基因测序数据压缩方法、系统及计算机可读介质与流程
本发明涉及基因测序和数据压缩技术,具体涉及一种基因测序数据压缩方法、系统及计算机可读介质。
背景技术:
近年来,随着下一代测序技术(nextgenerationsequence,ngs)的持续进步,基因测序的速度更快,成本更低,基因测序技术得以在更加广泛的生物、医疗、健康、刑侦、农业等等许多领域被推广应用,从而导致基因测序产生的原始数据量以每年3到5倍、甚至更快的速度爆炸式增长。而且,每个基因测序样本数据又很大,例如一个人的55x全基因组测序数据大约是400gb。因此,海量的基因测试数据的存储、管理、检索和传输面临技术和成本的挑战。
数据压缩(datacompression)就是缓解这个挑战的技术之一。数据压缩,是为了减少存储空间而把数据转换成比原始格式更紧凑形式的过程。原始的输入数据包含我们需要压缩或减小尺寸的符号序列。这些符号被压缩器编码,输出结果是编码过的数据。通常在之后的某个时间,编码后的数据会被输入到一个解压缩器,在这里数据被解码、重建,并以符号序列的形式输出原始数据。如果输出数据和输入数据始终完全相同,那么这个压缩方案被称为无损的(lossless),也称无损编码器。否则,它就是一个有损的(lossy)压缩方案。
目前,世界各国研究人员已经开发出多种用于基因测序数据的压缩方法。基于基因测序数据的用途,其压缩后必须随时可以重建、恢复成原始数据,因此,有实际意义的基因测序数据压缩方法都是无损压缩。如果按总的技术路线分类,可以将基因测序数据压缩方法分成三大类:通用(generalpurpose)压缩算法、有参考基因组(reference-based)的压缩算法和无参考基因组(reference-free)的压缩算法。
通用压缩算法,就是不考虑基因测序数据的特点,采用通用的压缩方法进行数据压缩。
无参考基因组压缩算法,就是不使用参考基因组,只是利用基因测序数据自身的特点,采用某种压缩方法对目标样本数据直接进行数据压缩。已有的无参考基因组压缩算法常用的压缩方法有霍夫曼编码、以lz77和lz78为代表的字典方法、算术编码等基础的压缩算法及其变种和优化。
有参考基因组压缩算法,就是选取某个基因组数据作为参考基因组,利用基因测序数据自身的特点,以及目标样本数据和参考基因组数据之间的相似性,间接进行数据压缩。已有的有参考基因组压缩算法常用的相似性表示、编码和压缩方法主要还是霍夫曼编码、以lz77和lz78为代表的字典方法、算术编码等基础的压缩算法及其变种和优化。
衡量压缩算法性能或效率的2个最常用的技术指标是:压缩率(compressionratio)或压缩比;压缩/解压时间或压缩/解压速度。压缩率=(压缩后数据大小/压缩前数据大小)*100%,压缩比=(压缩前数据大小/压缩后数据大小),即压缩率和压缩比互为倒数。压缩率和压缩比只和压缩算法本身有关,多种算法间可以直接进行比较,压缩率越小或压缩比越大,表明算法性能或效率越好;压缩/解压时间,即从读取原始数据到解压完成所需的机器运行时间;压缩/解压速度,即平均每单位时间可以处理压缩的数据量。压缩/解压时间和压缩/解压速度,既和压缩算法本身有关,也和使用的机器环境(包括硬件和系统软件)有关,因此,多种算法必须基于相同的机器环境运行,压缩/解压时间或压缩/解压速度的比较才有意义,在此前提下,压缩/解压时间越短,压缩/解压速度越快,表明算法性能或效率越好。另外,还有一个参考技术指标是运行时的资源消耗,主要是机器存储的峰值。在压缩率和压缩/解压时间相当的情况下,对存储的要求越少,表明算法性能或效率越好。
根据研究人员对已有的基因测序数据压缩方法的比较研究结果,无论是通用压缩算法、无参考基因组的压缩算法,还是有参考基因组压缩算法,都存在的问题有:1、压缩率还有进一步下降的空间;2、在获得相对较好的压缩率时,算法的压缩/解压时间相对较长,时间成本成为新的问题。此外,与通用压缩算法和无参考基因组压缩算法相比,有参考基因组压缩算法通常能获得更好的压缩率。但是,对于有参考基因组的压缩算法,参考基因组的选择会导致算法性能的稳定性问题,即处理相同的目标样本数据,当选择不同的参考基因组时,压缩算法性能可能存在明显差异;而使用相同的参考基因组选择策略,当处理同种的、不同的基因测序样本数据时,压缩算法的性能同样可能存在明显差异。
技术实现要素:
本发明要解决的技术问题:针对现有技术的上述问题,提供一种基因测序数据压缩方法、系统及计算机可读介质,本发明的基因测序数据压缩方法是一种无损的、有参考基因组的基因测序数据压缩方法,具有压缩率低,压缩时间短,压缩性能稳定的优点,不需要对基因数据进行精准比对,有较高的计算效率,预测数据模型p1的预测准确度越高,则可逆运算结果中的重复字符串就越多,从而压缩的压缩率就越低。
为了解决上述技术问题,本发明采用的技术方案为:
一方面,本发明提供一种基因测序数据压缩方法,实施步骤包括:
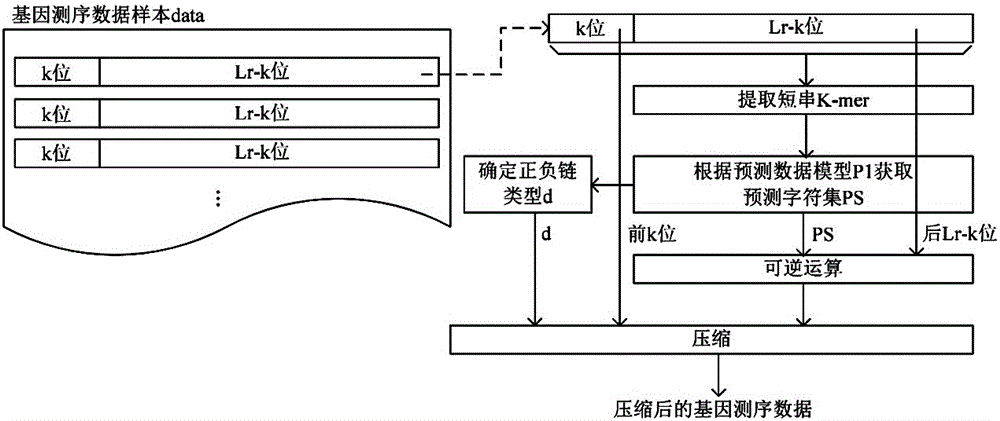
1)从基因测序数据样本data中遍历获取读长为lr的读序列r;
2)针对每一条读序列r,选择k位原始基因字母作为原始基因字符串cs0,从原始基因字符串cs0开始以k位长度作为滑动窗口顺序生成定长的k位字符串作为短串k-mer,根据短串k-mer确定读序列r的正负链类型d,且通过预设的预测数据模型p1获取每一个短串k-mer对应相邻位的预测字符c得到长度为lr-k位的预测字符集ps,所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其对应相邻位的预测字符c;将读序列r中不含k位原始基因字母的lr-k位原始基因字母、预测字符集ps编码后通过可逆函数进行可逆运算,所述可逆函数将任意一对相同的字符编码的运算输出结果相同;将读序列r的正负链类型d、k位原始基因字母以及可逆运算结果作为三条数据流压缩输出。
优选地,步骤2)的实施步骤包括:
2.1)从基因测序数据样本data中遍历获取一条读长为lr的读序列r,针对读序列r选择k位原始基因字母作为原始基因字符串cs0,从原始基因字符串cs0开始以k位长度作为滑动窗口顺序生成定长子字符串作为短串k-mer得到读序列短串集合kr;
2.2)按照顺序生成定长子字符串作为短串k-mer,根据短串k-mer确定读序列r的正负链类型d,且通过预设的预测数据模型p1获取每一个短串k-mer对应相邻位的预测字符c得到长度为lr-k位的预测字符集ps,所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其对应相邻位的预测字符c;
2.3)将读序列r中不含k位原始基因字母的lr-k位原始基因字母、预测字符集ps编码后通过可逆函数进行可逆运算,所述可逆函数将任意一对相同的字符编码的运算输出结果相同;
2.4)将读序列r的正负链类型d、原始基因字符串cs0以及可逆运算结果作为三条数据流压缩输出;
2.5)判断基因测序数据样本data中的读序列r是否遍历完毕,如果尚未遍历完毕,则跳转执行步骤2.1);否则结束并退出。
优选地,步骤2.2)的详细步骤包括:
2.2.1)针对读序列短串集合kr顺序提取短串k-mer,基于短串k-mer构造正链预测序列kp1,正链预测序列kp1中对于读序列短串集合kr中任意短串k-mer均存在一个对应的元组(k-mer,0),其中k-mer为短串k-mer,0表示假设该短串k-mer来自正链;
2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1获取其对应的预测字符c,得到所有预测字符c构成的正链预测字符序列ps1;所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其相邻位对应的预测字符c;
2.2.3)针对读序列短串集合kr顺序提取短串k-mer,基于短串k-mer构造负链预测序列kp2,负链预测序列kp2中对于读序列短串集合kr中任意短串k-mer均存在一个对应的元组(k-mer,1),其中k-mer为短串k-mer,1表示假设该短串k-mer来自负链;
2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1获取其相邻位对应的预测字符c,得到所有预测字符c构成的负链预测字符序列ps2;
2.2.5)计算正链预测字符序列ps1、读序列r中不含k位原始基因字母的lr-k位原始基因字母两者之间的编辑距离l1,计算负链预测字符序列ps2、读序列r中不含k位原始基因字母的lr-k位原始基因字母两者之间的编辑距离l2;
2.2.6)判断编辑距离为l1小于l2是否成立,如果成立则判定读序列r的正负链类别d为正链、将正链预测字符序列ps1作为lr-k位的预测字符集ps;否则,判定读序列r的正负链类别d为负链、将负链预测字符序列ps2作为lr-k位的预测字符集ps。
优选地,所述预测数据模型p1为长度为l的基因字符串,步骤2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1获取其相邻位对应的预测字符c的详细步骤包括:
2.2.2.1)使用预测数据模型p1对应的映射函数将正链预测序列kp1中的每一个元组(k-mer,0)映射到一个[0,l]范围的整数集合中的某一行,其中l是整数集合的上确界,整数集合的上确界l与预测数据模型p1的长度相同,0是整数集合的下确界,根据映射结果生成一维表t1,一维表t1的长度为(lr–k+1),一维表t1中的第i个元素t1[i]分别顺序存储映射至整数集合中第i行的元组(k-mer,0)对应的映射函数的值,i∈[0,lr-k];
2.2.2.2)根据一维表t1各个元组(k-mer,0)对应的映射函数的值从预测数据模型p1获取在其相邻位对应的预测字符c生成一维字符序列ps1,使得一维字符序列ps1的第i位ps1[i]的值等于预测数据模型p1中的第i位字符p1[t1[i]],预测数据模型p1中的第i位字符p1[t1[i]]为映射函数的值为ps1[i]对应的元组(k-mer,0)对应的预测字符c,其中i∈[0,lr-k],lr为读序列r的读长,k为短串k-mer的长度。
优选地,步骤2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1获取其相邻位对应的预测字符c的详细步骤包括:
2.2.4.1)使用预测数据模型p1对应的映射函数将负链预测序列kp2中的每一个元组(k-mer,1)映射到一个[0,l]范围的整数集合中的某一行,其中l是整数集合的上确界,整数集合的上确界l与预测数据模型p1的长度相同,0是整数集合的下确界,根据映射结果生成一维表t2,一维表t2中长度为(lr–k+1),一维表t2中的第i个元素t2[i]分别顺序存储映射至整数集合中第i行的元组(k-mer,1)对应的映射函数的值,i∈[0,lr-k];
2.2.4.2)根据一维表t2各个元组(k-mer,1)对应的映射函数的值从预测数据模型p1获取在其相邻位对应的预测字符c生成一维字符序列ps2,使得一维字符序列ps2的第i位ps2[i]的值等于预测数据模型p1中的第i位字符p1[t2[i]],预测数据模型p1中的第i位字符p1[t2[i]]为映射函数的值为ps2[i]对应的元组(k-mer,0)对应的预测字符c,其中i∈[0,lr-k],lr为读序列r的读长,k为短串k-mer的长度。
优选地,所述预测数据模型p1为预先基于参考基因组中的短串k-mer及其相邻位对应的碱基字母c0完成训练的神经网络模型;步骤2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1的映射函数映射获取其对应的预测字符c具体是指将正链预测序列kp1中的每一个元组(k-mer,0)输入神经网络模型,得到该元组(k-mer,0)对应的预测字符c;步骤2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1的映射函数映射获取其相邻位对应的预测字符c具体是指将正链预测序列kp2中的每一个元组(k-mer,1)输入神经网络模型,得到该元组(k-mer,1)对应的预测字符c。
优选地,步骤2)中的可逆函数具体采用xor异或函数或者位减法函数。
优选地,步骤2)中的压缩具体是指使用统计模型和熵编码进行压缩。
另一方面,本发明还提供一种基因测序数据压缩系统,包括计算机系统,所述计算机系统被编程以执行本发明前述基因测序数据压缩方法的步骤。
此外,本发明还提供一种计算机可读介质,所述计算机可读介质上存储有计算机程序,其特征在于,所述计算机程序使计算机执行本发明前述基因测序数据压缩方法的步骤。
本发明具有下述优点:
1、本发明的基因测序数据压缩方法是一种无损的、有参考基因组的基因测序数据压缩方法,通过利用参考基因组的预测数据模型p1,能够有效提升基因序列数据的压缩倍率,具有压缩率低,压缩时间短,压缩性能稳定的优点。
2、区别于现有技术使用参考序列进行基因序列精准比对后再进行数据压缩,本发明不需要对基因数据进行精准比对,有较高的计算效率;预测数据模型p1的预测准确度越高,则可逆运算结果中的重复字符串就越多,从而压缩的压缩率就越低。
附图说明
图1为本发明实施例一方法的原理示意图。
具体实施方式
实施例一:
参见图1,本实施例基因测序数据压缩方法的实施步骤包括:
1)从基因测序数据样本data中遍历获取读长为lr的读序列r;
2)针对每一条读序列r,选择k位原始基因字母作为原始基因字符串cs0,从原始基因字符串cs0开始以k位长度作为滑动窗口顺序生成定长的k位字符串作为短串k-mer,根据短串k-mer确定读序列r的正负链类型d,且通过预设的预测数据模型p1获取每一个短串k-mer对应相邻位的预测字符c得到长度为lr-k位的预测字符集ps,所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其对应相邻位的预测字符c;将读序列r中不含k位原始基因字母的lr-k位原始基因字母、预测字符集ps编码后通过可逆函数进行可逆运算,所述可逆函数将任意一对相同的字符编码的运算输出结果相同;将读序列r的正负链类型d、原始基因字符串cs0以及可逆运算结果作为三条数据流压缩输出。本实施例中,正负链类型d为0或1,0表示读序列r来自正链,1表示读序列r来自负链。
需要说明的是,获取相邻位的预测字符c时,相邻位的定义和选择原始基因字符串cs0的方式相关,如果选择原始基因字符串cs0为读序列r的前k位,则相邻位是指下一位;如果选择原始基因字符串cs0为读序列r的后k位,则相邻位是指上一位;如果选择原始基因字符串cs0为读序列r的中间k位,则相邻位包括上一位和下一位。参见图1,本实施例中相邻位具体指下一位,选择原始基因字符串cs0时选择前k位原始基因字母,读序列r中不含k位原始基因字母的lr-k位原始基因字母具体是指读序列r中的后lr-k位原始基因字母。
本实施例中,步骤2)的实施步骤包括:
2.1)从基因测序数据样本data中遍历获取一条读长为lr的读序列r,针对读序列r选择k位原始基因字母作为原始基因字符串cs0,从原始基因字符串cs0开始以k位长度作为滑动窗口顺序生成定长子字符串作为短串k-mer得到读序列短串集合kr;
2.2)按照顺序生成定长子字符串作为短串k-mer,根据短串k-mer确定读序列r的正负链类型d,且通过预设的预测数据模型p1获取每一个短串k-mer对应相邻位的预测字符c得到长度为lr-k位的预测字符集ps,所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其对应相邻位的预测字符c;
2.3)将读序列r中不含k位原始基因字母的lr-k位原始基因字母、预测字符集ps编码后通过可逆函数进行可逆运算,所述可逆函数将任意一对相同的字符编码的运算输出结果相同;
2.4)将读序列r的正负链类型d、原始基因字符串cs0以及可逆运算结果作为三条数据流压缩输出;
2.5)判断基因测序数据样本data中的读序列r是否遍历完毕,如果尚未遍历完毕,则跳转执行步骤2.1);否则结束并退出。
需要说明的是,读序列r的读长lr可以根据需要进行指定,例如在某些极限情况下甚至可以指定一个足够大的读长lr,从而将一个基因测序数据样本data指定在一个读序列r中。
本实施例中,步骤2.2)的详细步骤包括:
2.2.1)针对读序列短串集合kr顺序提取短串k-mer,基于短串k-mer构造正链预测序列kp1,正链预测序列kp1中对于读序列短串集合kr中任意短串k-mer均存在一个对应的元组(k-mer,0),其中k-mer为短串k-mer,0表示假设该短串k-mer来自正链;
2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1获取其对应的预测字符c,得到所有预测字符c构成的正链预测字符序列ps1;所述预测数据模型p1包含参考基因组的正链及负链中任意短串k-mer及其相邻位对应的预测字符c;
2.2.3)针对读序列短串集合kr顺序提取短串k-mer,基于短串k-mer构造负链预测序列kp2,负链预测序列kp2中对于读序列短串集合kr中任意短串k-mer均存在一个对应的元组(k-mer,1),其中k-mer为短串k-mer,1表示假设该短串k-mer来自负链;
2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1获取其相邻位对应的预测字符c,得到所有预测字符c构成的负链预测字符序列ps2;
2.2.5)计算正链预测字符序列ps1、读序列r中不含k位原始基因字母的lr-k位原始基因字母两者之间的编辑距离l1,计算负链预测字符序列ps2、读序列r中不含k位原始基因字母的lr-k位原始基因字母两者之间的编辑距离l2;
2.2.6)判断编辑距离为l1小于l2是否成立,如果成立则判定读序列r的正负链类别d为正链、将正链预测字符序列ps1作为lr-k位的预测字符集ps;否则,判定读序列r的正负链类别d为负链、将负链预测字符序列ps2作为lr-k位的预测字符集ps。
如果选择k位原始基因字母为读序列r的前k位,预测字符集ps实质上是读序列r的后lr-k位对应的预测字符集;如果选择k位原始基因字母为读序列r的后k位,则预测字符集ps实质上是读序列r的前lr-k位对应的预测字符集;如果选择k位原始基因字母为读序列r的中间k位,则预测字符集ps实质上包含了两部分预测字符集,两部分预测字符集的总长度为lr-k位,对应读序列r中不含k位原始基因字母的lr-k位原始基因字母。
本实施例中,所述预测数据模型p1为长度为l的基因字符串,步骤2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1获取其相邻位对应的预测字符c的详细步骤包括:
2.2.2.1)使用预测数据模型p1对应的映射函数将正链预测序列kp1中的每一个元组(k-mer,0)映射到一个[0,l]范围的整数集合中的某一行,其中l是整数集合的上确界,整数集合的上确界l与预测数据模型p1的长度相同,0是整数集合的下确界,根据映射结果生成一维表t1,一维表t1的长度为(lr–k+1),一维表t1中的第i个元素t1[i]分别顺序存储映射至整数集合中第i行的元组(k-mer,0)对应的映射函数的值,i∈[0,lr-k];
2.2.2.2)根据一维表t1各个元组(k-mer,0)对应的映射函数的值从预测数据模型p1获取在其相邻位对应的预测字符c生成一维字符序列ps1,使得一维字符序列ps1的第i位ps1[i]的值等于预测数据模型p1中的第i位字符p1[t1[i]],预测数据模型p1中的第i位字符p1[t1[i]]为映射函数的值为ps1[i]对应的元组(k-mer,0)对应的预测字符c,其中i∈[0,lr-k],lr为读序列r的读长,k为短串k-mer的长度。
本实施例中,步骤2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1获取其相邻位对应的预测字符c的详细步骤包括:
2.2.4.1)使用预测数据模型p1对应的映射函数将负链预测序列kp2中的每一个元组(k-mer,1)映射到一个[0,l]范围的整数集合中的某一行,其中l是整数集合的上确界,整数集合的上确界l与预测数据模型p1的长度相同,0是整数集合的下确界,根据映射结果生成一维表t2,一维表t2中长度为(lr–k+1),一维表t2中的第i个元素t2[i]分别顺序存储映射至整数集合中第i行的元组(k-mer,1)对应的映射函数的值,i∈[0,lr-k];
2.2.4.2)根据一维表t2各个元组(k-mer,1)对应的映射函数的值从预测数据模型p1获取在其相邻位对应的预测字符c生成一维字符序列ps2,使得一维字符序列ps2的第i位ps2[i]的值等于预测数据模型p1中的第i位字符p1[t2[i]],预测数据模型p1中的第i位字符p1[t2[i]]为映射函数的值为ps2[i]对应的元组(k-mer,0)对应的预测字符c,其中i∈[0,lr-k],lr为读序列r的读长,k为短串k-mer的长度。
需要说明的是,预测数据模型p1的长度l涉及压缩处理的性能和压缩率,预测数据模型p1的长度l越大,则将取出的每一个元组(k-mer,d,c0)的子元组(k-mer,d)映射到一个范围为[0,l]的整数集合中的某一行生成一个l行4列的二维统计表f时发生冲突的机会就越少,从而使得预测的准确率就会越高,但是这样会导致计算消费的资源较多;相反,预测数据模型p1的长度l越小,则将取出的每一个元组(k-mer,d,c0)的子元组(k-mer,d)映射到一个范围为[0,l]的整数集合中的某一行生成一个l行4列的二维统计表f时发生冲突的机会就越多,从而使得预测的准确率就会越低,计算消费的资源较少。
本实施例中,步骤2)中的可逆函数具体采用xor异或函数。本实施例中,a、c、g、t四种基因字母分别被编码为00、01、10和11四种字符编码,例如某一位基因字母为a,而预测字符c同样为a,则该位的xor异或操作结果(可逆运算结果)为00,否则xor异或操作结果根据预测字符c不同而有所不同;在解压时,针对预测字符c的字符编码和xor异或操作结果(可逆运算结果)再进行xor异或操作(通过可逆函数的反函数进行逆向操作),即可复原得到原始的基因字母。将a、c、g、t四种基因字母分别被编码为00、01、10和11四种字符编码是一种优选的比较精简的编码方式,此外也可以根据需要采用其他的二进制编码方式,同样也可以实现基因字母、预测字符、可逆运算结果三者的可逆转换。毫无疑问,除了xor异或运算以外,可逆函数也可以采用位减法函数,此时则可逆函数的反函数为位加法,同样也可以实现基因字母、预测字符、可逆运算结果三者的可逆转换。
本实施例中,步骤2)中的压缩具体是指使用统计模型和熵编码进行压缩。
本实施例中,步骤1)中的预测数据模型p1生成步骤包括:
a1)获取待处理的参考基因组数据dataref(参考基因组数据dataref为一个或多个物种的完整或部分参考基因组或物种中某一个具体个体测序得到的基因序列);
a2)获取参考基因组数据dataref中作为短串k-mer的任意定长子字符串及其预测字符c之间的映射关系,得到包含参考基因组的正链及负链中任意短串k-mer及其对应相邻位的预测字符c的预测数据模型p1。短串k-mer的长度k一般取值为16~32中的一个具体值。
其中,步骤a2)的实施步骤包括:
a2.1)按照指定间隔顺序提取参考基因组数据dataref的正链s1中的定长子字符串作为短串k-mer构造正链短串集合ks1,正链s1为原始顺序的参考基因组数据dataref本身;
a2.2)按照指定间隔顺序提取参考基因组数据dataref的负链s2中的定长子字符串作为短串k-mer构造负链短串集合ks2,负链s2为参考基因组数据dataref的逆序互补基因序列,逆序互补基因序列和参考基因组数据dataref之间碱基a和t互换、碱基c和g互换;
a2.3)根据正链短串集合ks1、负链短串集合ks2生成参考基因组数据dataref对应的预测数据模型p1,预测数据模型p1包含正链s1及负链s2中的任意短串k-mer及其对应的统计得到的最大可能相邻位的预测字符c之间的映射关系。
其中,步骤a2.3)的详细步骤包括:
a2.3.1)针对正链短串集合ks1顺序提取短串k-mer并构造正链预测集合kp1,正链短串集合ks1中的每一个元素在正链预测集合kp1中存在一个对应的元组,该元组至少包含三种信息:短串k-mer、来自正链的标记d、在正链s1中的下一个碱基字母c0;
a2.3.2)针对负链短串集合ks2顺序提取短串k-mer并构造负链预测集合kp2,负链短串集合ks2中的每一个元素在负链预测集合kp2中存在一个对应的元组,该元组至少包含三种信息:短串k-mer、自负链的标记d、元素在负链s2中的下一个碱基字母c0;
a2.3.3)将正链预测集合kp1和负链预测集合kp2中的元组映射到碱基字母a、c、g、t,统计正链s1及负链s2中的任意短串k-mer及其对应的统计得到的最大可能相邻位的碱基字母,得到包含正链s1及负链s2中的任意短串k-mer及其对应的统计得到的最大可能相邻位的预测字符c之间的映射关系预测数据模型p1。
其中,步骤a2.3.3)的详细步骤包括:
a2.3.3.1)逐个从正链预测集合kp1、负链预测集合kp2中取出每一个元组(k-mer,d,c0),其中k-mer为元组对应的短串k-mer,d为正负链类别,d=0表示为正链,d=1表示为负链,c0为元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母;
a2.3.3.2)使用预设的映射函数,将取出的每一个元组(k-mer,d,c0)的子元组(k-mer,d)映射到一个范围为[0,l]的整数集合中的某一行生成一个l行4列的二维统计表f且由元组(k-mer,d,c0)中元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母c0确定其命中行的对应列,其中l是整数集合的上确界,0是整数集合的下确界,统计整数集合中每个值对应出现的碱基字母a、c、g、t的数目,二维统计表f中的4列对应碱基字母a、c、g、t,二维统计表f中的元素fi,c存储了值为i的子元组(k-mer,d)所对应的碱基字母a、c、g、t出现的数目,其中,下标i∈[0,l],c∈{a、c、g、t};
a2.3.3.3)从0到l行遍历二维统计表f,将每行中最大值的元素fi,c所对应的碱基字母构成一维字符序列作为预测数据模型p1,预测数据模型p1的长度为l,l是整数集合的上确界,预测数据模型p1的第i位字符p1[i]表示命中整数集合第i行的元组对应的短串k-mer的预测字符c。
其中,步骤a2.3.3.2)中统计整数集合中每个值对应出现的a、c、g、t的数目具体是指:在将取出的每一个元组(k-mer,d,c0)的子元组(k-mer,d)映射到一个范围为[0,l]的整数集合时,针对二维统计表f的每一行的四个元素fi,c分别设置四个计数值的fi,a、fi,c、fi,g、fi,t,若命中整数集合第i行的元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母c0为a,则将第i行的fi,a加1;若命中整数集合第i行的元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母c0为c,则将第i行的fi,c加1;若命中整数集合第i行的元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母c0为g,则将第i行的fi,g加1;若命中整数集合第i行的元组对应的短串k-mer在正链s1或负链s2中对应的下一个碱基字母c0为t,则将第i行的fi,t加1;最终统计得到整数集合中每个值对应出现的a、c、g、t的数目fi,a、fi,c、fi,g、fi,t。
综上所述,本实施例的基因测序数据压缩方法能够满足基因测序数据压缩的无损、压缩率低、压缩时间短、性能稳定的要求,能够极大地缓解基因测试数据面临的存储、管理、检索和传输技术及成本的挑战。
本实施例还提供一种基因测序数据压缩系统,包括计算机系统,计算机系统被编程以执行本实施例前述基因测序数据压缩方法的步骤,在此不再赘述。此外,本实施例还提供一种计算机可读介质,计算机可读介质上存储有计算机程序,计算机程序使计算机执行本实施例前述基因测序数据压缩方法的步骤,在此不再赘述。
实施例二:
本实施例与实施例一基本相同,其主要区别点为步骤1)中的预测数据模型p1不同。
本实施例中,预测数据模型p1为预先基于参考基因组中的短串k-mer及其相邻位对应的碱基字母c0完成训练的神经网络模型;步骤2.2.2)针对正链预测序列kp1中的每一个元组(k-mer,0)通过预测数据模型p1的映射函数映射获取其对应的预测字符c具体是指将正链预测序列kp1中的每一个元组(k-mer,0)输入神经网络模型,得到该元组(k-mer,0)对应的预测字符c;步骤2.2.4)针对负链预测序列kp2中的每一个元组(k-mer,1)通过预测数据模型p1的映射函数映射获取其相邻位对应的预测字符c具体是指将正链预测序列kp2中的每一个元组(k-mer,1)输入神经网络模型,得到该元组(k-mer,1)对应的预测字符c。
对应地实施例一的步骤a2.3),本实施例生成的预测数据模型p1的详细步骤包括:
b2.3.1)针对正链短串集合ks1顺序提取短串k-mer并构造正链预测集合kp1,使得正链短串集合ks1中的每一个元素在正链预测集合kp1中存在一个对应的元组,该元组至少包含三种信息:短串k-mer、来自正链的标记d、在正链s1中的下一个碱基字母c0;
b2.3.2)针对负链短串集合ks2顺序提取短串k-mer并构造负链预测集合kp2,使得负链短串集合ks2中的每一个元素在负链预测集合kp2中存在一个对应的元组,该元组至少包含三种信息:短串k-mer、来自负链的标记d、负链s2中的下一个碱基字母c0;
b2.3.3)将正链预测集合kp1和负链预测集合kp2中的元组对应的短串k-mer及其在正链s1或负链s2中对应的下一个碱基字母c0生成训练集,通过训练集来对神经网络模型进行训练,并将完成训练的神经网络模型作为预测数据模型p1。
以上所述仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本发明领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!