用于刺激提前终止密码子的通读的方法和化合物与流程
本发明是在政府支持下根据合同号囊性纤维化基金会(cysticfibrosisfoundation)#rowe13xxo;nihp30#p30dk072482进行的。政府拥有本发明的某些权利。
背景技术:
囊性纤维化(cf)是一种由编码cf跨膜传导调节因子(cftr)的基因中的突变所引起的常染色体隐性病症,所述cftr是主要定位于沿呼吸道及多个器官排列的分泌上皮细胞的顶膜的阴离子通道。其中最常见的突变种类,cftr中的提前终止密码子(ptc)由于编码序列中的框内无义突变而导致翻译终止,产生无功能cftr蛋白(welshmj等人《细胞(cell)》;73:1251-1254;1993)。ptc是~11%的引起cf的等位基因和多种其它遗传疾病的近因(sloanepa等人《肺医学新见(currentopinioninpulmonarymedicine)》;16:591-597;2010)。
研发对具有无义突变的cf患者的治疗的成果集中在促进ptc的终止抑制(也称为翻译通读)的策略上。当通过近同源氨酰基trna携带的氨基酸被插入到错误终止密码子的多肽链中,使得翻译继续,并部分恢复全长功能蛋白时,完成翻译通读(bedwelldm等人,《自然医学(natmed)》;3:1280-1284;1997;howardm等人,《自然医学》;2:467-469;1996)。已发现数种诱导通读的药理学方法,但没有一种方法产生功效和安全性的最佳组合。举例来说,体外研究已经证明,某些氨基糖苷类可以促进通读并且已在具有混合结果的临床试验中得到测试(clancyjp等人,《美国呼吸与重症护理医学杂志(amjrespircritcaremed)》;163:1683-1692;2001;clancyjp等人,《美国呼吸系统细胞和分子生物学杂志(americanjournalofrespiratorycellandmolecularbiology)》;37:57-66;2007;wilschanskim等人,《新英格兰医学杂志(nengljmed)》;349:1433-1441;2003;wilschanskim等人,《新英格兰医学杂志》;349:1433-1441;2003;wilschanskim等人,《美国呼吸与重症护理医学杂志》;161:860-865;2000;sermet-gaudelusi等人,《bmc医学(bmcmed)》;5:5;2007),但其较不适合于长期使用。当在体外比较时,针对真核核糖体的翻译抑制而优化的合成氨基糖苷类衍生物展现出改善的通读和降低的毒性。阿他卢仑(ataluren)(先前ptc124)是一种诱导通读的口服生物可用小分子。在子组分析中,证明阿他卢仑在未使用经长期吸入的妥布霉素(其会干扰阿他卢仑的作用)的cf患者中有适度治疗益处(kereme等人,《柳叶刀呼吸医学(lancetrespirmed)》2014),当前正在对其进行前瞻性评估。
研发无毒、口服生物可用并且有效的通读剂的有吸引力的策略是评估已批准用于临床的化合物的效用,从而促进对人体中有前景的化合物的快速评估。本发明提供用于治疗至少部分地通过ptc的存在介导的疾病和病状的新型化合物。此类疾病和病状包括但不限于其中ptc密码子产生无功能多肽的遗传病状,如但不限于cf。本发明还提供用于增加至少部分地涉及疾病或病状的mrna转录物的稳定性水平的方法,其中mrna转录物含有由于通过本发明化合物的通读抑制导致的错义突变。此类疾病和病状包括但不限于其中ptc密码子产生无功能多肽的遗传病状,如但不限于cf。本发明还提供用于上述方法中的化合物和包含此类化合物的医药组合物。
附图说明
对于所有数字,如果没有特别提及,那么*p<0.05,**p<0.01,***p<0.001,****p<0.0001
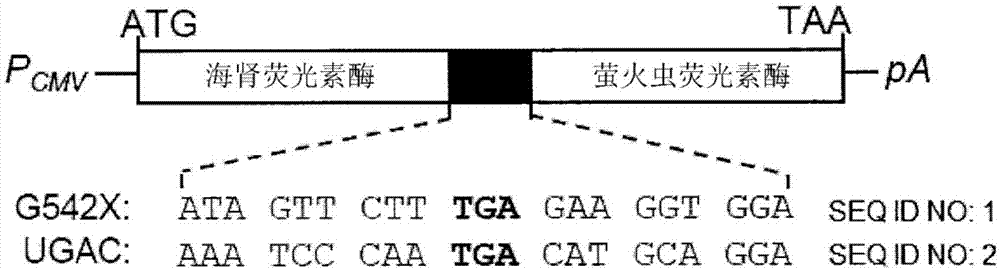
图1a显示双荧光素酶构建体的图,其显示在海肾与萤火虫荧光素酶之间插入的cftrptc情形(g542x和ugac)和wt。
图1b显示g418对ftrqxnugac细胞中荧光素酶活性的影响。将frtqxnugac细胞与g418(300至500μg/ml)或媒剂对照(0.3%dmso)一起培育24小时,然后测定荧光素酶活性(如与媒剂对照相比,****p<0.0001)。
图1c显示g418对ftrg542x细胞中荧光素酶活性的影响。将frtg542x细胞与g418(300至500μg/ml)或媒剂对照(0.3%dmso)一起培育24小时,然后测定荧光素酶活性(如与媒剂对照相比,****p<0.0001)。
图1d显示g418对ftrwt细胞中荧光素酶活性的影响。将frt野生型细胞与g418(300至500μg/ml)或媒剂对照(0.3%dmso)一起培育24小时,然后测定荧光素酶活性。
图2a显示g418对frtg542x细胞中cftr电导的时间依赖性影响。将frtg542x细胞与g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育0至72小时,然后测定电导。在测定电导之前,立即用fsk(20μm)处理细胞;在实验结束时加入cftrinh-172(10μm)以抑制cftr活性。图2a显示电导的代表性描记图。
图2b显示图2a中的数据的曲线图,其展示frtg542x细胞中g418对电导的刺激(如与媒剂对照相比,**p<0.01,***p<0.001)。
图2b显示图2a中的数据的曲线图,其展示cftrinh172对经g418刺激的电导的抑制(如与媒剂对照相比,***p<0.001)。
图3a显示散点图,其展示frtg542x细胞中存在毛喉素时cftr电导的百分比变化(归一化为g418对照,250μg/ml)。将frtg542x细胞与化合物(10μm)或媒剂对照(0.3%dmso)一起培育48小时。在分析期间,在急加入毛喉素(20μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。
图3b显示散点图,其展示frtqxnugac和frtg542x细胞中荧光素酶活性的最大活化百分比。将细胞与化合物(0.1μm-30μm)一起培育48小时,然后测定荧光素酶活性。
图4显示所选化合物对frtg542x细胞中cftr电导的影响。将frtg542x细胞与测试化合物(10μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时。在分析期间,在急加入毛喉素(20μm)之前首先测定基线电导(gt)以刺激cftr活性。
图5a显示所选化合物对frtg542x细胞中cftr电导的影响。将frtg542x细胞与测试化合物(1至30μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时。在分析期间,在急加入毛喉素(20μm)之前首先测定基线电导(gt)以刺激cftr活性。(*p<0.05,**p<0.01,***p<0.001,****p<0.0001)。
图5b显示所选化合物对frtg542x细胞中hrp活性的影响。将frtg542x细胞与化合物(10μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时,然后测定hrp活性(如与媒剂对照相比,*p<0.05,**p<0.01,***p<0.001,****p<0.0001)。
图5c显示所选化合物在frtqxnugac细胞中的双荧光素酶分析(萤火虫/海肾荧光素酶比)中的作用。将frtqxnugac细胞与化合物(0.01μm至10μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时,然后测定荧光素酶活性。
图5d显示通过双荧光素酶分析鉴定的8种先导化合物,表示为最高功效的萤火虫/海肾比的增加倍数。
图5e显示hrp分析和双荧光素酶分析之间的相关性。
图5f显示tecc分析和hrp分析之间的相关性。
图5f显示tecc分析和双荧光素酶分析之间的相关性。
图6显示g418、秋水仙碱、环己酰胺、奥苯达唑、泼尼卡酯、七叶皂苷、四磷酸咯萘啶、阿霉素和对氨基苯甲酸对源自g542x/δf508供体的原代hbe细胞中的短路电流(isc)的影响。将g542x/δf508细胞与测试化合物一起在指定浓度下培育48小时。为了测定isc,用cftr激动剂fsk(10μm)和vx-770(10μm)isc处理g542x/δf508细胞(如与媒剂对照相比,*p<0.05,**p<0.01,****p<0.0001)。
图7a显示七叶皂苷对frtg542x细胞中电导的影响。用七叶皂苷(10μm,实线)和媒剂对照(0.3%dmso,虚线)处理frtg542x细胞24小时。在分析期间,在急加入毛喉素(20μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。
图7b显示frtg542x细胞中七叶皂苷电导的剂量依赖性作用。如图7a中所述,在电导测量之前,用七叶皂苷(3和10μm)和媒剂对照(0.3%dmso)处理frtg542x细胞24小时(如与媒剂对照相比,*p<0.05,***p<0.001)。
图7c显示七叶皂苷对hrp分析中cftr蛋白表达的影响。在检测细胞表面cftr蛋白之前,用七叶皂苷(0.001μm至20μm)和媒剂对照(0.3%dmso)处理frtg542xhrp细胞48小时。测定七叶皂苷对ctfr蛋白表达的半数最大有效浓度(ec50)。
图7d显示七叶皂苷对hbeg542x/δf508细胞中的短路电流(isc)的影响。将hbe细胞与七叶皂苷(10μm)或媒剂对照(0.1%dmso)一起培育48小时。为了测定isc,用氨氯吡脒(100μm)处理hbe细胞以阻断上皮钠通道的活性,随后加入cftr激动剂fsk(10μm)和vx-770(10μm)以及cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。
图7e显示七叶皂苷对hbeg542x/δf508细胞中的短路电流(isc)的影响。实验设置与图7d中所述相同。七叶皂苷显著增加原代hbe细胞中经fsk刺激的isc,其经vx-770进一步增加。
图7f显示七叶皂苷对hbeg542x/δf508细胞中cftrmrna水平的影响。七叶皂苷(10μm)、g4129(250μg/ml)和媒剂对照(0.1%dmso)在hbe细胞中培育48小时。从hbe细胞中分离rna,并使用如本文所述的实时pcr测定g542xmrna的水平。将g542xmrna水平归一化为frt野生型mrna水平。
图7g显示七叶皂苷对frtw1282x细胞中电导的影响。用七叶皂苷(10μm)和媒剂对照(0.3%dmso)处理frtw1282x细胞48小时。在测定电导之前立即加入毛喉素(20μm)和vx-770(10μm)以刺激cftr(如与媒剂对照相比,****p<0.001)。
图7h显示七叶皂苷对原代hbew1282x/δf508细胞中的短路电流(isc)的影响。用七叶皂苷(10μm)和媒剂对照(0.1%dmso)处理hbew1282x/δf508细胞48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。
图7i显示七叶皂苷对原代hbeδf508/δf508细胞中的短路电流(isc)的刺激作用。用七叶皂苷(10μm)和媒剂对照(0.1%dmso)处理hbeδf508/δf508细胞48小时。在测定isc之前立即加入毛喉素(20μm)和vx-770(10μm)以刺激cftr(如与媒剂对照相比,***p<0.001)。
图7j显示七叶皂苷对原代hbeg542x/δf508细胞中的短路电流(isc)的影响。用七叶皂苷(10μm)和媒剂对照(0.1%dmso)处理hbeg542x/δf508细胞48小时。在测定isc之前立即加入毛喉素(20μm)和vx-770(10μm)以刺激cftr(如与媒剂对照相比,***p<0.001)。
图8显示两种商业来源的七叶皂苷对hbeg542x/δf508细胞中的短路电流(isc)的影响。将hbeg542x/δf508细胞与七叶皂苷(两种来源均呈10μm)和媒剂对照(0.1%dmso)一起培育48小时。在测定七叶皂苷活性之前立即加入毛喉素(20μm)和vx-770(10μm)以刺激cftr(如与媒剂对照相比,*p<0.05,**p<0.01,***p<0.001)。
图9显示七叶皂苷与cftr增效剂和cftr校正剂的组合对frtw1282x中电导的影响。将单独或呈所示组合的七叶皂苷(10μm)、g418(250μg/ml)、vx-770(10μm)和vx-809(6μm)与frtw1282x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性(如与媒剂对照相比,*p<0.05,****p<0.0001)。
图10显示nmdi-1和nmdi-14的结构。
图10b显示与nmdi-1或nmdi-14的组合对frtg542x细胞中电导的影响。将escin(10μm)、nmdi-1(80μg/ml)和nmdi-14(20μg/ml)(单独或呈所示组合)与frtg542x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性(如与媒剂对照相比,**p<0.01,***p<0.001)。
图10c显示七叶皂苷与nmdi-14的组合对frtw1282x细胞中电导的影响。如图10b所述,将单独或呈所示组合的七叶皂苷(10μm)和nmdi-14(20μg/ml)与frtw1282x细胞一起预培育48小时,然后测定电导(如与媒剂对照相比,**p<0.01,****p<0.0001)。
图11显示七叶皂苷与氨基糖苷类妥布霉素或庆大霉素的组合对frtw1282x细胞中电导的影响。将单独或呈所示组合的七叶皂苷(10μm)、妥布霉素(150μg/ml)、庆大霉素(250μg/ml)和媒剂对照(0.3%dmso)与frtw1282x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性(如与媒剂对照相比,****p<0.0001)。
图12a显示七叶皂苷与cftr增效剂(gp-5)和校正剂(vx-809和c1和c2的组合)对frtg542x细胞中电导的影响。将七叶皂苷(10μm)、vx-908(3μm)、gp-5(10μm)、c1+c2(10μm)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtg5422x细胞一起预培育培养48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性(如与媒剂对照相比,****p<0.0001)。
图12b显示七叶皂苷与cftr增效剂(gp-5)和校正剂(vx-809和c1和c2的组合)对frtw1282x细胞中电导的影响。将七叶皂苷(10μm)、vx-809(3μm)、gp-5(10μm),c1+c2(10μm)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtw1282x细胞一起预培育48小时,之后加入毛喉素(20μm)或vx-770(10μm)以刺激cftr(如与媒剂对照相比,*p<0.05,**p<0.01,***p<0.001,****p<0.0001)。
图13a显示七叶皂苷和cftr校正剂vx-809对frtg542x细胞中cftr电导的影响。将七叶皂苷(10μm)、vx-809(3μm)、g418(250μg/ml)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtg542x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性(如与媒剂对照相比,****p<0.0001)。
图13b显示七叶皂苷和cftr校正剂vx-809对frty122x细胞中cftr电导的影响。如图13a中所述,将七叶皂苷(10μm)、vx-809(3μm)、g418(250μg/ml)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frty122x细胞一起预培育48小时,然后测定电导(如与媒剂对照相比,*p<0.05,***p<0.001)。
图13c显示七叶皂苷和cftr校正剂vx-809对frtw1282x细胞中cftr电导的影响。如图13a中所述,将七叶皂苷(10μm)、vx-809(3μm)、g418(250μg/ml)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtw1282x细胞一起预培育48小时,然后测定电导(如与媒剂对照相比,*p<0.05,**p<0.01,****p<0.0001)。
图14a显示七叶皂苷和cftr校正剂vx-809对frtg542xhrp细胞中cftr表达的影响。将七叶皂苷(10μm)、vx-809(3μm)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtg542xhrp细胞一起预培育48小时(如与媒剂对照相比,****p<0.0001)。
图14b显示七叶皂苷和cftr校正剂vx-809对frty122xhrp细胞中cftr表达的影响。将七叶皂苷(10μm)、vx-809(3μm)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frty122xhrp细胞一起预培育48小时(如与媒剂对照相比,**p<0.01,****p<0.0001)。
图14c显示七叶皂苷和cftr校正剂vx-809对frtw1282xhrp细胞中cftr表达的影响。将七叶皂苷(10μm)、vx-809(3μm)和媒剂对照(0.3%dmso)(单独或呈所示组合)与frtw1282xhrp细胞一起预培育48小时(如与媒剂对照相比,***p<0.001,****p<0.0001)。
图15a显示代表性描记线,其展示七叶皂苷与cftr校正剂vx-809和g418的组合对hbeg542x/δf508细胞中isc的影响。用七叶皂苷(10μm)、g418(250μg/ml)、vx-809(3μm)或媒剂对照(0.1%dmso)(单独或以所示组合)处理hbeg542x/δf508细胞48小时。在测定isc之前立即加入毛喉素(10μm)和vx-770(10μm)以刺激cftr。
图15b显示七叶皂苷与cftr校正剂vx-809和g418的组合对hbeg542x/δf508细胞中isc的影响。实验设置与图15a中所述相同(如与媒剂对照相比,**p<0.01,***p<0.001,****p<0.0001)。
图16a显示七叶皂苷对源自纯合idua-w402x小鼠的小鼠胚胎成纤维细胞(mef)细胞中α-l-艾杜糖苷酶活性的影响。mef细胞在七叶皂苷(0.25至160μm)或媒剂对照(0.1%dmso)存在下生长48小时。48小时后,获得细胞裂解物并测定mefα-l-艾杜糖苷酶的活性。
图16b显示阿霉素对源自纯合idua-w402x小鼠的小鼠胚胎成纤维细胞(mef)细胞中α-l-艾杜糖苷酶活性的影响。mef细胞在阿霉素(5.0至160μm)或媒剂对照(0.1%dmso)存在下生长48小时。48小时后,获得细胞裂解物并测定mefα-l-艾杜糖苷酶的活性。
具体实施方式
定义
如本文所用,术语“烷基”,无论是单独使用还是作为取代基的一部分使用,均包括包含1-20个碳原子的直链烃基。因此,所述短语包括如甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、癸基、十一烷基、十二烷基等的直链烷基。所述短语还包括直链烷基的支链异构体,包括但不限于通过举例提供的以下:-ch(ch3)2、-ch(ch3)(ch2ch3)、-ch(ch2ch3)2、-c(ch3)3、-c(ch2ch3)3、-ch2ch(ch3)2、-ch2ch(ch3)(ch2ch3)、-ch2ch(ch2ch3)2、-ch2c(ch3)3、-ch2c(ch2ch3)3、-ch(ch3)ch(ch3)(ch2ch3)、-ch2ch2ch(ch3)2、-ch2ch2ch(ch3)(ch2ch3)、-ch2ch2ch(ch2ch3)2、-ch2ch2c(ch3)3、-ch2ch2c(ch2ch3)3、-ch(ch3)ch2ch(ch3)2、-ch(ch3)ch(ch3)ch(ch3)ch(ch3)2、-ch(ch2ch3)ch(ch3)ch(ch3)(ch2ch3)以及其它。所述短语还包括环烷基,如环丙基、环丁基、环戊基、环己基、环庚基和环辛基,以及被如上定义的直链和支链烷基取代的此类环。所述短语还包括多环烷基,如但不限于金刚烷基降冰片基和双环[2.2.2]辛基以及被如上定义的直链和支链烷基取代的此类环。
如本文所用,无论是单独使用还是作为取代基的一部分使用,术语“烯基”包括具有在任意两个相邻碳原子之间的至少一个双键的烷基。
如本文所用,术语“未取代的烷基”和“未取代的烯基”是指不含杂原子的烷基、烯基和炔基。
如本文所用,术语“取代的烷基”和“取代的烯基”是指如上定义的烷基烯基和炔基,其中与碳或氢的一个或多个键被键取代为非氢或非碳原子,如但不限于,基团,如烷氧基和芳氧基中的氧原子;基团,如烷基和芳基硫醚基团、砜基团、磺酰基和亚砜基团中的硫原子;基团,如三烷基甲硅烷基、二烷基芳基甲硅烷基、烷基二芳基甲硅烷基和三芳基甲硅烷基中的硅原子;和其它各种基团中的其它杂原子。
如本文所用,术语“烷酰基”是指烷基羰基,其中烷基部分是取代的或未取代的。
如本文所用,术语“烯酰基”是指烯基羰基,其中烯基部分是取代的或未取代的。
如本文所用,术语“治疗(treatment/treat/treating)”是指在疾病或病状的症状、示象或特征出现后开始的以便消除或减少此类症状、示象或特征的作用过程(如给予缀合物或医药组合物)。此类治疗不一定是绝对有用的。
如本文所用,术语“需要治疗”是指护理人员对患者需要或将从治疗中获益的判断。这一判断是基于护理人员专业知识领域中的诸多因素做出,但其包括患者生病或将生病的知识,其作为可通过本发明方法或化合物治疗的疾病或病状的结果。
如本文所用,术语“需要预防”是指护理人员对患者需要或将从预防中获益的判断。这一判断是基于护理人员专业知识领域中的诸多因素做出,但其包括患者将生病或可能生病的知识,其作为可通过本发明方法或化合物预防的疾病或病状的结果。
如本文所用,术语“个体”、“受试者”或“患者”是指任何动物,包括哺乳动物,如小鼠、大鼠、其它啮齿动物、兔、狗、猫、猪、牛、绵羊、马或灵长类动物和人类。所述术语可以指定男性或女性或两者,或排除男性或女性。
如本文所用,术语“治疗有效量”是指单独或作为医药组合物的一部分的缀合物的量,其能够对疾病或病状的任何症状、示象或特征具有任何可检测的积极作用。此类效果不一定是绝对有益的。
当在本说明书中提及时,以下取代基被认为具有以下结构:
背景
在本研究中,使用高通量筛选(hts)评估了1600个临床批准的化合物库,以鉴定抑制cftr-ptc无义突变的药剂。进行两次主要的平行分析(一次为机理分析,另一次为表型分析),然后在一系列二级分析中确认选定的化合物,这些化合物对cftr无义突变的通读具有特异性,这些突变也经证明对临床成功具有预测性。通过这一过程,鉴定出几种诱导通读的化合物,包括七叶皂苷,其为一种在表达cftr提前终止等位基因的各种原代人类气道细胞中有效且强效的天然产物。这些结果表明,鉴定的化合物可用于治疗无义介导的遗传疾病,如cf。
抑制ptc和诱导通读的药剂是不同医药类别的混合物(参见表1)。由多于一种药剂代表的类别是翻译抑制剂(环己酰胺)、抗炎药(秋水仙碱、七叶皂苷)、抗蠕虫药(奥苯达唑)和皮质类固醇(泼尼卡酯、数种相关药剂)。在cftr表达和功能分析中有效增强ptc抑制的翻译抑制剂的鉴定表明其降低翻译精确度的能力发生在完全翻译抑制之前的剂量下。
本发明使用两种互补分析,其与侧重于cftr表达和功能的稳健的一系列二级测试相结合。在这样做时,避免了荧光素酶稳定化所遇到的问题,其已被报道增强萤火虫荧光素酶信号而不依赖于ptc的通读活性。在二级筛选中,双荧光素酶通读分析用于验证在hts中获得的初始数据,并允许萤火虫荧光素酶内部归一化为海肾荧光素酶,以消除mrna丰度的变化或翻译起始。药剂的二次评估显示,分析之间的一致性通常很高。通读(tecc)和功能(双荧光素酶)分析之间相对较弱的相关性可能是由于细胞生长和翻译如何影响分析中检测到的读数的差异。例如,在tecc分析中,当细胞生长和翻译最小时,在单层形成后加入药物。相反,在双荧光素酶分析中,在细胞接种期间添加药物,同时生长和翻译都快速发生。此外,在翻译和cftr依赖性离子转运之间存在多个可能影响精确相关性的干预步骤(即er保留、camp依赖性门控等)。然而,每次分析产生的相对平衡的化合物数量表明,当与适当的二级评估策略配对时,任一种方法都可以充当主要筛选。
在八种先导化合物中,七叶皂苷展现出一种引起关注的特征,并且是初始筛选中少数几种对添加cftrinh-172有强烈反应的药剂之一。有趣的是,与其通读功效相比,七叶皂苷处理后frt和hbe细胞中的功能性cftr活性相对较强。这种效果可以通过mrna转录物的稳定化和非有义介导的rna衰变(nmd)的抑制来解释,这可预期在ptc抑制的情况下协同地改善功能。另一种可能性是七叶皂苷作为cftr激动剂的双重作用。由于七叶皂苷对一氧化氮(no)代谢的影响,其改善了慢性静脉功能不全患者的结果。no还参与人类肺上皮细胞中cftr通道和氯电流的激活,这可能是所述化合物的潜在优势。本发明评估了cftr非依赖性通读构建体、cftr缺陷亲本细胞和δf508纯合hbe细胞中的七叶皂苷。这些实验结果清楚地表明,七叶皂苷的作用并非由于在通读之外刺激cftr活性。
先前有使用草药制剂治疗cf的经验。例如,已显示姜黄素在体外激活cftr通道,并且还可具有物种特异性校正剂活性。来自中药植物的两种白藜芦醇寡聚物被鉴定为cftr抑制剂;其它已从草本植物狭叶红景天(rhodiolakirilowii)中分离出来。这种经验使得对七叶皂苷可能改善cf结果的可能性持乐观态度。七叶皂苷高度适于人类给药,并先前已有效地用于慢性静脉功能不全,展现出合理的安全性和药物动力学特性(sutera等人,《先进疗法(advther)》;23:179-190;2006)。
方法
如上所论述,ptc密码子的存在导致多肽过早截短并且全部或部分缺乏其野生型对应物的功能活性。当例如用本发明的化合物刺激ptc的通读时,由近同源氨酰基trna携带的氨基酸,或在一些情况下,同源氨酰基trna,被插入到错误终止密码子的多肽链中,使得翻译继续,并部分恢复全长功能蛋白。因此,蛋白质的表达增加。当并有近同源氨酰基trna时,所得多肽含有野生型多肽中不存在的氨基酸。此外,由于ptc的存在,mrna转录物本身可能受到nmd的影响。nmd是存在于所有真核生物中的一种监视途径。其主要功能是通过消除含有ptc的mrna转录物来减少基因表达错误。通过减少nmd的过程,含有ptc的mrna转录物的水平增加,因此在本发明的通读刺激化合物存在下可用于翻译。本发明证明,所公开的某些化合物不仅会刺激ptc密码子的通读,而且还会抑制nmd,从而增加含有ptc的mrna转录物的稳定性和/或水平。
在一个实施例中,本发明提供用于治疗至少部分地通过ptc的存在介导的疾病或病状的方法。此类疾病和病状包括但不限于其中编码多肽的基因含有无义突变,导致多肽中存在ptc密码子,引起多肽无功能或功能降低的那些疾病和病状。这些疾病和病状包括但不限于cf、杜氏肌营养不良(duchennemusculardystrophy)、贝克肌营养不良(beckermusculardystrophy)、脊髓性肌萎缩,、粘多糖贮积症i(包括赫尔勒综合征(hurlersyndrome)、赫尔勒-沙伊综合征(hurler-scheiesyndrome)和沙伊综合征(scheiesyndrome))、血友病、b型血友病、尤塞氏综合征(ushersyndrome)和癌症。癌症中的ptc包括但不限于肿瘤抑制基因(如但不限于p53)中的ptc。代表的p53中的ptc密码子包括但不限于w53x、q144x、w146x、q192x、r196x、r213x、e298x和r306x。
本文所述的方法包含向受试者给予本发明化合物的步骤。这种给药会刺激ptc的通读和/或增加含有ptc的mrna的稳定性。在本实施例的一个方面,这种给药会抑制天然存在的ptc并刺激含有ptc的mrna转录物的通读。在本实施例的另一方面,这种给药会增加含有ptc密码子的mrna转录物的水平和/或稳定性,并抑制天然存在的ptc并刺激含有ptc的mrna转录物的通读。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。在所述方法中,ptc可以是所属领域已知的任何ptc。在另一个实施例中,ptc已知或怀疑至少部分地引起疾病或病状或与疾病或病状相关。在另一个实施例中,ptc是与cf相关或引起cf的一种,如但不限于r553x、g542x、w1282x、r1162x和y122x。在另一个实施例中,ptc是与赫尔勒综合征相关或引起赫尔勒综合征的一种,如但不限于w402x、q70x、r628x、q148x或e404x。
在另一个实施例中,本发明提供治疗受试者中cf的方法,其中cf至少部分地通过ptc的存在介导。在本实施例的某些方面,ptc密码子产生无功能多肽,如但不限于ctfr。所述方法包含向受试者给予本发明化合物的步骤。这种给药会刺激ptc的通读和/或增加含有ptc的mrna的稳定性。在本实施例的一个方面,这种给药会抑制天然存在的ptc并刺激含有ptc的mrna转录物的通读。在本实施例的另一方面,这种给药会增加含有ptc密码子的mrna转录物的水平和/或稳定性,并抑制天然存在的ptc并刺激含有ptc的mrna转录物的通读。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。
在另一个实施例中,本发明提供治疗受试者中cf的方法,其中cf至少部分地通过cftr中ptc的存在介导。在本实施例的某些方面,ptc密码子产生无功能多肽cftr。这种方法包含向受试者给予本发明化合物并进一步向受试者给予以下至少一种的步骤:i)增加cftr多肽功能的药剂(例如cftr增效剂);ii)增加cftr多肽运输的药剂(例如cftr校正剂);iii)抑制nmd的药剂(例如nmd抑制剂);iv)增加靶mrna的mrna水平的药剂(例如组蛋白脱乙酰基酶抑制剂);v)增加一般肺功能的药剂;或vi)前述的组合(前述中的每一种均被认为是第二药剂)。在本方法中,化合物的给予会刺激ptc的通读和/或增加mrna转录物的稳定性,从而增加受试者中cftr多肽的水平,同时给予增加cftr多肽功能的药剂会改善由ptc通读产生的cftr的功能,使得可改善cftr多肽运输的药剂会增加由功能相关位置(例如细胞表面)处的ptc通读产生的cftr多肽水平,并且抑制nmd和/或增加mrna水平的药剂会为本发明化合物的作用提供增加的mrna水平。在本方法的一个方面,仅将增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在本实施例的另一方面,仅将增加cftr多肽向细胞表面运输的药剂(例如cftr校正剂)与本发明化合物一起给予。在本实施例的另一方面,将增加cftr多肽功能的药剂和增加cftr多肽向细胞表面运输的药剂(例如cftr增效剂和cftr校正剂)与本发明化合物一起给予。在本实施例的另一方面,将抑制nmd的药剂(例如nmd抑制剂)与本发明化合物一起给予。在本实施例的另一方面,将抑制nmd的药剂和增加cftr多肽向细胞表面运输的药剂(例如cftr校正剂)与本发明化合物一起给药。在本实施例的另一方面,将增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在本实施例的另一方面,将增加cftr多肽向细胞表面的运输的药剂(例如cftr校正剂)和增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在任何前述实施例中,增加mrna水平的药剂可以与本发明化合物一起给予。在任何前述实施例中,增加一般肺功能的药剂可以与本发明化合物一起给予。
在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。在本实施例的一个方面,增加cftr多肽功能的药剂是vx-770、gp-5或前述的组合。在本实施例的一个方面,增加cftr多肽运输的药剂是vx-809、c1、c2、c1+c2或前述的组合。在本实施例的一个方面,抑制nmd的药剂是nmdi-1、nmdi-9、nmdi-25或nmdi-14。一种或多种药剂的给予可以在给予本发明化合物之前、同时或之后进行。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于治疗受试者中的粘多糖贮积症i(包括但不限于赫尔勒综合征、赫尔勒-沙伊综合征和沙伊综合征)的方法,其中前述中的任一种至少部分地由α-l艾杜糖苷酶中ptc的存在介导。在本实施例的某些方面,ptc密码子产生无功能α-l艾杜糖苷酶多肽。此类方法包含向受试者给予本发明化合物并任选地进一步向受试者给予抑制nmd的药剂的步骤。在本方法中,化合物的给予会刺激ptc的通读和/或增加mrna转录物的稳定性,从而增加受试者中α-l艾杜糖苷酶多肽的水平。当给予抑制nmd的药剂时,这种药剂会增加编码α-l艾杜糖苷酶多肽的mrna的水平。在一个实施例中,粘多糖贮积症i是赫尔勒综合征。
在本实施例的一个方面,ptc是w402x、q70x、r628x、q148x、e404x、y343x或q548x。在本实施例的另一方面,ptc是w402x。在本实施例的另一方面,ptc是q70x。在本实施例的一个方面,抑制nmd的药剂是nmdi-1、nmdi-9、nmdi-25或nmdi-14。一种或多种药剂的给予可以在给予本发明化合物之前、同时或之后进行。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于在受试者中药理学抑制ptc的方法。所述方法包含向受试者给予本发明化合物的步骤。这种给药会刺激ptc的通读。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于药理学抑制受试者中与cf相关的ptc的方法。所述方法包含向受试者给予本发明化合物的步骤。这种给药会刺激与cf相关的ptc的通读。在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于药理学抑制受试者中与cf相关的ptc的方法,其中ptc存在于cftr中。在本实施例的某些方面,ptc密码子产生无功能多肽ctfr。这种方法包含向受试者给予本发明化合物并进一步向受试者给予以下至少一种的步骤:i)增加cftr多肽功能的药剂(例如cftr增效剂);ii)增加cftr多肽运输的药剂(例如cftr校正剂);iii)抑制nmd的药剂(例如nmd抑制剂);iv)增加靶mrna的mrna水平的药剂(例如组蛋白脱乙酰基酶抑制剂);v)增加一般肺功能的药剂;或vi)前述的组合(前述中的每一种均被认为是第二药剂)。在本方法中,化合物的给予会刺激ptc的通读和/或增加mrna转录物的稳定性,从而增加受试者中cftr多肽的水平,同时给予增加cftr多肽功能的药剂会改善由ptc通读产生的cftr的功能,使得可改善cftr多肽运输的药剂会增加由功能相关位置(例如细胞表面)处的ptc通读产生的cftr多肽水平,并且抑制nmd和/或增加mrna水平的药剂会为本发明化合物的作用提供增加的mrna水平。在本方法的一个方面,仅将增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在本实施例的另一方面,仅将增加cftr多肽向细胞表面运输的药剂(例如cftr校正剂)与本发明化合物一起给予。在本实施例的另一方面,将增加cftr多肽功能的药剂和增加cftr多肽向细胞表面运输的药剂(例如cftr增效剂和cftr校正剂)与本发明化合物一起给予。在本实施例的另一方面,将抑制nmd的药剂(例如nmd抑制剂)与本发明化合物一起给予。在本实施例的另一方面,将抑制nmd的药剂和增加cftr多肽向细胞表面运输的药剂(例如cftr校正剂)与本发明化合物一起给药。在本实施例的另一方面,将增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在本实施例的另一方面,将增加cftr多肽向细胞表面的运输的药剂(例如cftr校正剂)和增加cftr多肽功能的药剂(例如cftr增效剂)与本发明化合物一起给予。在任何前述实施例中,增加mrna水平的药剂可以与本发明化合物一起给予。在任何前述实施例中,增加一般肺功能的药剂可以与本发明化合物一起给予。
在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。在本实施例的一个方面,增加cftr多肽功能的药剂是vx-770、gp-5或前述的组合。在本实施例的一个方面,增加cftr多肽运输的药剂是vx-809、c1、c2、c1+c2或前述的组合。在本实施例的一个方面,抑制nmd的药剂是nmdi-1、nmdi-9、nmdi-25或nmdi-14。一种或多种药剂的给予可以在给予本发明化合物之前、同时或之后进行。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于稳定含有ptc的mrna转录物的方法,其包括通过抑制无义介导的衰变。所述方法包含向受试者给予本发明化合物的步骤。在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。在本实施例的一个方面,ptc是w402x、q70x、r628x、q148x、e404x、y343x或q548x。在本实施例的另一方面,ptc是w402x。在本实施例的另一方面,ptc是q70x。在本实施例的另一方面,ptc是q70x。在本实施例的一个方面,ptc与选自由以下组成的组的疾病或病状相关:囊性纤维化、杜氏肌营养不良、贝克肌营养不良、脊髓性肌萎缩、赫尔勒综合征、血友病、b型血友病、尤塞氏综合征和癌症。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在另一个实施例中,本发明提供用于增加含有ptc密码子的mrna转录物的水平的方法,其包括通过抑制无义介导的衰变。所述方法包含向受试者给予本发明化合物的步骤。在本实施例的一个方面,ptc是r553x、g542x、w1282x、r1162x、y122x、q637x、q2x、s4x、q39x或q1313x。在本实施例的另一方面,ptc是g542x。在本实施例的另一方面,ptc是w1282x。在本实施例的一个方面,ptc是w402x、q70x、r628x、q148x、e404x、y343x或q548x。在本实施例的另一方面,ptc是w402x。在本实施例的另一方面,ptc是q70x。在本实施例的一个方面,ptc与选自由以下组成的组的疾病或病状相关:囊性纤维化、杜氏肌营养不良、贝克肌营养不良、脊髓性肌萎缩、赫尔勒综合征、血友病、b型血友病、尤塞氏综合征和癌症。在本实施例的一个方面,化合物以治疗有效量给予。此外,此类方法还可包含鉴定需要这种治疗的受试者,如通过鉴定受试者中ptc的存在。
在一个优选的实施例中,上述方法还可包含给予可以在本发明化合物之前或与其同时给予的第二药剂。在某些实施例中,第二药剂为:i)增加cftr多肽功能的药剂(例如cftr增效剂);ii)增加cftr多肽运输的药剂(例如cftr校正剂);iii)抑制nmd的药剂(例如nmd抑制剂);iv)增加靶mrna的mrna水平的药剂(例如组蛋白脱乙酰基酶抑制剂);v)增加一般肺功能的药剂;或vi)前述的组合(前述中的每一种均被认为是第二药剂)。在某些实施例中,所述第二药剂是cftr增效剂、cftr校正剂、nmd抑制剂、增加mrna水平的药剂或前述中的任一种的组合。在某些实施例中,cftr增效剂是vx-770、gp-5或前述的组合。在某些实施例中,cftr校正剂是vx-809、c1、c2或前述的组合。在某些实施例中,nmd抑制剂是nmdi-1、nmdi-9、nmdi-25、nmdi-14或前述的组合。在某些实施例中,增加mrna水平的药剂是组蛋白脱乙酰基酶抑制剂。在某些实施例中,增加一般肺功能的药剂是黄酮、异黄酮、乳香酸、毛喉素、egcg、胡椒碱(bioperine)、槲皮素、穗花杉双黄酮(amentoflavone)、反式白藜芦醇、姜黄或前述的组合。
在前述方法中,治疗上适用的化合物的代表性实例包括但不限于七叶皂苷、七叶皂苷的纯化或部分纯化的异构体、式i化合物、式iia化合物、式iib化合物、七叶皂苷1a、七叶皂苷1b、异七叶皂苷1a、异七叶皂苷1b、秋水仙碱、环己酰胺、阿霉素、四磷酸咯萘啶(pt)、泼尼卡酯、奥苯达唑、阿苯达唑、甲苯咪唑和对氨基苯甲酸钾(paba)。在某些优选的实施例中,所述化合物是式ia化合物。在某些优选的实施例中,所述化合物是式ib化合物。在某些优选的实施例中,所述化合物是式iia化合物。在某些优选的实施例中,所述化合物是式iib化合物,其中r4是h。在某些优选的实施例中,化合物是七叶皂苷1a。在某些优选的实施例中,化合物是七叶皂苷1b。在某些优选的实施例中,化合物是异七叶皂苷1a。在某些优选的实施例中,化合物是异七叶皂苷1b。在某些优选的实施例中,所述化合物是对氨基苯甲酸钾(paba)。
本发明化合物的给药可以如所属领域已知的那样完成,因为所公开的化合物都已知在治疗上适用于人类的其它病状。这包括口服给药方案,以在人类血浆中提供最有效浓度。这还包括采用皮下或静脉内给药途径以达到足够的水平的给药方案。
用于本发明的化合物
如上所论述,本发明提供用于增加ptc密码子通读的方法,用于稳定含有ptc密码子的mrna转录物的方法和用于通过给予本发明化合物来增加含有ptc密码子的mrna的水平的方法。
本文公开多种可用于所公开的方法中的化合物。本文描述的或包含在本发明组合物中的某些化合物可以以特定的几何或立体异构形式存在。此外,本发明化合物还可以是光学活性的。本发明涵盖所有此类化合物,包括其顺式和反式异构体、(r)-和(s)-对映异构体、非对映异构体、(d)-异构体、(l)-异构体、外消旋混合物以及其它混合物,其均属于本发明的范围内。另外的不对称碳原子可以存在于取代基,如烷基中。所有这些异构体及其混合物都包括在本发明中。在某些情况下,可以指定特定的几何或立体异构形式。
在一个实施例中,化合物是七叶皂苷(也称为七叶素)。七叶皂苷是欧洲七叶树(aesculushippocastanum)(马栗树)中发现的皂苷的混合物。七叶皂苷是马栗树中的主要活性化合物,并且是其大部分药用特性的原因所在。欧洲七叶树种子的提取物在中国传统上被用作胃用镇痛剂、退烧药和抗痔剂(matsuda等人,1997)。从种子中分离的皂苷混合物是五环三萜,并被称为以α和β形式存在的七叶皂苷。这是已经被描述为展现消炎(matsuda等人,1997;rothkopf及vogel,1976);抗水肿、毛细血管保护性、降血糖(kimura等人,2006)、抗肥胖(hu等人,2008)和乙醇吸收抑制(sirtori,2001;yoshikawa等人,1996)活性的七叶皂苷的β形式。七叶皂苷被发现会抑制小鼠中由乙酸所致和大鼠中由组胺所致的急性炎症(matsuda等人,1998);抑制大鼠中的创伤性脑损伤(xiao及wei,2005);减轻术后粘连(fu等人,2005年);加速胃肠传输(matsuda等人,1999);抑制大鼠中的脑缺血损伤所致凋亡(hu等人,2004);消除大鼠中的经卵巢切除引起的骨质减少(pytlik等人,2000;pytlik等人,1999);显示降血糖活性(yoshikawa等人,1996);并展现抗溃疡作用(marhuenda等人,1994)。这种三萜也被发现会抑制大鼠中的慢性异常灶形成,并诱导人类结肠癌ht29细胞凋亡(patlolla等人,2006)。七叶皂苷在hiv-1患者的临床试验(grases等人,2004)中治疗钝物撞击伤(wetzel等人,2002)以及皮肤瘙痒(li等人,2004)。此外,发现这种三萜会抑制内皮细胞上粘附分子的表达(hu等人,2004;montopoli等人,2007),预防缺氧引起的中性粒细胞与内皮细胞的粘附(arnould等人,1996),并抑制hiv-1蛋白酶(yang等人,1999)。临床证据表明七叶皂苷会安全有效地治疗慢性静脉功能不全(sirtoricr,《药理学研究(pharmacol.res.)》44(3):183-193,2001年九月;pittlermh等人;《考科蓝系统综述数据库(cochranedatabasesystrev)》(1):cd003230,2006;diehmc.等人;《柳叶刀(lancet)》347(8997):292-294,1996年二月)。
在一个实施例中,化合物是七叶皂苷。当指定七叶皂苷没有任何进一步的特征界定时,其指的是从欧洲七叶树(如从种子)纯化的七叶皂苷,并且被理解为包括五环三萜的混合物并且包括α和β形式并且包括七叶皂苷1a、七叶皂苷1b、异七叶皂苷1a和异七叶皂苷1b。在本实施例的一个方面,七叶皂苷包含通式ia化合物或其药学上可接受的盐、水合物或溶剂合物的混合物:
其中:
r1选自由以下组成的组:当归酰基和顺芷酰基;
r2选自由以下组成的组:乙酰基(-c(o)ch3)和-h;并且
r3选自由以下组成的组:乙酰基(-c(o)ch3)和-h。
在本实施例的一个方面,化合物的混合物包括至少一种下列化合物:
i)式ia化合物,其中r1是顺芷酰基,r2是乙酰基并且r3为-h(七叶皂苷1a);
ii)式ia化合物,其中r1是当归酰基,r2是乙酰基并且r3是-h(七叶皂苷1b);
iii)式ia化合物,其中r1是顺芷酰基,r2是-h并且r3是乙酰基(异七叶皂苷1a);和
iv)式ia化合物,其中r1是当归酰基,r2是-h并且r3是乙酰基(异七叶皂苷1b)。
在某些实施例中,混合物含有全部4种化合物i)至iv)。在某些实施例中,混合物含有化合物i)和任选的化合物ii)至iv)中的一种或多种。在某些实施例中,混合物含有化合物ii)和任选的化合物i)或iii)至iv)中的一种或多种。
在本实施例的另一方面,七叶皂苷包含通式ib化合物或其药学上可接受的盐、水合物或溶剂合物的混合物,其中r1、r2和r3如关于通式ia化合物所定义。
在本实施例的一个方面,化合物的混合物包括至少一种下列化合物:
v)式ib化合物,其中r1是顺芷酰基,r2是乙酰基并且r3为-h(七叶皂苷1a);
vi)式ib化合物,其中r1是当归酰基,r2是乙酰基并且r3是-h(七叶皂苷1b);
vii)式ib化合物,其中r1是顺芷酰基,r2是-h并且r3是乙酰基(异七叶皂苷1a);和
viii)式ib化合物,其中r1是当归酰基,r2是-h并且r3是乙酰基(异七叶皂苷1b)。
在某些实施例中,混合物含有全部4种化合物v)至viii)。在某些实施例中,混合物含有化合物v)和任选的化合物vi)至viii)中的一种或多种。在某些实施例中,混合物含有化合物vi)和任选的化合物v)和vii)至viii)中的一种或多种。在某些实施例中,混合物含有化合物vii)和任选的化合物v)、vi)和viii)中的一种或多种。在某些实施例中,混合物含有化合物viii)和任选的化合物v)至vii)中的一种或多种。
在另一个实施例中,所述化合物是七叶皂苷的纯化异构体。合适的异构体包括但不限于:
七叶皂苷1a,其中r1是顺芷酰基,r2是乙酰基并且r3是式ia或ib中的-h;
七叶皂苷1b,其中r1是当归酰基,r2是乙酰基并且r3是式ia或ib中的-h;
异七叶皂苷1a,其中r1是顺芷酰基,r2是-h并且r3是式ia或ib中的乙酰基;
异七叶皂苷1b,其中r1是当归酰基,r2是-h并且r3是式ia或ib中的乙酰基;和
前述的混合物。
在某些实施例中,混合物含有全部4种化合物。在某些实施例中,混合物含有七叶皂苷1a和任选的七叶皂苷1b、异七叶皂苷1a和异七叶皂苷1b中的一种或多种。在某些实施例中,混合物含有七叶皂苷1b和任选的七叶皂苷1a、异七叶皂苷1a和异七叶皂苷1b中的一种或多种。在某些实施例中,混合物含有异七叶皂苷1a和任选的七叶皂苷1a、七叶皂苷1b和异七叶皂苷1b中的一种或多种。在某些实施例中,混合物含有异七叶皂苷1b和任选的七叶皂苷1a、七叶皂苷1b和异七叶皂苷1a中的一种或多种。在某些实施例中,混合物含有七叶皂苷1a和七叶皂苷1b以及任选的异七叶皂苷1a和异七叶皂苷1b中的一种或多种。
在一个具体实施例中,七叶皂苷的异构体是七叶皂苷1a。在另一个具体实施例中,七叶皂苷的异构体是异七叶皂苷1a。在一个具体实施例中,七叶皂苷的异构体是七叶皂苷1b。在另一个具体实施例中,七叶皂苷的异构体是异七叶皂苷1b。
当关联七叶皂苷的异构体使用时,术语“纯化的”意指指定的异构体以相对于所存在的化合物的总重量,等于或大于85wt%(如90wt%、95wt%、98wt%或99wt%)存在。当关联七叶皂苷的异构体使用时,术语“部分纯化的”意指指定的异构体以相对于所存在的化合物的总重量,等于或大于50wt%(如60wt%、70wt%、80wt%或90wt%)存在。
在另一个实施例中,所述化合物具有式iia或式iib的结构或其药学上可接受的盐、水合物或溶剂合物
其中:
r1、r2和r3各自独立地选自由以下组成的组:-h、烷基、烯基、烷酰基和烯酰基;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的一个方面,烷基具有长度为1至10个碳的烷基部分,并且烷基部分是取代或未取代的。在本实施例的另一方面,烷基具有长度为1至10个碳的烷基部分,并且烷基部分是未取代的。在本实施例的另一方面,烷基具有长度为1至5个碳的烷基部分,并且烷基部分是未取代的。
在本实施例的一个方面,烯基具有长度为1至10个碳的烷基部分,并且烯基部分是取代或未取代的。在本实施例的另一方面,烯基具有长度为1至10个碳的烷基部分,并且烯基部分是未取代的。在本实施例的另一方面,烯基具有长度为1至5个碳的烷基部分,并且烯基部分是未取代的。
在本实施例的一个方面,烷酰基具有长度为1至10个碳的烷基部分,并且烷基部分是取代或未取代的。在本实施例的另一方面,烷酰基具有长度为1至10个碳的烷基部分,并且烷基部分是未取代的。在本实施例的另一方面,烷酰基具有长度为1至5个碳的烷基部分,并且烷基部分是未取代的。代表性的烷酰基包括但不限于乙酰基(acetyl)、己酰基、苯甲酰基、乙基丁酰基、乙酰基(ethanoyl)、丙酰基、异丁酰基和丁酰基。
在本实施例的一个方面,烯酰基具有长度为1至10个碳的烯基部分,并且烯基部分是取代或未取代的。在本实施例的另一方面,烯酰基具有长度为1至10个碳的烯基部分,并且烯基部分是未取代的。在本实施例的另一方面,烯酰基具有长度为1至5个碳的烯基部分,并且烯基部分是未取代的。代表性的烯酰基包括但不限于当归酰基、顺芷酰基、千里酰基、巴豆酰基、3,3-二甲基丙烯酰基、桂皮酰基、戊烯酰基、2-丙烯酰基、(2e)-2-戊烯酰基和4-戊烯酰基。
在本实施例的另一方面:
r1选自由以下组成的组:-h、烷基、烯基、乙酰基(acetyl)、己酰基、苯甲酰基、乙基丁酰基、乙酰基(ethanoyl)、丙酰基、异丁酰基、丁酰基、当归酰基、顺芷酰基、千里酰基、巴豆酰基、3,3-二甲基丙烯酰基、桂皮酰基、戊烯酰基、2-丙烯酰基、(2e)-2-戊烯酰基和4-戊烯酰基;
r2是乙酰基;
r3是-h;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的另一方面:
r1选自由以下组成的组:-h、当归酰基、顺芷酰基、千里酰基、巴豆酰基、3,3-二甲基丙烯酰基、桂皮酰基、戊烯酰基、2-丙烯酰基、(2e)-2-戊烯酰基和4-戊烯酰基;
r2是乙酰基;
r3是-h;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的另一方面:
r1选自由以下组成的组:当归酰基和顺芷酰基;
r2是乙酰基;
r3是-h;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的另一方面:
r1选自由以下组成的组:-h、烷基、烯基、乙酰基(acetyl)、己酰基、苯甲酰基、乙基丁酰基、乙酰基(ethanoyl)、丙酰基、异丁酰基、丁酰基、当归酰基、顺芷酰基、千里酰基、巴豆酰基、3,3-二甲基丙烯酰基、桂皮酰基、戊烯酰基、2-丙烯酰基、(2e)-2-戊烯酰基和4-戊烯酰基;
r2是-h;
r3是乙酰基;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的另一方面:
r1选自由以下组成的组:-h、当归酰基、顺芷酰基、千里酰基、巴豆酰基、3,3-二甲基丙烯酰基、桂皮酰基、戊烯酰基、2-丙烯酰基、(2e)-2-戊烯酰基和4-戊烯酰基;
r2是-h;
r3是乙酰基;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在本实施例的另一方面:
r1选自由以下组成的组:当归酰基和顺芷酰基;
r2是-h;
r3是乙酰基;并且
r4选自由以下组成的组:1-11个糖单元的线性糖链、1-11个糖单元的分支糖链和-h。
在前述实施例的一个方面,r4是1至5个糖单元的线性糖链。在本实施例的另一方面,r4是3个糖单元的线性糖链。
在前述实施例的一个方面,r4是1至5个糖单元的分支糖链。在前述实施例的另一方面,r4是3个糖单元的分支糖链。在前述实施例的又一方面,r4是针对式iia化合物的结构iiia分支糖链或针对式iib化合物的结构iiib分支糖链:
在本实施例的一个方面,r4为-h并且化合物可被认为是皂苷元(sapogenin)。
在本实施例的第一优选方面,所述化合物具有通式iia,其中r1是当归酰基,r2是乙酰基,r3为-h并且r4为-h。
在本实施例的第二个优选方面,所述化合物具有通式iia,其中r1是顺芷酰基,r2是乙酰基,r3为-h并且r4为-h。
在本实施例的第三个优选方面,所述化合物具有通式iia,其中r1是当归酰基,r2是-h,r3为乙酰基并且r4是-h。
在本实施例的第四优选方面,所述化合物具有通式iia,其中r1是顺芷酰基,r2是-h,r3为乙酰基并且r4是-h。
在本实施例的第五优选方面,所述化合物具有通式iia,其中r1是当归酰基,r2是乙酰基,r3为-h并且r4是1-5个糖单元的分支糖链。
在本实施例的第六优选方面,所述化合物具有通式iia,其中r1是顺芷酰基,r2是乙酰基,r3为-h并且r4是1-5个糖单元的分支糖链。
在本实施例的第七个优选方面,所述化合物具有通式iia,其中r1是当归酰基,r2是-h,r3为乙酰基并且r4是1-5糖单元的支链糖链。
在本实施例的第八优选方面,所述化合物具有通式iia,其中r1是顺芷酰基,r2是-h,r3为乙酰基并且r4是1-5糖单元的支链糖链。
在本实施例的第九优选方面,所述化合物具有通式iib,其中r1是当归酰基,r2是乙酰基,r3为-h并且r4为-h。
在本实施例的第十优选方面,所述化合物具有通式iib,其中r1是顺芷酰基,r2是乙酰基,r3为-h并且r4为-h。
在本实施例的第十一优选方面,所述化合物具有通式iib,其中r1是当归酰基,r2是-h,r3为乙酰基并且r4是-h。
在本实施例的第十二优选方面,所述化合物具有通式iib,其中r1是顺芷酰基,r2是-h,r3为乙酰基并且r4是-h。
在本实施例的第十三优选方面,所述化合物具有通式iib,其中r1是当归酰基,r2是乙酰基,r3为-h并且r4是1-5个糖单元的分支糖链。
在本实施例的第十四优选方面,所述化合物具有通式iib,其中r1是顺芷酰基,r2是乙酰基,r3为-h并且r4是1-5个糖单元的分支糖链。
在本实施例的第十五优选方面,所述化合物具有通式iib,其中r1是当归酰基,r2是-h,r3为乙酰基并且r4是1-5糖单元的支链糖链。
在本实施例的第十六优选方面,所述化合物具有通式iib,其中r1是顺芷酰基,r2是-h,r3为乙酰基并且r4是1-5糖单元的支链糖链。
在另一个实施例中,化合物是阿苯达唑或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是秋水仙碱或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是环己酰胺或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是奥苯达唑或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是甲苯咪唑或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是四磷酸咯萘啶或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是泼尼卡酯或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是对氨基苯甲酸钾(paba)或其药学上可接受的盐、水合物或溶剂合物。
在另一个实施例中,化合物是阿霉素或其药学上可接受的盐、水合物或溶剂合物。
物质组合物、组合和调配物
本发明还提供物质组合物、组合和调配物,其包含纯化或部分纯化的七叶皂苷异构体的混合物,基本上由其组成或由其组成。此类物质组合物、组合和调配物提供优于所属领域化合物的优点,所述优点在于七叶皂苷的特定异构体以纯化或部分纯化的形式组合,而现有技术的物质组合物、组合和调配物含有来自植物来源的未分级的提取物。
在本实施例的一个方面,物质组合物、组合或调配物包含通式iia或1b化合物或其药学上可接受的盐、水合物或溶剂合物的混合物,基本上由其组成或由其组成,其中r1、r2和r3如关于通式ia化合物所定义。
在本实施例的一个方面,物质组合物、组合或调配物包含七叶皂苷1a、七叶皂苷1b、异七叶皂苷1a和异七叶皂苷1b;基本上由其组成或由其组成。在本实施例的另一方面,物质组合物、组合或调配物包含七叶皂苷1a和任选的七叶皂苷1b、异七叶皂苷1a和异七叶皂苷1b中的一种或多种,基本上由其组成或由其组成。在本实施例的另一方面,物质组合物、组合或调配物包含七叶皂苷1b和任选的七叶皂苷1a、异七叶皂苷1a和异七叶皂苷1b中的一种或多种,基本上由其组成或由其组成。在本实施例的另一方面,物质组合物、组合或调配物包含异七叶皂苷1a和任选的七叶皂苷1a、七叶皂苷1b和异七叶皂苷1b中的一种或多种,基本上由其组成或由其组成。在本实施例的另一方面,物质组合物、组合或调配物包含异七叶皂苷1b和任选的七叶皂苷1a、七叶皂苷1b和异七叶皂苷1a中的一种或多种,基本上由其组成或由其组成。在某些实施例中,所述混合物含有七叶皂苷1a和七叶皂苷1b以及任选的异七叶皂苷1a和异七叶皂苷1b中的一种或多种。
当关联物质组合物、组合或调配物中的七叶皂苷的异构体使用时,术语“纯化的”意指指定的异构体以相对于所存在的化合物的总重量,等于或大于85wt%(如90wt%、95wt%、98wt%或99wt%)存在。当关联物质组合物、组合或调配物中的七叶皂苷的异构体使用时,术语“部分纯化的”意指指定的异构体以相对于所存在的化合物的总重量,等于或大于50wt%(如60wt%、70wt%、80wt%或90wt%)存在。
此类物质组合物、组合或调配物可任选地包括一种或多种本文公开的第二药剂。此类物质组合物、组合或调配物可任选地与一种或多种本文公开的第二药剂结合给药。这种第二药剂的给药可以在给予物质组合物、组合或调配物的同时、之前或之后。
医药组合物
可以将本发明化合物配制成医药组合物,用于以适于体内给药的生物相容形式给予受试者。本发明提供一种医药组合物,其包含与药学上可接受的赋形剂混合的本发明化合物。在与组合物的其它成分相容并且对其接受者无害的意义上,药学上可接受的赋形剂必须是可接受的。本文所用的药学上可接受的赋形剂可选自各种有机或无机材料,其用作医药调配物的材料并且以镇痛剂、缓冲剂、粘合剂、崩解剂、稀释剂、乳化剂、赋形剂、增量剂、助流剂、增溶剂、稳定剂、悬浮剂、张力剂、媒剂和增粘剂形式并入。还可以添加医药添加剂,如抗氧化剂、芳香剂、着色剂、风味改善剂、防腐剂和甜味剂。可接受的医药赋形剂的实例包括羧甲基纤维素、结晶纤维素、甘油、阿拉伯胶、乳糖、硬脂酸镁、甲基纤维素、粉剂、盐水、海藻酸钠、蔗糖、淀粉、滑石和水等。在一些实施例中,术语“药学上可接受的”意指由联邦或州政府的管理机构批准或在美国药典或其它公认的药典中列出用于动物,更特别是人类。
通常,药学上可接受的赋形剂对活性化合物是化学惰性的,并且在使用条件下是无毒的。药学上可接受的赋形剂的实例可包括例如水或盐水溶液、聚合物(如聚乙二醇)、碳水化合物及其衍生物、油、脂肪酸或醇。在一些实施例中,赋形剂是盐水或水。在一些实施例中,赋形剂是盐水。在一些实施例中,赋形剂是水。药学上可接受的赋形剂也是所属领域技术人员所熟知的。赋形剂的选择将部分地由特定化合物以及用于给予组合物的特定方法确定。因此,存在多种适合的本发明医药组合物的配方。以下方法和赋形剂仅是示范性的,决不是限制性的。
表面活性剂,如但不限于洗涤剂,也适用于配方中。表面活性剂的具体实例包括聚乙烯吡咯烷酮、聚乙烯醇、乙酸乙烯酯和乙烯基吡咯烷酮的共聚物、聚乙二醇、苯甲醇、甘露醇、甘油、山梨糖醇或脱水山梨糖醇的聚氧乙烯化酯;卵磷脂或羧甲基纤维素钠;或丙烯酸衍生物,如甲基丙烯酸酯等,阴离子表面活性剂,如碱性硬脂酸盐,特别是硬脂酸钠、硬脂酸钾或硬脂酸铵、硬脂酸钙或硬脂酸三乙醇胺;烷基硫酸盐,特别是月桂基硫酸钠和鲸蜡基硫酸钠;十二烷基苯磺酸钠或二辛基磺基丁二酸钠;或脂肪酸,特别是那些由椰子油衍生的脂肪酸,阳离子表面活性剂,如式n+r'r″r″′r″″y-的水溶性季铵盐,其中r基团是相同或不同的任选羟基化烃基,并且y-是强酸的阴离子,如卤化物、硫酸根和磺酸根阴离子;十六烷基三甲基溴化铵是可以使用的阳离子表面活性剂之一,式n+r'r″r″′的胺盐,其中r基团是相同或不同的任选羟基化烃基;十八烷基胺盐酸盐是可以使用的阳离子表面活性剂之一,非离子表面活性剂,如任选的脱水山梨糖醇的聚氧乙烯化酯,特别是聚山梨醇酯80,或聚氧乙烯化烷基醚;聚乙二醇硬脂酸酯、蓖麻油的聚氧乙烯化衍生物、聚甘油酯、聚氧乙烯化脂肪醇、聚氧乙烯化脂肪酸或环氧乙烷和环氧丙烷的共聚物,两性表面活性剂,如甜菜碱的经取代月桂基化合物。
当被给予受试者时,本发明化合物和药学上可接受的赋形剂可以是无菌的。在一些实施例中,当静脉内给予本发明化合物时,水溶液是赋形剂。在一些实施例中,当静脉内给予本发明化合物时,赋形剂是盐水溶液。葡萄糖水溶液和甘油溶液也可用作液体赋形剂,特别是用于可注射溶液。合适的医药赋形剂还可包括如淀粉、葡萄糖、乳糖、蔗糖、明胶、麦芽、稻米、面粉、白垩、硅胶、硬脂酸钠、甘油单硬脂酸酯、滑石、氯化钠、脱脂乳、甘油、丙烯、乙二醇、聚乙二醇300、水、乙醇、聚山梨醇酯20等的赋形剂。如果需要,本发明组合物还可含有少量的润湿剂或乳化剂,或ph缓冲剂。
本发明的医药调配物通过医药领域熟知的方法制备。例如,将本发明化合物与赋形剂结合而呈悬浮液或溶液。任选地,还添加一种或多种辅助成分(例如缓冲剂、调味剂、表面活性剂等)。赋形剂的选择取决于化合物的溶解度和化学性质、选择的给药途径和标准医药实践。在一些实施例中,调配物包含本发明化合物和水。在一些实施例中,调配物包含本发明化合物和盐水。
另外,本发明化合物通过已知程序给予受试者,如人类或动物受试者,所述已知程序包括但不限于口服、舌下或口腔给药、肠胃外给药、皮下给药、透皮给药、通过吸入或鼻内给药、阴道、直肠和肌肉内。本发明化合物可以胃肠外,通过筋膜上、囊内、颅内、皮内、鞘内、肌肉内、眶内、腹膜内、脊柱内、胸骨内、血管内、静脉内、实质、皮下或舌下注射或通过导管给药。在一些实施例中,通过肌肉内递送将本发明化合物给予受试者。在一些实施例中,通过腹膜内递送将本发明化合物给予受试者。在一些实施例中,通过静脉内递送将本发明化合物给予受试者。在一些实施例中,口服给予本发明化合物。在某些实施例中,本发明化合物通过推注给药来给予,例如iv或im推注给药。
对于口服给药,本发明化合物的调配物可以以胶囊、片剂、粉末、颗粒或以悬浮液或溶液形式存在。胶囊调配物可以是明胶、软凝胶或固体。片剂和胶囊调配物还可含有一种或多种佐剂、粘合剂、稀释剂、崩解剂、赋形剂、填充剂或润滑剂,其各自是所属领域已知的。这样的实例包括碳水化合物(如乳糖或蔗糖)、无水磷酸氢钙、玉米淀粉、甘露醇、木糖醇、纤维素或其衍生物、微晶纤维素、明胶、硬脂酸盐、二氧化硅、滑石、羟基乙酸淀粉钠、阿拉伯胶、调味剂、防腐剂、缓冲剂、崩解剂和着色剂。口服给药的组合物可含有一种或多种任选的药剂,如但不限于甜味剂,如果糖、阿斯巴甜或糖精;调味剂,如薄荷、冬青油或樱桃;着色剂;和防腐剂,以提供药学上适口的制剂。
对于肠胃外给药(即,通过除消化道以外的途径注射给药),本发明化合物可以与与受试者的血液等渗的无菌水溶液组合。这样的调配物是通过将固体活性成分溶解在含有生理上相容的物质,如氯化钠、甘氨酸等的水中,并具有与生理条件相容的缓冲ph,以产生水溶液,然后使溶液无菌而制备。调配物可以存在于单剂量或多剂量容器,如密封的安瓿或小瓶中。调配物可以通过任何注射方式递送,包括但不限于,筋膜上、囊内、颅内、皮内、鞘内、肌肉内、眶内、腹膜内、脊柱内、胸骨内、血管内、静脉内、实质、皮下或舌下或通过导管进入受试者的体内。
肠胃外给药包括水基和非水基溶液。其实例包括例如水、盐水、水性糖或糖醇溶液、醇(如乙醇、异丙醇、乙二醇)、醚、油、甘油酯、脂肪酸和脂肪酸酯。在一些实施例中,水用于肠胃外给药。在一些实施例中,盐水用于肠胃外给药。用于肠胃外注射的油包括动物油、植物油、合成油或石油基油。用于溶液的糖的实例包括蔗糖、乳糖、右旋糖、甘露糖等。油的实例包括矿物油、石蜡油、大豆油、玉米油、棉籽油、花生油等。脂肪酸和酯的实例包括油酸、肉豆蔻酸、硬脂酸、异硬脂酸及其酯。
对于透皮给药,本发明化合物与皮肤渗透促进剂组合,如丙二醇、聚乙二醇、异丙醇、乙醇、油酸、n-甲基吡咯烷酮等,其增加皮肤对本发明化合物的渗透性并允许化合物渗透皮肤并进入血液。化合物/促进剂组合物还可以进一步与聚合物质结合,如乙基纤维素、羟丙基纤维素、乙烯/乙酸乙烯酯、聚乙烯吡咯烷酮等,以提供呈凝胶形式的组合物,将其溶解在溶剂,如二氯甲烷中,蒸发至所需粘度,然后施加到背衬材料上以提供贴剂。
在一些实施例中,本发明化合物呈单位剂量形式,如片剂、胶囊或单剂量小瓶。合适的单位剂量,即有效量,可以在临床试验期间确定,所述临床试验被适当地设计成用于指定所选化合物给药的每种病状,并且当然将根据所需的临床终点而变化。
本发明还提供用于治疗和预防受试者中本文所述病症的制品。制品包含本发明化合物或包含本发明化合物的医药组合物,任选地还含有如本文所述的第二药剂。制品可以包装有本发明化合物或包含本发明化合物的医药组合物能够治疗和/或预防的各种病症的说明。例如,制品可包含单位剂量的能够治疗或预防某种病症的本发明化合物或包含本发明化合物的医药组合物,和单位剂量能够治疗或预防某种疾病,例如囊性纤维化的说明。
剂量和给药
根据本发明方法,以治疗有效量向受试者给予本发明化合物(或与受试者的细胞接触)。基于已知程序,所属领域技术人员容易确定这一量,包括分析体内确立的滴定曲线和本文公开的方法和分析。在一些实施例中,治疗有效量会增加受试者中ptc密码子的通读。在一些实施例中,治疗有效量会增加受试者中含有ptc的mrna的稳定性。
在一个实施例中,本发明化合物以治疗有效量给予,无论是单独的还是作为医药组合物的一部分。当然,治疗有效量和给药剂量将根据已知因素而变化,如特定药剂的药效学特征和其给药方式和途径;接受者的年龄、健康和体重;疾病状态或病状的严重程度和阶段;同时治疗的种类;治疗频率;和所需效果。
所给予的化合物的总量也将通过给药途径、时间和频率以及可能伴随化合物给药的任何不良副作用的存在、性质和程度以及所需的生理作用来确定。所属领域技术人员将理解,各种病症或疾病状态,特别是慢性病症或疾病状态,可能需要涉及多次给药的长时间治疗。在这些医药组合物中,以组合物的总重量计,本发明化合物通常以约0.5-95重量%的量存在。多剂量形式可以作为单一治疗的一部分给予。
在一些实施例中,治疗有效量的本发明化合物的范围为约0.1毫克/千克/天至约50毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.5毫克/千克/天至约30毫克/千克/天。在一些实施例中,治疗有效量的范围为约1毫克/千克/天至约25毫克/千克/天。在一些实施例中,治疗有效量的范围为约2毫克/千克/天至约20毫克/千克/天。在一些实施例中,治疗有效量的范围为约3毫克/千克/天至约15毫克/千克/天。在一些实施例中,治疗有效量的范围为约4毫克/千克/天至约10毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约20毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约15毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约10毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约5毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约6.5毫克/千克/天。在一些实施例中,治疗有效量的范围为约0.1毫克/千克/天至约9毫克/千克/天。在一些实施例中,治疗有效量的范围为约1毫克/千克/天至约14毫克/千克/天。
在某些实施例中,治疗有效量根据治疗过程以一个或多个剂量给予(其中剂量是指在一天内给予的本发明化合物的量)。在某些实施例中,每天给予剂量一次(每天1次/给药)。在某些实施例中,每天给予剂量两次(每天2次/给药;例如,每天两次给药中给予有效量的一半)。在某些实施例中,每天给予剂量三次(每天三次/给药;例如,每天两次给药中给予有效量的三分之一)。当剂量被分成每天多次给药时,可以将剂量等分,或者可以在每次给药时不均等地划分剂量。任何给定剂量可以以单一剂型或多于一种剂型(例如片剂)递送。
与非活性成分组合以产生本文所述医药组合物的活性成分的量将取决于所治疗的宿主和特定的给药方式而变化。例如,在一些实施例中,用于口服给予人类的剂型含有约1至1000mg本发明化合物,其与合适且适宜量的药学上可接受的赋形剂一起配制。在一些实施例中,本文所述的医药组合物含有约1至800mg、1至600mg、1至400mg、1至200mg、1至100mg或1至50mg本发明化合物。
材料和方法
化合物
由(康涅狄格州盖洛兹维尔(gaylordsville,ct)的microsourcediscoverysystems公司)获得pharmakon1600库。以dmso储备溶液形式供应化合物。另外的化合物从市售来源获得。用于进一步表征实验的七叶皂苷以粉末形式获自西格玛-奥德里奇公司(sigma-aldrich)(密苏里州圣路易斯(st.louis,mo.);目录号e1378)。另外的七叶皂苷由microsourcediscovery系统公司(康涅狄格州盖洛兹维尔)获得,用于进行另外验证。
细胞培养
用ugac终止密码子、cftrg542x和w1282x终止密码子稳定转导费雪(fischer)大鼠甲状腺(frt)细胞,并使用flp-intm系统按照制造商的方案(英杰(invitrogen))匹配cftrwt对照cdna。维持细胞并在补充有2.68g/l碳酸氢钠和10%fbs的营养混合物f-12ham(sigmaf6636)培养基中生长。
使用条件性重编程(conditionalreprogramming),来源于肺外植体的携带无义等位基因(如g542x或w1282x)的人类支气管上皮(hbe)细胞单独或与δf508cftr(g542x/δf508、w1282x/δf508)反式扩增(liux等人,《美国病理学杂志(amjpathol)》;180:599-607,2012),接种到可渗透的支持物上并在分化培养基中生长至少6-8周,直到终末分化。
荧光素酶hts报告基因分析
荧光素酶分析在pharmakon1600库上进行,以确定每种化合物在促进通读方面的功效。将5μl呈dmso储备溶液的每种化合物溶解在f-12ham培养基中,并使用biomekfx分配到384孔板(康宁(corning))的独立孔中。然后使用matrixwellmate添加含有报告基因构建体(qxnugac)的frt细胞(10,000个细胞/孔)。将板在37℃、5%co2和95%湿度下培育24小时,之后通过将板平衡到室温,加入等体积的promegabrightglo(普洛麦格(promega)),并且使用envision板读取器(珀金埃尔默(perkinelmer))在5分钟内读取增强的荧光来测定萤火虫荧光素酶活性。将dmso浓度保持恒定在0.3%,以10个浓度(100nmto30μm)以堆叠的板内形式测试化合物,每个板的320个化合物样品依次在10个板上以1:2连续稀释。g418(500μg/ml)用作阳性对照。
跨上皮氯离子电导分析
将如本文所述的稳定转染的frt细胞以~2×106个细胞/过滤器的密度接种到costar24孔0.4μm可渗透支持物上,并在37℃下用5%co2维持。使用上皮电压计评定跨上皮电阻,以确保形成紧密的单层,如电阻>6kcm2所限定。在药剂的初始评估期间,在评定跨上皮电导(gt)之前,用化合物(10μm)、媒剂对照(0.3%dmso)或阳性对照(250μg/mlg418)处理细胞48小时;后续研究使用其它浓度的化合物。测量是在基线,在加入20μm的毛喉素(fsk,刺激cftr活性的camp激动剂)和10μmcftrinh-172(一种cftr特异性抑制剂)之后进行。
验证性双荧光素酶分析
双荧光素酶报告基因系统的基本特征先前已描述(grentzmanng等人,《rna》4:479-486,1998)。将本研究中所用的含有uga的通读盒(5'-aaatcccaatgacatgcagga-3';seqidno:1)插入表达质粒中的海肾和萤火虫荧光素酶基因之间并使用flipintm系统制备稳定的frt细胞系。这些构建体在先导化合物、媒剂对照(0.3%dmso)或阳性对照(250μg/mlg418)的存在下生长24个小时并如先前所述(xuex等人,《美国呼吸系统细胞和分子生物学杂志》;50:805-816;2014)进行分析。
在原代细胞单层中进行的尤斯室(ussingchamber)研究
如先前所述,在电压钳条件下测量短路电流(isc)(rowesm等人,《分子医药杂志(jmolmed)》(柏林);89:1149-1161;2011)。简要地说,在用成纤维细胞条件培养基涂覆后,将如本文所述的原代hbe细胞接种在costar0.4-mm可渗透支持物(5.3×105个细胞/过滤器,6.5mm直径;马里兰州贝塞斯达(bethesda,md))上(liux等人,《美国病理学杂志(amjpathol)》;180:599-607,2012)。用化合物处理分化良好的细胞48小时,并通过使用尤斯室(美国生理仪器(physiologicinstruments))中的上皮电压钳装置获得isc测量值。在测量基线isc后,加入氨氯吡脒(100μm)以阻断上皮钠通道活性,然后加入cftr激动剂(20μmfsk和10μmvx-770)。在实验结束时加入cftrinh-172(10μm)以阻断cftr依赖性isc。dmso(0.1%)和g418(250μg/ml)分别用作媒剂和阳性对照。
辣根过氧化物酶(hrp)cftr表达分析
为了测量cftr的细胞表面表达,将表达g542xcftr和hrp的融合蛋白的frt细胞用10μm的先导化合物在37℃和5%co2下处理48小时。将dmso浓度保持在0.3%,将frt细胞在补充有1mmmgcl2和0.1mmcacl2的pbs中洗涤4次,然后在室温下与hrp底物(费舍尔(fisher),#pi-34096,15微升/孔)一起培育10分钟。通过synergyneo微板读数仪(伯腾仪器(biotekinstruments))分析荧光hrp信号。
实时聚合酶链式反应(rt-pcr)
使用qiagenrnaeasy分离试剂盒(凯杰(qiagen))使rna与用七叶皂苷(10μm)处理的frtg542x细胞分离,并如先前所述,使用taqman一步pcr预混液试剂(taqmanonesteppcrmastermixreagent)(应用生物系统公司(appliedbiosystems))进行实时pcr(sloanepa等人,《科学公共图书馆综合卷(plosone)》;7:e39809;2012)。简要地说,使用qiagenrneasy微型试剂盒根据制造商的说明书分离总rna。为了防止出现可能的dna污染,用不含rna酶的dna酶(加利福尼亚州巴伦西亚的凯杰(qiagen,valencia,ca))预处理样品。用于人类cftr的序列特异性引物和探针购自assaysondemand(应用生物系统公司;针对cftr的分析id:hs00357011_m1)。所述探针跨越人类cftr序列的外显子21/22边界延伸。taqman一步pcr预混液试剂试剂盒(应用生物系统公司)用于逆转录及pcr。反应体积为25μl并含有12.5μl不含尿嘧啶-n-糖基化酶的2×预混液、0.625μl40×multiscribe和rna酶抑制剂混合物、1.25μl20×目标引物和探针、5.625μl无核酸酶水(德克萨斯州奥斯汀的安必逊(ambion,austin,tx))和5μlrna样品。用光学帽盖住反应板并短暂离心以除去气泡。热循环仪条件如下:阶段1:48℃持续30分钟;阶段2:95℃持续10分钟;阶段3:95℃持续15秒,重复40个循环,60℃持续1分钟。一式三份地运作所有实验以进行验证。相对转录物水平归一化为frtwtmrna水平。
α-l-艾杜糖苷酶活性分析
α-l-艾杜糖苷酶活性如下测定。细胞提取物获自针对w402x突变纯合的小鼠胚胎成纤维细胞(mef)细胞,并使用慢病毒表达系统使用sv40大t抗原永生化。如wang等人所述测定α-l-艾杜糖苷酶活性(wang,d.等人,2012;《分子遗传学和代谢(molgenetmetab)》105:116-125)。简要地说,在低ph下将4-甲基伞形酮艾杜糖苷(fmu)加入细胞裂解物中作为底物(例如,~130mm甲酸钠缓冲液,ph3.5)。将反应在37℃下培育72小时,然后通过加入高ph缓冲液(例如甘氨酸-naoh缓冲液,ph10.8)淬灭反应。使用荧光计在365nm激发和450nm发射下立即测量裂解的游离fmu分子的荧光,并且活性艾杜糖苷酶的量表示为每毫克蛋白质每小时释放的fmu纳摩尔数。
统计
hts分析命中识别的阈值为>95%阴性对照的ci。对于tecc、isc、双荧光素酶和基于hrp细胞的分析,视需要使用t测试或anova比较描述性统计数据(平均值、sd和sem),所述t测试或anova使用graphpadprism软件(加利福尼亚州拉荷亚(lajolla,ca))和microsoftexcel(华盛顿州西雅图(seattle,wa))进行。所有统计学测试均为双侧,并以5%显著性水平(即α=0.05)进行。
实例
实例1-用于荧光素酶hts分析的稳定细胞系的研发和验证
在先前的出版物中,申请人已经表明,哺乳动物细胞中的四种最常见的cf相关的无义突变(g542x、r553x、r1162x和w1282x)都对利用可供使用的药物进行的通读诱导具有相对地抗性(xuex等人,《美国呼吸系统细胞和分子生物学杂志》;50:805-816;2014)。因此,目标是通过hts鉴定将有效促进ptc的通读的化合物。为了测量通读活性,使用由海肾和萤火虫荧光素酶基因构成的双荧光素酶报告基因构建体产生稳定的frt细胞系,所述构建体环绕具有g542x或ugac序列(图1a)(图1a中称为seqidno:1和2的序列)的cftr通读盒。选择g542x是因为其是最常见的cftr-ptc无义突变,并且申请人之前已经确认了frtg542x用于测量cftr功能(xuex等人,《美国呼吸系统细胞和分子生物学杂志》;50:805-816;2014;rowesm等人;《分子医药杂志》(柏林);89:1149-1161;2011)。选择ugac是因为其是对阳性对照药剂最敏感的ptc。使用两种不同的构建体可以最大限度地减少偏倚并使识别有效药物的潜力达到最大。
为了验证这些细胞系,用剂量高达300至500μg/ml的g418处理细胞系,g418是一种在体外具有确定功效的通读剂(bedwelldm等人;《自然医学》;3:1280-1284;1997)。荧光素酶活性的增加表明g418刺激了所测试构建体中ptc的通读。在测定荧光素酶活性之前,将g418或媒剂对照(0.3%dmso)与细胞一起预培育24小时。与frtuqxngac细胞和frtg542x细胞中dmso处理的媒剂(p<0.0001)(图1b和1c)相比,g418以剂量依赖性方式显著增加萤火虫荧光素酶活性,但在匹配的frt野生型cftr对照细胞(图1d)中没有显著影响,证实frtg542x和frtqxnugac细胞系将是用于筛选的信息模型。出于筛选目的,测量萤火虫荧光素酶水平而不评定海肾表达作为总构建体表达的指示。
为了评定时间依赖性,通过测定g418在毛喉素存在下对cftr电导的影响,g418在frtg542x细胞中的功效。在这一分析中,cftr电导的增加表明g418刺激了所测试构建体中ptc的通读。在毛喉素存在下的cftr电导指示frtg542x细胞与g418(250μg/ml)或媒剂对照(0.3%dmso)培育0至72小时,然后测定电导。在测定电导之前,立即用fsk(20μm)处理细胞;在实验结束时加入cftrinh-172(10μm)以抑制cftr活性。在毛喉素诱导的电导中存在时间依赖性增加,在72小时时观察到最大效果(图2a和2b);毛喉素诱导的gt的这种增加也对cftrinh-172敏感(图2a和2c)。为方便起见,选择与测试化合物预培育48小时用于后续研究,因为这一时间点引起>75%的信号强度并促进高通量评估。双荧光素酶报告基因也观察到类似的时间依赖性增加(数据未显示)。
实例2-高通量筛选
在确定对通读剂敏感的稳定的cftrg542x和ugac构建体系后,进行1600个临床批准的化合物的hts。两种独立的方法并行进行。采用cftr介导的跨上皮氯离子电导(tecc)分析和高度敏感的基于萤光素酶的报告基因分析(xuex等人;《美国呼吸系统细胞和分子生物学杂志》;50:805-816;2014)。tecc分析提供cftr活性的读数,从而确保生理相关性。基于荧光素酶的报告基因分析能够以更高的通量同时测试多种浓度的测试化合物。
在tecc分析中,用单次剂量(10μm)的每种化合物处理frtg542x细胞。将frtg542x细胞与之前的化合物或媒剂对照(0.3%dmso)一起培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。在这些实验中,cftr电导的增加表明化合物刺激了所测试构建体中ptc的通读并且cftr是有功能的。
在1600种化合物中,有58种在毛喉素存在下引起cftr电导增加(如标准化为250μg/mlg418),命中率呈现为3.6%(图3a)。归一化为g418阳性对照的刺激毛喉素活性≥20%的化合物被认为是有活性的。在没有cftr表达的frt细胞(即亲代细胞)中测量在毛喉素活化后也对cftrinh-172反应的化合物。30个药剂使毛喉素刺激的gt增加超过20%阈值,因此出于其非特异性活性而从筛选中剔除(数据未显示)。
在萤火虫荧光素酶分析中,使用10点,连续2倍稀释浓度反应分析(0.1μm-30μm),用相同的1600种化合物处理frtqxnugac和frtg542x荧光素酶报告基因。荧光素酶活性的增加表明化合物刺激了所测试构建体中ptc的通读。hts分析是稳健的,z值平均为0.7。在frtugac细胞中,初始筛选产生由相对于g418阳性对照,荧光素酶的最大活化≥20%(图3b)所限定的115种活性命中物。基于不利的医药特性排除了这些化合物中的90种。
在考虑特异性、药物特性和其它因素后,tecc和荧光素酶筛选共计产生了48种初始命中物。在这一阶段,五种化合物是两种分析的常见命中物。
实例3-先导化合物的评估
为了优先考虑所鉴定的48种通读阳性化合物中哪一种在增强ptc通读和恢复蛋白质功能方面最有效,使用frtg542x细胞和不含cftr的frt细胞(亲代细胞)重复tecc分析(一式三份)。将化合物(10μm)或媒剂对照(0.3%dmso)与细胞一起培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。g418(250μg/ml)用作阳性对照。在这一分析中,cftr电导的增加表明化合物刺激了所测试构建体中ptc的通读并且cftr是有功能的。所测试的48种化合物提供在下表1中(数字标记对应于图4中所示的那些)。粗体化合物表示在所述一种或多种分析中显示出优良特性的化合物,并且可用作通读刺激剂。
评定相对于媒剂对照的电导变化。消除了未引起cftr电导增加的化合物。如图4所示,13种化合物始终诱导经毛喉素刺激的frtg542x细胞中的电导的增加(0.1±0.005)。那些化合物也增加了没有cftr的frt细胞的电导(数据未显示),因此出于特异性差而被排除。其余化合物被认为是先导化合物。在上述分析中鉴定的先导化合物是秋水仙碱、环己酰胺、阿霉素、七叶皂苷、四磷酸咯萘啶(pt)、泼尼卡酯、奥苯达唑和对氨基苯甲酸钾(paba);在另外的实验中,阿苯达唑和甲苯咪唑被鉴定为先导化合物(数据未显示)。
进行了一系列功能分析以更详细地评估这些先导化合物的功效。使用frtg542x细胞,测定每种化合物在增加氯离子电导方面的最佳剂量(图5a)。将frtg542x细胞与化合物(1μm至30μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时。如上文实例2中所述进行电导分析。在这一分析中,cftr电导的增加表明化合物在所测试构建体中刺激ptc的通读并且cftr是有功能的。当用秋水仙碱、环己酰胺、七叶皂苷、pt、泼尼卡酯、奥苯达唑和paba处理时,frtg542x细胞展现出显著更高(p<0.05)的经毛喉素刺激的cftr电导(图5a)。
使用frtqxnugac细胞,测定双荧光素酶分析中每种化合物的最佳剂量(萤火虫/海肾荧光素酶比)(图5c)。在这一分析中,荧光素酶比的增加表明化合物刺激了所测试构建体中ptc的通读。将frtqxnugac细胞与化合物(0.01μm至10μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时。相对于媒剂对照,frtg542x细胞在10μm剂量下显示出最佳功效(图5c)。应注意,对于七叶皂苷,~2μm的活性与10μm的活性相似,并且对于阿霉素,活性在~1μm达到峰值(图5c和5d)。
为了获得cftr功能的额外读数,使用hrp分析评估先导化合物对cftr细胞表面表达的影响。在测定hrp活性之前,将ftrg542x细胞与化合物(10μm)、g418(250μg/ml)或媒剂对照(0.3%dmso)一起培育48小时。在这一分析中,hrp活性的增加表明化合物刺激了所测试构建体中ptc的通读。与媒剂对照相比,用秋水仙碱、环己酰胺、奥苯达唑、七叶皂苷和阿霉素处理的frtg542x细胞中的hrp活性显著更高,通过cftr表达的生物化学分析验证了上述功能测量(图5b)。
cftr表达也与萤火虫荧光素酶活性(p=0.02)(图5e)并与经毛喉素刺激的电导增加正相关(图5f)。还观察到双荧光素酶活性和经毛喉素刺激的电导增加之间的中度相关性(图5g)。总的来说,这些结果表明,恢复通读活性伴随着cftr功能的恢复,进一步表明这八种先导化合物的潜在功效。
表2总结了上述8种先导化合物的结果。对于tecc分析,结果显示为平均gt值(ms/cm2±标准误差)。对于双荧光素酶活性分析,结果显示为平均萤火虫/海肾比(±标准偏差)。对于hrp分析,结果表示为以平均相对光单位(rlu)(±标准误差)检测的蛋白质表达表示。对于hbe尤斯室分析,结果以平均isc(ua/cm2)(±标准误差)表示。除非另有说明,否则化合物在10μm下测试。
表2
a-1μm;b-3μm;c-30μm;
*-p<0.05,**-p<0.01,***-p<0.001,****-p<0.0001
实例4-原代hbe细胞中先导化合物的评估
基于先前研究,原代hbe细胞已成为预测cf临床试验结果的有价值的临床前工具(vangoorf等人;《美国国家科学院院刊(procnatlacadsciusa)》;106:18825-18830;2009;ramseybw等人;《新英格兰医学杂志》;365:1663-1672;2011)。功能分析用源自g542x/δf508-cftr杂合的患者的hbe细胞进行。先前显示,艾维卡福(ivacaftor)(先前vx-770),一种通过增加细胞表面处的cftr的开放概率来起作用的cftr增效剂,会增强合成性通读药物在恢复cftr功能方面的能力(xuex等人;《美国呼吸系统细胞和分子生物学杂志》;50:805-8163;2014)。测定艾维卡福与每种先导化合物的最佳剂量组合的效果,如图5a中所测定。为了测定短路电流(isc),用胺氯吡脒(100μm)处理hbe细胞以阻断上皮钠通道活性,之后加入cftr激动剂fsk(10μm)和vx-770(10μm);在实验结束时加入cftrinh-172(10μm)以阻断cftr依赖性isc。如图6所示,与媒剂对照相比,艾维卡福在用pt(6.2±0.3ua/cm2)、泼尼卡酯(4.9±1.2ua/cm2)、paba(6.6±1.2ua/cm2)和七叶皂苷(7.1±0.3ua/cm2)处理的细胞中显著增加了毛喉素刺激后的isc。增强通常与毛喉素反应成比例,与此设置中的增效剂活性相容。总之,这些结果与先前的研究一致,表明将ptc抑制化合物与艾维卡福结合的治疗策略的前景,并提供支持筛选中鉴定的化合物的实用性的额外数据。
实例5-七叶皂苷的评估
基于上述结果,在八种先导化合物中,七叶皂苷成为一种特别引人注目的化合物。七叶皂苷单独以及与艾维卡福组合引发最高水平的通读(图5d),细胞表面cftr表达(图5b)和毛喉素依赖性isc(图6)。重要的是,七叶皂苷已被用于如上所论述的消炎、抗水肿和血管保护特性,并且是一种容易获得的没有已知的副作用的化合物。因此,与其它展现出有利反应的先导药剂相比,七叶皂苷对于进一步测试是特别有吸引力的化合物。
在七叶皂苷的独立评估中,七叶皂苷在用20μm七叶皂苷预处理24小时的frtg542x细胞中显示经毛喉素刺激(20μm)的电导(gt)显著增加。通过加入cftrinh-172(10μm),七叶皂苷的刺激作用完全减弱(图7a)。图7b显示七叶皂苷的作用是剂量依赖性的(3μm和10μm的七叶皂苷)。虽然在电导分析中观察到3μm和10μm浓度的七叶皂苷的功效,但是使用hrp分析(测量细胞表面蛋白表达)在更宽的剂量范围(0.001μm至20μm;培育48小时)下测试七叶皂苷以进一步评定低剂量七叶皂苷的作用。.图7c显示hrp分析(frtg542x细胞)中的七叶皂苷的ec50为2.3μm,表明对人类用途具有足够的效力。
在来自g542x/δf508cf供体的原代人类支气管上皮(hbe)细胞中进行另外的研究。将hbe细胞与七叶皂苷(10μm)或媒剂对照(0.1%dmso)一起培育48小时。为了测定短路电流(isc),用胺氯吡脒(100μm)处理hbe细胞以阻断上皮钠通道活性,之后加入cftr激动剂fsk(10μm)和vx-770(10μm);在实验结束时加入cftrinh-172(10μm)以阻断cftr依赖性isc。图7d显示,如与媒剂对照相比,七叶皂苷(实线)诱导毛喉素依赖性isc显著增加,其随着vx-770的添加而进一步增加。经七叶皂苷刺激的isc在给予cftrinh-172后被抑制。如申请人实验室中所测定,10μm(11.9±2.5μa/cm2)下单独用毛喉素刺激后的cftr活性是野生型cftr活性的~46%,其进一步受vx-770刺激(图7e)。此外,七叶皂苷还增强了cftrmrna表达水平。将七叶皂苷(10μm)、g4129(250μg/ml)和媒剂对照(0.1%dmso)在hbe细胞中培育48小时。从hbe细胞中分离rna,并使用实时pcr测定g542xmrna的水平,并如本文所述将其归一化为frt野生型mrna水平。图7f显示,如与媒剂对照相比,七叶皂苷使g542xmrna水平增加~2倍,并且显示出比g418更大的增强。这一发现证实了isc结果,并证明七叶皂苷可以诱导ptc的通读并抑制nmd。
为了探索七叶皂苷对于治疗具有其它常见的无义突变的cf患者的潜在益处,在frtw1282x细胞、来自w1282x/δf508杂合供体的hbe细胞、来自δf508/δf508纯合供体的hbe细胞和来自g542x/δf508杂合供体的hbe细胞中测试七叶皂苷。在这些实验中,将frt和hbe细胞与七叶皂苷(10μm)、vx-809(6μm)和媒剂对照(对于frt细胞为0.3%dmso,并且对于hbe细胞为0.1%dmso)一起培育48小时。如本文所述进行单独分析。结果如图7g到7j所示。
图7g显示七叶皂苷在frtw1282x细胞中在毛喉素的存在下显著增强cftr功能;vx-770进一步增强了cftr功能。图7h显示在源自w1282x/δf508杂合供体的原代hbe中七叶皂苷的相同结果。为了确定七叶皂苷对刺激cftr活性的特异性,参见图7g和7h,在源自针对δf508突变纯合的患者的hbe细胞中测试七叶皂苷。如所预期,如由毛喉素/vx-770依赖性isc没有发生增加所指示,七叶皂苷在原代hbeδf508/δf508细胞中没有作用(因为这些细胞不含ptc)。相比之下,vx-809显示出cftr活性显著增加,如通过毛喉素依赖性isc的增加所示,其随着添加vx-770而进一步增加。已显示vx-809在具有δf508突变的患者中会部分恢复cftr功能(vangoorf等人,《美国国家科学院院刊》;108:18843-18848;2011)。这一结果表明,七叶皂苷的作用是由于诱导通读和/或抑制无义介导的mrna衰变。
在来自g542x/δf508供体的原代hbe中进行另外的实验,以进一步分析七叶皂苷和vx-809对cftr活性的影响。七叶皂苷刺激cftr活性,如毛喉素依赖性isc的增加所示,其随着添加vx-770而进一步增加,如先前图7e中所见。vx-809诱导cftr活性适度增加,如毛喉素依赖性isc的略微增加所示,其随着添加vx-770而进一步增加。
为了比较七叶皂苷的商业来源,在来自g542x/δf508cf供体的原代hbe中比较从两个不同的商业供应商(西格玛-奥德里奇公司和microsourcediscoverysystems公司)获得的七叶皂苷。将hbeg542x/δf508细胞与七叶皂苷(两种来源均为10μm)和媒剂对照(0.1%dmso)一起培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。来自任一来源的七叶皂苷均诱导cftr活性显著增加,如通过毛喉素依赖性isc的增加所示,其随着添加vx-770而进一步增加(图8)。
实例6-七叶皂苷与cftr校正剂和增效剂协同作用
如先前在文献中所报道,cftr校正剂(如vx-809)和cftr增效剂(如vx-770)的组合协同作用以改善cftr功能。为了研究本发明的通读刺激化合物与cftr校正剂和增效剂的相互作用,在frtw1282x细胞中进行的三重组合研究中研究七叶皂苷与cftr校正剂和cftr增效剂组合的作用。在这些实验中,使用10μm七叶皂苷、250μg/mlg418、10μmvx-770和6μmvx-809。将单独或呈所示组合的化合物与frtw1282x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。本发明化合物对ptc通读的增强通过cftr电导的增加来确定。cftr电导还表明,由于ptc的通读,cftr具有功能。
结果如图9所示。单独使用每种化合物均可不同程度地增强cftr功能。vx-809和vx-770的组合提供了cftr功能的显著增加,其随着进一步添加vx-770而没有增加。vx-809和七叶皂苷的组合也提供了cftr功能的显著增加,其随着进一步添加vx-770而增加。对于g418(遗传霉素,一种刺激通读的氨基糖苷类化合物)和vx-809的组合,观察到类似的结果,但程度小于七叶皂苷和vx-809的组合。当七叶皂苷与vx-809和vx-770组合时,观察到针对三重组合的cftr功能协同增加。进一步添加vx-770还进一步增强cftr功能。虽然对于g418、vx-809和vx-770的三重组合,也观察到cftr功能的显著增强,但所述增强比伴随七叶皂苷、vx-809和vx-770观察到的增强小约3倍。
这些结果表明,在cftr校正剂和cftr增效剂存在下,七叶皂苷协同增强cftr功能,并且在cftr校正剂和cftr增效剂单独存在下,也增强cftr功能。
实例7-七叶皂苷显示与nmd抑制剂的协同作用
包括ptc的转录本可能在通过nmd翻译为蛋白质之前迅速降解。nmd是一种存在于所有真核生物中的用以通过消除含有ptc的mrna转录物来减少基因表达错误的监视途径。通过减少nmd的过程,含有ptc的mrna转录物的水平增加。因此,含有ptc的转录物可在本文公开的通读刺激化合物存在下进行翻译。已知多种小分子nmd抑制剂(nmdi),包括nmdi-1、nmdi-9、nmdi-25和nmdi-14。在某些情况下,nmdi抑制nmd所需的蛋白质复合物的形成。例如,nmdi-1抑制nmd所需的hsmg5-hupf1复合物,nmdi-14抑制nmd所需的upf1-smg7复合物。
为了研究所公开的通读刺激化合物与nmdi之间的相互作用,研究七叶皂苷与两种示范性nmdi,nmdi-1和nmdi-14组合的作用。在这些实验中,frtg542x细胞与七叶皂苷(10μm)、nmdi-1(80μg/ml)和nmdi-14(20μg/ml)一起使用。将单独或呈所示组合的化合物与frtg542x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。在仅含有七叶皂苷和nmdi-14的frtw1282x细胞中进行相同的实验。本发明化合物对ptc通读的增强通过cftr电导的增加来确定。cftr电导还表明,由于ptc的通读,cftr具有功能。
结果如图10b和10c所示。图10b显示frtg542x细胞中的结果。如前所示,在vx-770和毛喉素的存在下,七叶皂苷单独刺激cftr功能。仅nmdi-1对cftr功能几乎没有影响。与单独的七叶皂苷相比,nmdi-1和七叶皂苷的组合引起cftr功能改善,特别是在vx-770存在下。单独的nmdi-14在vx-770存在下显著增加cftr功能。与单独的七叶皂苷相比,nmdi-14和七叶皂苷的组合不会引起cftr功能的进一步改善。
图10c显示frtw1282x细胞中的结果。如前所示,在vx-770和毛喉素的存在下,七叶皂苷单独刺激cftr功能。单独的nmdi-14在vx-770和毛喉素存在下显著增加cftr功能。与单独的七叶皂苷相比,nmdi-14和七叶皂苷的组合显示在vx-770和毛喉素存在下,cftr功能协同增加。
这些结果证实,本发明化合物可以刺激cftrptc的通读,并且与nmdi的组合可以进一步增强本发明的通读刺激化合物的活性。
实例8-氨基糖苷类基本上不会拮抗七叶皂苷的作用
氨基糖苷类是一类主要用于治疗革兰氏阴性细菌感染的抗菌剂。已显示氨基糖苷类通过抑制核糖体校正(proofreading)来降低翻译精确度,允许在无义密码子处错误地插入氨基酸并继续翻译至基因的3'末端。氨基糖苷类与原核生物和真核生物的核糖体rna的解码位点结合,并产生构象变化,其降低同源和近同源trna之间的区别,准许氨基酸插入终止密码子。净效果是继续翻译到天然终止密码子。
据报道,氨基糖苷类干扰通读刺激化合物的作用。为了研究所公开的通读刺激化合物和氨基糖苷类之间的相互作用,研究七叶皂苷与两种示范性氨基糖苷类,妥布霉素和庆大霉素组合的作用。在这些实验中,frtw1282x细胞与七叶皂苷(10μm)、妥布霉素(150μg/ml)和庆大霉素(250μg/ml)一起使用。将单独或呈所示组合的化合物与frtw1282x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和vx-770(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。本发明化合物对ptc通读的增强通过cftr电导的增加来确定。cftr电导还表明,由于ptc的通读,cftr具有功能。
提供于图11中的结果显示,妥布霉素和庆大霉素与七叶皂苷组合都不会在很大程度上干扰七叶皂苷对cftr活性的刺激,也不会增加七叶皂苷对cftr活性的刺激。如前所示,在vx-770和毛喉素的存在下,七叶皂苷单独刺激cftr功能。相比之下,妥布霉素和庆大霉素单独在vx-770或毛喉素存在下不刺激cftr功能。在毛喉素存在下,七叶皂苷和妥布霉素或七叶皂苷和庆大霉素的组合证明,与单独的七叶皂苷相比,氨基糖苷类并未干扰七叶皂苷刺激cftr活性或进一步增加七叶皂苷刺激cftr活性。与单独的七叶皂苷相比,在vx-770存在下,七叶皂苷和妥布霉素或七叶皂苷和庆大霉素的组合显示出略微降低的cftr刺激,但伴随组合观察到的cftr刺激是稳健并且统计学显著的。至于与毛喉素的组合,妥布霉素和庆大霉素都没有进一步增加七叶皂苷对cftr活性的刺激。
这些结果证实,本发明化合物可以刺激cftrptc的通读,并且与氨基糖苷类的组合不会显著抑制本发明的通读刺激化合物的活性。
实例9-七叶皂苷与cftr校正剂和增效剂协同作用
如实例6中所论述,cftr增效剂和校正剂已显示在cf的治疗中是有效的。在这一实例中,研究另外的cftr增效剂(gp-5)和另外的cftr校正剂(c1与c2组合)以及vx-809以确定其与本发明的通读刺激化合物的相互作用。
图12a显示七叶皂苷与cftr校正剂vx-809和c1+c2的组合的组合在frtg542x细胞中的作用。本发明化合物对ptc通读的增强通过cftr电导的增加来确定。cftr电导还表明,由于ptc的通读,cftr具有功能。将单独或呈所示组合的七叶皂苷(10μm)、vx-809(3μm)、gp-5(10μm)、c1+c2(10μm)和媒剂对照(0.3%dmso)与frtg542x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和gp-5(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。在frtg542x细胞中,vx-809、gp-5和c1+c2在毛喉素和gp-5存在下对cftr刺激显示出非显著影响,而在毛喉素和gp-5存在下,七叶皂苷对cftr刺激显示出显著影响。在毛喉素和gp-5的存在下,如与媒剂对照相比,七叶皂苷和vx-809或七叶皂苷和c1+c2的组合显示出协同相互作用。
图12b显示七叶皂苷与cftr校正剂vx-809和c1+c2的组合以及cftr增效剂gp-5组合的作用,以及七叶皂苷与所有三种化合物组合在frtw1282x细胞中的作用。cftr活性的刺激通过cftr电导的增加来确定。将单独或呈所示组合的七叶皂苷(10μm)、vx-809(3μm)、gp-5(10μm)、c1+c2(10μm)和媒剂对照(0.3%dmso)与frtw1282x细胞一起预培育48小时。在分析期间,在急加入毛喉素(20μm)和gp-5(10μm)之前首先测定基线电导(gt)以刺激cftr活性;在分析结束时加入cftr抑制剂cftrinh-172(10μm)以抑制cftr活性。在frtw1282x细胞中,在毛喉素和gp-5存在下,七叶皂苷、vx-809、c1+c2和gp-5对cftr刺激显示出不同的刺激作用,其中c1+c2对gp-5的添加显示出显著影响。在毛喉素和gp-5存在下,gp-5和c1+c2的组合显著增加cftr活性。然后测定七叶皂苷与这些化合物在刺激cftr活性中的组合。当在仅gp-5存在下与vx-809和c1+c2组合时,七叶皂苷显示cftr活性显著增加,并且当在毛喉素和gp-5存在下与c1+c2和gp-5组合时,cftr活性显著增加。七叶皂苷、c1+c2和gp-5的组合显示出任何测试组合的cftr活性的最大刺激(在毛喉素和gp-5存在下)。
这些结果证实,本发明化合物可以刺激cftrptc的通读,并且cftr增效剂和活化剂的组合可以进一步增强本发明的通读刺激化合物的活性。
实例10-用七叶皂苷与vx-809的组合增强ptc通读
在具有g542x、y122x和w1282x突变的frt细胞中进一步检查本发明的通读刺激化合物与cftr校正剂vx-809组合的作用。本发明化合物对ptc通读的增强通过cftr电导的增加(图13a-13c)或细胞表面cftr蛋白表达的增加(图14a-14c)来确定。cftr电导还表明,由于ptc的通读,cftr具有功能。将单独或呈所示组合的七叶皂苷(10μm)、vx-809(3μm)、g418(250μg/ml)和媒剂对照(0.3%dmso)与frtg542x、frty122x和frtw1282x细胞一起预培育48小时(在这一实例中,在确定化合物的作用之前不加入毛喉素和vx-770/gp-5)。结果显示在图13a和14a(frtg542x细胞)、13b和14b(frty122x细胞)和13c和14c(frtw1282x细胞)中。
在frtg542x细胞中,七叶皂苷和g418对cftr电导显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr电导的刺激作用的协同增加(图13a)。在frty122x细胞中,七叶皂苷和g418对cftr电导显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr电导的进一步增加的刺激作用(图13b)。在frtw1282x细胞中,七叶皂苷和g418对cftr电导显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr电导的刺激作用的协同增加(图13c)。
在frtg542xhrp细胞中,七叶皂苷和vx-809对cftr蛋白表达显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr蛋白质表达的进一步增加的刺激作用(图14a)。在frty122xhrp细胞中,七叶皂苷和vx-809对cftr蛋白表达显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr蛋白质表达的进一步增加的刺激作用(图14b)。在frtw1282xhrp细胞中,七叶皂苷和vx-809对cftr蛋白表达显示出显著的刺激作用。与单独的vx-809和七叶皂苷相比,七叶皂苷和vx-809的组合显示出对cftr蛋白表达的进一步增加的刺激作用(图14c)。
在来自具有杂合g542x/δf508突变的供体的原代hbe细胞中进行另外的实验。本发明化合物对ptc通读的增强通过cftrisc的增加来确定。为了测定短路电流(isc),用胺氯吡脒(100μm)处理hbe细胞以阻断上皮钠通道活性,之后加入cftr激动剂fsk(10μm)和vx-770(10μm);在实验结束时加入cftrinh-172(10μm)以阻断cftr依赖性isc。
图15a显示测试化合物和组合对isc的影响的代表性描记线。图15b显示vx-809和七叶皂苷和g418的组合在毛喉素和vx-809存在下显著增加isc,七叶皂苷、七叶皂苷和vx-809的组合以及七叶皂苷和g418的组合在vx-770存在下显著增加isc。
这些结果证实,本发明化合物可以刺激cftrptc的通读,并且cftr增效剂的组合可以进一步增强本发明的通读刺激化合物的活性。
实例11-七叶皂苷刺激赫尔勒综合征中的ptc的通读
赫尔勒综合征是溶酶体贮积病的最严重形式,并且是由酶α-l-艾杜糖苷酶(由idua基因编码)的丧失引起的。α-l-艾杜糖苷酶参与溶酶体内葡糖胺聚糖(gag)的降解。在某些人群中,ptc突变占导致赫尔勒综合征的突变的大约三分之二。此外,甚至在α-l-艾杜糖苷酶活性的小恢复体中也显示出降低赫尔勒综合征的作用。赫尔勒综合征中最常见的两种突变是q70x和w402x突变。
为了研究本发明的通读刺激化合物在赫尔勒综合征中的作用,在七叶皂苷(0.25至160μm)、阿霉素(50至160μm)或媒剂对照(0.1%dmso)存在下使源自纯合idua-w402x小鼠的永生化小鼠胚胎成纤维细胞(mef)生长48小时。48小时后,获得细胞裂解物并测定mefα-l-艾杜糖苷酶的活性。本发明化合物对ptc通读的增强通过α-l-艾杜糖苷酶活性的增加来确定。对于七叶皂苷,结果如图16a中所示,并且对于阿霉素,结果如图16b中所示。虚线(上部)表示0.06的比活性对应于野生型mefα-l-艾杜糖苷酶活性的约0.2%,虚线(下部)表示仅在媒剂对照存在下的活性。
如与媒剂对照相比,七叶皂苷处理引起α-l-艾杜糖苷酶活性的剂量依赖性增加(图16a)。在七叶皂苷浓度低至5至10μm时,观察到α-l-艾杜糖苷酶活性显著增加。七叶皂苷处理并没有显著降低总蛋白,尽管在大于10μm的七叶皂苷浓度下观察到总蛋白一定程度上降低。
如与媒剂对照相比,阿霉素处理引起α-l-艾杜糖苷酶活性的剂量依赖性增加(图16b)。在5至160μm的阿霉素浓度下看到α-l-艾杜糖苷酶活性显著增加,同时在较低的阿霉素浓度下观察到最大刺激。在所有测试浓度下,阿霉素处理确实使总蛋白质浓度降低约40%。
这些结果证实,本发明化合物不仅可以刺激cftrptc的通读,还可以刺激其它疾病中存在的ptc的通读,也表明本发明化合物在刺激ptc通读中的广泛用途。
- 还没有人留言评论。精彩留言会获得点赞!