的免疫组化染色证实了该结果并表明了作为cx43或pcdh7缺失结果的显著的病变尺寸的减小(图10b)。在尾静脉注射之后,cx43或pcdh7的缺失并未通过mda231-brm2细胞影响肺部病变的形成(图10c)。
因为连接蛋白可以独立于通道功能介导细胞间的相互作用,我们采用了缺乏通道功能但仍组装半通道的cx43(t154a)突变体(图2e)(beahm,oshima等人2006)。野生型或t154a突变体的cx43在cx43缺失的脑转移瘤的癌细胞中再表达(图10d)。突变体cx43无法介导钙粘附蛋白从星形胶质细胞转移至mda231-brm细胞(图2e)。野生型cx43挽回了cx43缺失的mda231-brm和h2030-brm细胞的脑转移活性,而cx43(t154a)并没有(图2f,图10e)。同时,这些观察支持了以下模型,其中pcdh7直接且特异性地与cx43相互作用,以选择性地促进癌细胞与星形胶质细胞之间的功能性间隙连接形成(图2g)。
为了明确pcdh7和cx43对脑转移瘤形成起作用的阶段,我们用mda231-brm2细胞进行了短期转移试验。在这个模型中,跨过bbb的外渗是在接种后7天完成的,第14天发生血管粘连和明显的过度增生(valiente,obenauf等人,2014)。接种7天后癌细胞中cx43或pcdh7的缺失并未显著减少脑实质中gfp+癌细胞的数量(图11a)。接种后14天,由cx43或pcdh7缺失的细胞产生的微转移显示出减少的增殖,如通过ki67染色所测定(图11b)。在离体脑切片试验中测定脑转移瘤细胞的凋亡(valiente,obenauf等人2014)。通过这种方法,我们发现cx43或pcdh7缺失的细胞中半胱天冬酶3染色增加,这与凋亡增加一致(图11c)。值得注意的是,cx43缺失的或pcdh7缺失的细胞仍然能够与毛细血管进行紧密的相互作用(图11d)。因此,癌细胞-星形胶质细胞间隙连接支持初次外渗和血管粘连后的脑转移瘤发展。
癌细胞间隙连接触发星形胶质细胞细胞因子释放.为了确定这种cx43介导的脑转移瘤生长背后的机制,我们采用翻译核糖体亲和纯化(trap)(heiman,schaefer等人2008)来测定混合共培养物中的癌细胞基因表达(图12a)。我们在具有基本的或降低的cx43表达的mda231-brm2细胞中表达egfp标签的l10a核糖体亚基。在癌细胞与星形胶质细胞共培养24小时后,通过全局转录组测序(trap-rnaseq)进行egfp免疫沉淀反应并从癌细胞收获多核糖体(polysome)相关的mrna(图12b、c)。基因特征分析揭示,在与星形胶质细胞共培养后,干扰素(ifn)和nf-κb通路是脑转移瘤细胞中最活跃的通路,并且这些效应需要cx43(图3a)。其他上调的通路包括her2/akt和tgfβ。如通过stat1和nf-κbp65的增加的磷酸化所确定的,来自星形胶质细胞-mda231-brm2共培养物的条件培养基足以激活癌细胞中的ifn和nf-κb信号转导(图3b,图12d)。来自星形胶质细胞与cx43缺失的或cx43(t154a)重建的癌细胞的共培养物的条件培养基没有观察到这种效应(图3c)。
对在mda231-brm2-星形胶质细胞共培养物中产生的条件培养基的分析(图3d)表明i型干扰素ifnα和tnfα以间隙连接依赖性方式的积累(图3e,图13a-b);没有检测到ii型干扰素ifnγ(数据未显示)。单独或与星形胶质细胞共培养的mda231-brm2如通过trap-rnaseq所检测到的不表达这些细胞因子(数据未显示)。在共培养后在重新分离的星形胶质细胞中检测到infα和tnfαmrna的上调(图3f)。这些结果表明,异型细胞的间隙连接通讯引起星形胶质细胞中ifnα和tnfα的产生,触发癌细胞中stat1和nf-κb通路的活化。ifnα和tnfα的添加抑制了体外脑转移瘤的癌细胞对细胞毒性化疗的凋亡应答(图3g,图13c)。为了评估脑转移瘤中这些通路的功能重要性,我们在脑转移瘤细胞中通过shrna(图3h,图13d)敲减stat1或通过iκbα超抑制子(sr-iκbα)的过表达来抑制nf-κb(boehm,zhao等人2007)(图3i)。当接种到小鼠中时,这些细胞产生比对照对应物更少的脑转移瘤(图3j,图13e),这表明stat1和nf-rb激活剂为脑中的转移细胞提供存活优势。
癌细胞间隙连接激活星形胶质细胞中的细胞溶质dsdna应答.鉴于ifnα和tnfα可以通过不同的投入物被单独诱导,两种细胞因子的联合上调让人联想到对细胞溶质双链dna(dsdna)的细胞应答(cai,chiu等人2014)。细胞溶质dsdna触发cgas-sting通路,其中环状gmp-amp合成酶(cgas)感测细胞溶质dsdna并合成第二信使2’3’-环状gmp-amp(cgamp)。cgamp结合至sting触发tbk1和irf3的磷酸化和活化、irf3的细胞核积累和irf3靶基因ifna和tnfa的转录激活(wu,sun等人2013)。这一通路代表了一种古老的抗病毒先天免疫应答(cai,chiu等人2014)。
mda231-brm2细胞与星形胶质细胞的共孵育以cx43依赖性方式触发tbk1和irf3的磷酸化(图4a,图14a)。irf3的细胞核积累仅在共培养物中的星形胶质细胞中发生,而不是在单独培养的星形胶质细胞或癌细胞中发生(图4b)。使用lc-ms/ms,我们在mda231-brm2细胞中检测到cgamp,但在单独培养的星形胶质细胞中检测不到(图4c-d,图14b)。固定数量的mda231-brm2细胞与星形胶质细胞的共培养导致cgamp水平的cx43依赖性增加(图4c-d)。使用将线粒体dsdna释放到细胞溶质中的压力条件,我们证实星形胶质细胞能够对细胞溶质dsdna进行应答以产生cgamp(rongvaux,jackson等人2014)。
亚细胞分级表明,这些脑转移瘤细胞和其他人类癌细胞系含有细胞溶质dsdna,而星形胶质细胞和其他非肿瘤人类细胞则不含有(图4e,图14c,d)。通过免疫荧光,我们在脑转移瘤的癌细胞中检测到细胞溶质dsdna(图4f,图9e),但未在星形胶质细胞中检测到(图14f)。为了确定癌细胞dna是否通过cx43间隙连接传递到星形胶质细胞,我们用5-乙炔基-2’-脱氧尿苷(edu)标记癌细胞dna,将细胞与星形胶质细胞共培养,并通过显微镜(图4g,图14g)或流式细胞仪(图4h)分析标记的dna的分布。两种方法都证实了以cx43依赖性的方式的dna从癌细胞至星形胶质细胞的转移。
总之,这些结果支持一个模型,其中脑转移瘤的癌细胞含有细胞溶质dsdna和cgamp,并采用pcdh7以在基于cx43的间隙连接中与星形胶质细胞紧密相连。间隙连接允许细胞溶质dsdna(和cgamp)从癌细胞通过至星形胶质细胞,以引发额外的cgamp、tbk1和irf3活化的产生以及ifnα和tnfα的产生。作为旁分泌因子,这些细胞因子在癌细胞中激活stat1和nf-κb信号转导,其支持癌细胞在面对微环境和化疗压力下的生长和存活(图4i)。
间隙连接活性的药理学抑制
间隙连接组分的基因抑制降低脑转移瘤过度增生的证据为检测针对脑转移瘤的间隙连接活性的药理抑制因子提供依据。为此,我们选择了两种口服生物可利用的化合物进行临床前试验。除抗炎活性外,甲氯芬那酸盐抑制cx43间隙连接门控(harks,deroos等人2001),在动物模型中抑制癫痫(jin,dai等人2013),在全身给药后穿过bbb(harks,deroos等人2001),全身耐受性良好(holmes1966)并且目前是fda批准的非甾体抗炎药(nsaid)。托纳博沙是一种在星型胶质细胞中与特有的立体选择性结合位点相结合的苯并吡喃衍生物(herdon,jerman等人1997,chan,evans等人1999),其抑制间隙连接介导的病理生理过程,包括皮层传播抑制(read,smith等人2000)和动物模型中的三叉神经节神经元卫星细胞信号转导(damodaram,thalakoti等人2009),并且在偏头痛患者中全身耐受性良好且安全(dahlof,hauge等人,2009)。如通过流式细胞仪所检测的,托纳博沙和甲氯芬那酸盐都抑制染料从星形胶质细胞转移至癌细胞(图5a),并且在这些细胞的共培养物中释放ifnα和tnfα(图5b),重演了在cx43或pcdh7的敲减中所见的表型。在免疫缺陷小鼠中动脉接种mda231-brm2细胞或h2030-brm3细胞,或在免疫活性小鼠中动脉接种kras/p53-393n1细胞后第一天起,用载体或这些化合物处理小鼠(图5c,图15a、b)。这两种药物防止脑转移瘤的出现,这与我们的间隙连接活性与转移性过度增生相关的证据是一致的。然而,这种治疗并没有限制mda231-brm2细胞作为肺转移性病变或作为原位肿瘤的生长(图15c、d)。
针对间隙连接的疗法.为了测试cx43或pcdh7缺失在已建立的转移瘤中的作用,我们用tet-可诱导的shrna表达载体转导mda231-brm2细胞(图5e)。在相同启动子控制下的红色荧光蛋白(rfp)在体内提供发夹表达的标志物(图10e)。用可诱导的cx43或pcdh7shrna载体转导的细胞分别显示cx43或pcdh7的多西环素依赖性缺失(图15f)。将这些细胞心脏内地注射并在14天形成脑转移瘤。在这个阶段,在所有小鼠中通过bli的脑病变都是明显的(图15g);侵袭性病变吞噬微血管(图5d),并将导致动物在2-3周内死亡(bos,zhang等人2009,valiente,obenauf等人2014)。与对照相比,在第14天开始的多西环素给药在三周后导致脑转移瘤负荷降低(图5f、g)。
脑转移瘤的特点是化疗耐药性明显(zhang,price等人1992,deeken和loscher2007)。卡铂越过bbb(pitz,desai等人2011),在患有来自乳腺癌(lim和lin2014)或肺癌(taimur和edelman2003)的脑转移瘤的患者中的总体存活率得到适度改善。从第14天开始的单独的卡铂(50mg/kg/5天)将脑转移瘤抑制到与cx43或pcdh7的缺失相似的程度(图5f、g);卡铂和多西环素的组合进一步降低转移瘤负荷(图5f、g)。因此,我们评估了将间隙连接调节治疗与化疗相结合的有效性(图5h)。单独用卡铂治疗最低限度地抑制脑转移瘤生长(图5i)。托那博沙(10mg/kg)或甲氯芬那酸盐(20mg/kg)作为单一药剂(图5i)在35天终点时显著抑制转移性病变的进展。卡铂与托那博沙或甲氯芬那酸盐的联合明显地抑制了脑转移瘤(图5i)。
3讨论
大脑代表独特而难以对付的转移瘤靶标,星形胶质细胞是微环境的主要特征。我们提供证据表明,癌细胞采用pcdh7在至关重要的cx43间隙连接中选择性地与星形胶质细胞紧密结合。钙粘附蛋白家族成员是发育和组织稳态中细胞间通讯的重要介质(yagi和takeichi2000),特别是在神经系统中(hirano,suzuki等人2003)。引人注目的是,脑转移瘤细胞采用该家族的特定成员,其正常表达主要限于脑部(yoshida,yoshitomo-nakagawa等人1998)。因此,pcdh7与作为大脑限制组件的来自乳腺癌和肺癌的脑转移瘤选择性地表达的以定植大脑的st6galnac5(bos,zhang等人2009)和神经丝氨酸蛋白酶抑制剂(neuroserpin)(valiente,obenauf等人2014)相结合。
pcdh7和cx43有助于脑转移瘤的定植和化疗耐药性。癌细胞与星形胶质细胞之间的基于cx43的功能性间隙连接可使癌细胞将细胞溶质dsdna扩散到星形胶质细胞网络。这激活了星形胶质细胞cgas-sting通路,最终导致包括ifnα和tnfα在内的细胞因子的释放。这些细胞因子通过针对生理和化疗应激原的保护为脑转移瘤细胞提供生长优势。其他上调的通路包括her2/akt和tgfβ。因此,我们的结果为以前观察到的在体外星形胶质细胞对癌细胞的化疗保护作用提供体内证据和机理的支撑(kim,kim等人2011)。目前的证据与在先的工作一起表明,癌细胞以两种方式保护自身免受星形胶质细胞攻击,首先,通过产生细胞毒性纤溶酶生成的丝氨酸蛋白酶抑制剂,其次,通过间隙连接与星形胶质细胞紧密连接并占有dsdna应答。
细胞溶质dsdna首先被定义为抗病毒感染的先天免疫激活剂(stetson和medzhitov2006)。在癌细胞中,存在许多可能的dsdna来源,包括基因组不稳定性、线粒体应激和暴露于dna损伤剂。dna触发的先天免疫应答,并且特别是cgamp可通过间隙连接穿过至其它细胞(patel,king等人2009,ablasser,schmid-burgk等人2013)。配合这些观察结果,我们发现,与星形胶质细胞和其他基质细胞相比,包括脑转移瘤衍生物在内的恶性细胞含有高水平的细胞溶质dsdna和cgamp。重要的是,在脑转移瘤中,dsdna应答在癌细胞中的固有的细胞溶质dsdna出现,其是cx43依赖性的,并涉及宿主组织星形胶质细胞,因此代表了前所未有的促转移过程。
脑转移瘤是癌症患者发病率和死亡率的主要原因,几乎没有可用的治疗选择。脑转移瘤级联的早期步骤,包括通过bbb的癌细胞传播和外渗,尚不能适应于治疗(maher,mietz等人2009,eichler,chung等人2011)。然而,对于转移瘤病变的存活和过度增生,癌细胞对cx43/pcdh7间隙连接的依赖性表明了治疗机会。我们使用化疗和间隙连接调节剂相结合的临床前结果为这些针对脑转移瘤的干预的治疗性潜力提供原理证明。
表1.shrna的靶标序列(seqidno:1至seqidno:14,顶部至底部)
plko.1慢病毒载体-人类基因
tripz可诱导型慢病毒载体-人类基因
gipz慢病毒载体-小鼠基因
对照sh
参考文献
axelsen,l.n.,calloe,k.,holstein-rathlou,n.h.,andnielsen,m.s.(2013).managingthecomplexityofcommunication:regulationofgapjunctionsbypost-translationalmodification.frontpharmacol4,130.
bartzatt,r.(2012).anti-inflammatorydrugsandpredictionofnewstructuresbycomparativeanalysis.antiinflammantiallergyagentsmedchem11,151-160.
bennett,m.v.,andgoodenough,d.a.(1978).gapjunctions,electrotoniccoupling,andintercellularcommunication.neurosciresprogrambull16,1-486.
bos,p.d.,zhang,x.h.,nadal,c.,shu,w.,gomis,r.r.,nguyen,d.x.,minn,a.j.,vandevijver,m.j.,gerald,w.l.,foekens,j.a.,etal.(2009).genesthatmediatebreastcancermetastasistothebrain.nature459,1005-1009.
bradley,d.p.,smith,m.i.,netsiri,c.,smith,j.m.,bockhorst,k.h.,hall,l.d.,huang,c.l.,leslie,r.a.,parsons,a.a.,andjames,m.f.(2001).diffusion-weightedmriusedtodetectinvivomodulationofcorticalspreadingdepression:comparisonofsumatriptanandtonabersat.expneurol172,342-353.
chambers,a.f.,groom,a.c.,andmacdonald,i.c.(2002).disseminationandgrowthofcancercellsinmetastaticsites.natrevcancer2,563-572.
chan,w.n.,evans,j.m.,hadley,m.s.,herdon,h.j.,jerman,j.c.,parsons,a.a.,read,s.j.,stean,t.o.,thompson,m.,andupton,n.(1999).identificationof(-)-cis-6-acetyl-4s-(3-chloro-4-fluoro-benzoylamino)-3,4-dihydro-2,2-dimethyl-2h-benzo[b]pyran-3s-olasapotentialantimigraineagent.bioorgmedchemlett9,285-290.
dahlof,c.g.,hauge,a.w.,andolesen,j.(2009).efficacyandsafetyoftonabersat,agapjunctionmodulator,intheacutetreatmentofmigraine:adouble-blind,parallel-group,randomizedstudy.cephalalgia29suppl2,7-16.
damodaram,s.,thalakoti,s.,freeman,s.e.,garrett,f.g.,anddurham,p.l.(2009).tonabersatinhibitstrigeminalganglionneuronal-satelliteglialcellsignaling.headache49,5-20.
deangelis,l.m.,andposner,j.b.(2009).intracranialmetastasis.inneurologiccomplicationsofcancer,s.gilman,ed.(oxforduniversitypress),pp.141-274.
deeken,j.f.,andloscher,w.(2007).theblood-brainbarrierandcancer:transporters,treatment,andtrojanhorses.clincancerres13,1663-1674.
eichler,a.f.,chung,e.,kodack,d.p.,loeffler,j.s.,fukumura,d.,andjain,r.k.(2011).thebiologyofbrainmetastases-translationtonewtherapies.natrevclinoncol8,344-356.
eugenin,e.a.,basilio,d.,saez,j.c.,orellana,j.a.,raine,c.s.,bukauskas,f.,bennett,m.v.,andberman,j.w.(2012).theroleofgapjunctionchannelsduringphysiologicandpathologicconditionsofthehumancentralnervoussystem.jneuroimmunepharmacol7,499-518.
gaspar,l.e.,gay,e.g.,crawford,j.,putnam,j.b.,herbst,r.s.,andbonner,j.a.(2005).limited-stagesmall-celllungcancer(stagesi-iii):observationsfromthenationalcancerdatabase.clinlungcancer6,355-360.
gaspar,l.e.,scott,c.,murray,k.,andcurran,w.(2000).validationofthertogrecursivepartitioninganalysis(rpa)classificationforbrainmetastases.intjradiatoncolbiolphys47,1001-1006.
gavrilovic,i.t.,andposner,j.b.(2005).brainmetastases:epidemiologyandpathophysiology.jneurooncol75,5-14.
gilula,n.b.,reeves,o.r.,andsteinbach,a.(1972).metaboliccoupling,ioniccouplingandcellcontacts.nature235,262-265.
goadsby,p.j.,ferrari,m.d.,csanyi,a.,olesen,j.,mills,j.g.,andtonabersat,t.o.n.s.g.(2009).randomized,double-blind,placebo-controlled,proof-of-conceptstudyofthecorticalspreadingdepressioninhibitingagenttonabersatinmigraineprophylaxis.cephalalgia29,742-750.
goldberg,g.s.,lampe,p.d.,andnicholson,b.j.(1999).selectivetransferofendogenousmetabolitesthroughgapjunctionscomposedofdifferentconnexins.natcellbiol1,457-459.
gupta,g.p.,nguyen,d.x.,chiang,a.c.,bos,p.d.,kim,j.y.,nadal,c.,gomis,r.r.,manova-todorova,k.,andmassague,j.(2007).mediatorsofvascularremodellingco-optedforsequentialstepsinlungmetastasis.nature446,765-770.
harks,e.g.,deroos,a.d.,peters,p.h.,dehaan,l.h.,brouwer,a.,ypey,d.l.,vanzoelen,e.j.,andtheuvenet,a.p.(2001).fenamates:anovelclassofreversiblegapjunctionblockers.jpharmacolexpther298,1033-1041.
herdon,h.j.,jerman,j.c.,stean,t.o.,middlemiss,d.n.,chan,w.n.,vong,a.k.,evans,j.m.,thompson,m.,andupton,n.(1997).characterizationofthebindingof[3h]-sb-204269,aradiolabelledformofthenewanticonvulsantsb-204269,toanovelbindingsiteinratbrainmembranes.brjpharmacol121,1687-1691.
heyn,c.,ronald,j.a.,ramadan,s.s.,snir,j.a.,barry,a.m.,mackenzie,l.t.,mikulis,d.j.,palmieri,d.,bronder,j.l.,steeg,p.s.,etal.(2006).invivomriofcancercellfateatthesingle-celllevelinamousemodelofbreastcancermetastasistothebrain.magnresonmed56,1001-1010.
hirano,s.,suzuki,s.t.,andredies,c.(2003).thecadherinsuperfamilyinneuraldevelopment:diversity,functionandinteractionwithothermolecules.frontbiosci8,d306-355.
holder,j.w.,elmore,e.,andbarrett,j.c.(1993).gapjunctionfunctionandcancer.cancerres53,3475-3485.
holmes,e.l.(1966).experimentalobservationsonflufenamic,mefenamic,andmeclofenamicacids.iv.tolerationbynormalhumansubjects.annphysmedsuppl,36-49.
hsu,m.,andl,t.,li,g.,meinkoth,j.l.,andherlyn,m.(2000).cadherinrepertoiredeterminespartner-specificgapjunctionalcommunicationduringmelanomaprogression.jcellsci113(pt9),1535-1542.
jin,m.,dai,y.,xu,c.,wang,y.,wang,s.,andchen,z.(2013).effectsofmeclofenamicacidonlimbicepileptogenesisinmicekindlingmodels.neuroscilett543,110-114.
joyce,j.a.,andpollard,j.w.(2009).microenvironmentalregulationofmetastasis.natrevcancer9,239-252.
juszczak,g.r.,andswiergiel,a.h.(2009).propertiesofgapjunctionblockersandtheirbehavioural,cognitiveandelectrophysiologicaleffects:animalandhumanstudies.progneuropsychopharmacolbiolpsychiatry33,181-198.
juul,m.h.,rivedal,e.,stokke,t.,andsanner,t.(2000).quantitativedeterminationofgapjunctionintercellularcommunicationusingflowcytometricmeasurementoffluorescentdyetransfer.celladhescommun7,501-512.
kienast,y.,vonbaumgarten,l.,fuhrmann,m.,klinkert,w.e.,goldbrunner,r.,herms,j.,andwinkler,f.(2010).real-timeimagingrevealsthesinglestepsofbrainmetastasisformation.natmed16,116-122.
kim,s.j.,kim,j.s.,park,e.s.,lee,j.s.,lin,q.,langley,r.r.,maya,m.,he,j.,kim,s.w.,weihua,z.,etal.(2011).astrocytesupregulatesurvivalgenesintumorcellsandinduceprotectionfromchemotherapy.neoplasia13,286-298.
kim,s.y.,chung,h.s.,sun,w.,andkim,h.(2007).spatiotemporalexpressionpatternofnonclusteredprotocadherinfamilymembersinthedevelopingratbrain.neuroscience147,996-1021.
li,b.,wang,c.,zhang,y.,zhao,x.y.,huang,b.,wu,p.f.,li,q.,li,h.,liu,y.s.,cao,l.y.,etal.(2013).elevatedplgfcontributestosmall-celllungcancerbrainmetastasis.oncogene32,2952-2962.
lim,e.,andlin,n.u.(2014).updatesonthemanagementofbreastcancerbrainmetastases.oncology(willistonpark)28(7).
lorger,m.,andfelding-habermann,b.(2010).capturingchangesinthebrainmicroenvironmentduringinitialstepsofbreastcancerbrainmetastasis.amjpathol176,2958-2971.
loscher,w.,andpotschka,h.(2005).drugresistanceinbraindiseasesandtheroleofdrugeffluxtransporters.natrevneurosci6,591-602.
luker,k.e.,smith,m.c.,luker,g.d.,gammon,s.t.,piwnica-worms,h.,andpiwnica-worms,d.(2004).kineticsofregulatedprotein-proteininteractionsrevealedwithfireflyluciferasecomplementationimagingincellsandlivinganimals.procnatlacadsciusa101,12288-12293.
maassenvandenbrink,a.,vandenbroek,r.w.,devries,r.,upton,n.,parsons,a.a.,andsaxena,p.r.(2000).thepotentialanti-migrainecompoundsb-220453doesnotcontracthumanisolatedbloodvesselsormyocardium;acomparisonwithsumatriptan.cephalalgia20,538-545.
maher,e.a.,mietz,j.,arteaga,c.l.,depinho,r.a.,andmohla,s.(2009).brainmetastasis:opportunitiesinbasicandtranslationalresearch.cancerres69,6015-6020.
miyamoto,s.,yagi,h.,yotsumoto,f.,kawarabayashi,t.,andmekada,e.(2006).heparinbindingepidermalgrowthfactor-likegrowthfactorasanoveltargetingmoleculeforcancertherapy.cancersci97,341-347.
nguyen,d.x.,chiang,a.c.,zhang,x.h.,kim,j.y.,kris,m.g.,ladanyi,m.,gerald,w.l.,andmassague,j.(2009).wnt/tcfsignalingthroughlef1andhoxb9mediateslungadenocarcinomametastasis.cell138,51-62.
nilsen,k.e.,kelso,a.r.,andcock,h.r.(2006).antiepilepticeffectofgap-junctionblockersinaratmodelofrefractoryfocalcorticalepilepsy.epilepsia47,1169-1175.
noy,r.,andpollard,j.w.(2014).tumor-associatedmacrophages:frommechanismstotherapy.immunity41,49-61.
oberheim,n.a.,goldman,s.a.,andnedergaard,m.(2012).heterogeneityofastrocyticformandfunction.methodsmolbiol814,23-45.
oshima,a.(2014).structureandclosureofconnexingapjunctionchannels.febslett588,1230-1237.
pekny,m.,andnilsson,m.(2005).astrocyteactivationandreactivegliosis.glia50,427-434.
pitz,m.w.,desai,a.,grossman,s.a.,andblakeley,j.o.(2011).tissueconcentrationofsystemicallyadministeredantineoplasticagentsinhumanbraintumors.jneurooncol104,629-638.
read,s.j.,hirst,w.d.,upton,n.,andparsons,a.a.(2001).corticalspreadingdepressionproducesincreasedcgmplevelsincortexandbrainstemthatisinhibitedbytonabersat(sb-220453)butnotsumatriptan.brainres891,69-77.
read,s.j.,smith,m.i.,hunter,a.j.,upton,n.,andparsons,a.a.(2000).sb-220453,apotentialnovelantimigraineagent,inhibitsnitricoxidereleasefollowinginductionofcorticalspreadingdepressionintheanaesthetizedcat.cephalalgia20,92-99.
sarrouilhe,d.,dejean,c.,andmesnil,m.(2014).involvementofgapjunctionchannelsinthepathophysiologyofmigrainewithaura.frontphysiol5,78.
silberstein,s.d.,schoenen,j.,gobel,h.,diener,h.c.,elkind,a.h.,klapper,j.a.,andhoward,r.a.(2009).tonabersat,agap-junctionmodulator:efficacyandsafetyintworandomized,placebo-controlled,dose-rangingstudiesofacutemigraine.cephalalgia29suppl2,17-27.
smith,m.i.,read,s.j.,chan,w.n.,thompson,m.,hunter,a.j.,upton,n.,andparsons,a.a.(2000).repetitivecorticalspreadingdepressioninagyrencephalicfelinebrain:inhibitionbythenovelbenzoylamino-benzopyransb-220453.cephalalgia20,546-553.
solan,j.l.,andlampe,p.d.(2009).connexin43phosphorylation:structuralchangesandbiologicaleffects.biochemj419,261-272.
stelzer,k.j.(2013).epidemiologyandprognosisofbrainmetastases.surgneurolint4,s192-202.
taimur,s.,andedelman,m.j.(2003).treatmentoptionsforbrainmetastasesinpatientswithnon-small-celllungcancer.curroncolrep5,342-346.
theis,m.,andgiaume,c.(2012).connexin-basedintercellularcommunicationandastrocyteheterogeneity.brainres1487,88-98.
upton,n.,blackburn,t.p.,campbell,c.a.,cooper,d.,evans,m.l.,herdon,h.j.,king,p.d.,ray,a.m.,stean,t.o.,chan,w.n.,etal.(1997).profileofsb-204269,amechanisticallynovelanticonvulsantdrug,inratmodelsoffocalandgeneralizedepilepticseizures.brjpharmacol121,1679-1686.
valiente,m.,obenauf,a.c.,jin,x.,chen,q.,zhang,x.h.,lee,d.j.,chaft,j.e.,kris,m.g.,huse,j.t.,brogi,e.,etal.(2014).serpinspromotecancercellsurvivalandvascularco-optioninbrainmetastasis.cell156,1002-1016.
winslow,m.m.,dayton,t.l.,verhaak,r.g.,kim-kiselak,c.,snyder,e.l.,feldser,d.m.,hubbard,d.d.,dupage,m.j.,whittaker,c.a.,hoersch,s.,etal.(2011).suppressionoflungadenocarcinomaprogressionbynkx2-1.nature473,101-104.
xing,f.,kobayashi,a.,okuda,h.,watabe,m.,pai,s.k.,pandey,p.r.,hirota,s.,wilber,a.,mo,y.y.,moore,b.e.,etal.(2013).reactiveastrocytespromotethemetastaticgrowthofbreastcancerstem-likecellsbyactivatingnotchsignallinginbrain.embomolmed5,384-396.
yagi,t.,andtakeichi,m.(2000).cadherinsuperfamilygenes:functions,genomicorganization,andneurologicdiversity.genesdev14,1169-1180.
yoshida,k.,yoshitomo-nakagawa,k.,seki,n.,sasaki,m.,andsugano,s.(1998).cloning,expressionanalysis,andchromosomallocalizationofbh-protocadherin(pcdh7),anovelmemberofthecadherinsuperfamily.genomics49,458-461.
zhang,r.d.,price,j.e.,fujimaki,t.,bucana,c.d.,andfidler,i.j.(1992).differentialpermeabilityoftheblood-brainbarrierinexperimentalbrainmetastasesproducedbyhumanneoplasmsimplantedintonudemice.amjpathol141,1115-1124.
ablasser,a.,j.l.schmid-burgk,i.hemmerling,g.l.horvath,t.schmidt,e.latzandv.hornung(2013)."cellintrinsicimmunityspreadstobystandercellsviatheintercellulartransferofcgamp."nature503(7477):530-534.
anders,s.,d.j.mccarthy,y.chen,m.okoniewski,g.k.smyth,w.huberandm.d.robinson(2013)."count-baseddifferentialexpressionanalysisofrnasequencingdatausingrandbioconductor."natprotoc8(9):1765-1786.
beahm,d.l.,a.oshima,g.m.gaietta,g.m.hand,a.e.smock,s.n.zucker,m.m.toloue,a.chandrasekhar,b.j.nicholsonandg.e.sosinsky(2006)."mutationofaconservedthreonineinthethirdtransmembranehelixofalpha-andbeta-connexinscreatesadominant-negativeclosedgapjunctionchannel."jbiolchem281(12):7994-8009.
boehm,j.s.,j.j.zhao,j.yao,s.y.kim,r.firestein,i.f.dunn,s.k.sjostrom,l.a.garraway,s.weremowicz,a.l.richardson,h.greulich,c.j.stewart,l.a.mulvey,r.r.shen,l.ambrogio,t.hirozane-kishikawa,d.e.hill,m.vidal,m.meyerson,j.k.grenier,g.hinkle,d.e.root,t.m.roberts,e.s.lander,k.polyakandw.c.hahn(2007)."integrativegenomicapproachesidentifyikbkeasabreastcanceroncogene."cell129(6):1065-1079.
bos,p.d.,d.x.nguyenandj.massague(2010)."modelingmetastasisinthemouse."curropinpharmacol10(5):571-577.
cai,j.,w.g.jiangandr.e.mansel(1998)."gapjunctionalcommunicationandthetyrosinephosphorylationofconnexin43ininteractionbetweenbreastcancerandendothelialcells."intjmolmed1(1):273-278.
cai,x.,y.h.chiuandz.j.chen(2014)."thecgas-cgamp-stingpathwayofcytosolicdnasensingandsignaling."molcell54(2):289-296.
gaspar,l.e.,k.chansky,k.s.albain,e.vallieres,v.rusch,j.j.crowley,r.b.livingstonandd.r.gandara(2005)."timefromtreatmenttosubsequentdiagnosisofbrainmetastasesinstageiiinon-small-celllungcancer:aretrospectivereviewbythesouthwestoncologygroup."jclinoncol23(13):2955-2961.
gatza,m.l.,j.e.lucas,w.t.barry,j.w.kim,q.wang,m.d.crawford,m.b.datto,m.kelley,b.mathey-prevot,a.pottiandj.r.nevins(2010)."apathway-basedclassificationofhumanbreastcancer."procnatlacadsciusa107(15):6994-6999.
haydon,p.g.andm.nedergaard(2015)."howdoastrocytesparticipateinneuralplasticity?"coldspringharbperspectbiol7(3):a020438.
heiman,m.,a.schaefer,s.gong,j.d.peterson,m.day,k.e.ramsey,m.suarez-farinas,c.schwarz,d.a.stephan,d.j.surmeier,p.greengardandn.heintz(2008)."atranslationalprofilingapproachforthemolecularcharacterizationofcnscelltypes."cell135(4):738-748.
kim,d.,g.pertea,c.trapnell,h.pimentel,r.kelleyands.l.salzberg(2013)."tophat2:accuratealignmentoftranscriptomesinthepresenceofinsertions,deletionsandgenefusions."genomebiol14(4):r36.
love,m.i.,w.huberands.anders(2014)."moderatedestimationoffoldchangeanddispersionforrna-seqdatawithdeseq2."genomebiol15(12):550.
patel,s.j.,k.r.king,m.casaliandm.l.yarmush(2009)."dna-triggeredinnateimmuneresponsesarepropagatedbygapjunctioncommunication."procnatlacadsciusa106(31):12867-12872.
pollmann,m.a.,q.shao,d.w.lairdandm.sandig(2005)."connexin43mediatedgapjunctionalcommunicationenhancesbreasttumorcelldiapedesisinculture."breastcancerres7(4):r522-534.
read,s.j.,m.i.smith,a.j.hunter,n.uptonanda.a.parsons(2000)."sb-220453,apotentialnovelantimigraineagent,inhibitsnitricoxidereleasefollowinginductionofcorticalspreadingdepressionintheanaesthetizedcat."cephalalgia20(2):92-99.
rongvaux,a.,r.jackson,c.c.harman,t.li,a.p.west,m.r.dezoete,y.wu,b.yordy,s.a.lakhani,c.y.kuan,t.taniguchi,g.s.shadel,z.j.chen,a.iwasakiandr.a.flavell(2014)."apoptoticcaspasespreventtheinductionoftypeiinterferonsbymitochondrialdna."cell159(7):1563-1577.
sharma,v.,t.abraham,a.so,m.f.allardandj.h.mcneill(2010)."functionaleffectsofproteinkinasesandperoxynitriteoncardiaccarnitinepalmitoyltransferase-1inisolatedmitochondria."molcellbiochem337(1-2):223-237.
stetson,d.b.andr.medzhitov(2006)."recognitionofcytosolicdnaactivatesanirf3-dependentinnateimmuneresponse."immunity24(1):93-103.
stoletov,k.,j.strnadel,e.zardouzian,m.momiyama,f.d.park,j.a.kelber,d.p.pizzo,r.hoffman,s.r.vandenbergandr.l.klemke(2013)."roleofconnexinsinmetastaticbreastcancerandmelanomabraincolonization."jcellsci126(pt4):904-913.
wilson,a.a.,l.w.kwok,e.l.porter,j.g.payne,g.s.mcelroy,s.j.ohle,s.r.greenhill,m.t.blahna,k.yamamoto,j.c.jean,j.p.mizgerdandd.n.kotton(2013)."lentiviraldeliveryofrnaiforinvivolineage-specificmodulationofgeneexpressioninmouselungmacrophages."molther21(4):825-833.
wu,j.,l.sun,x.chen,f.du,h.shi,c.chenandz.j.chen(2013).″cyclicgmp-ampisanendogenoussecondmessengerininnateimmunesignalingbycytosolicdna.″science339(6121):826-830.
zhang,q.,n.iwakuma,p.sharma,b.m.moudgil,c.wu,j.mcneill,h.jiangands.r.grobmyer(2009).″goldnanoparticlesasacontrastagentforinvivotumorimagingwithphotoacoustictomography.″nanotechnology20(39):395102.
zhang,x.h.,x.jin,s.malladi,y.zou,y.h.wen,e.brogi,m.smid,j.a.foekensandj.massague(2013).″selectionofbonemetastasisseedsbymesenchymalsignalsintheprimarytumorstroma.″cell154(5):1060-1073.
本文引用了各种参考文献,其内容整体并入本文以供参考。本文引用了各种核酸和氨基酸序列登录号,并且参考这些登录号的完整序列整体并入本文以供参考。
- 还没有人留言评论。精彩留言会获得点赞!