包含血管破坏剂的用于治疗肝癌的组合物的制作方法
本公开涉及用于治疗肝癌的组合,其包含血管破坏剂(vda),以及使用其治疗肝癌的方法。
背景技术:
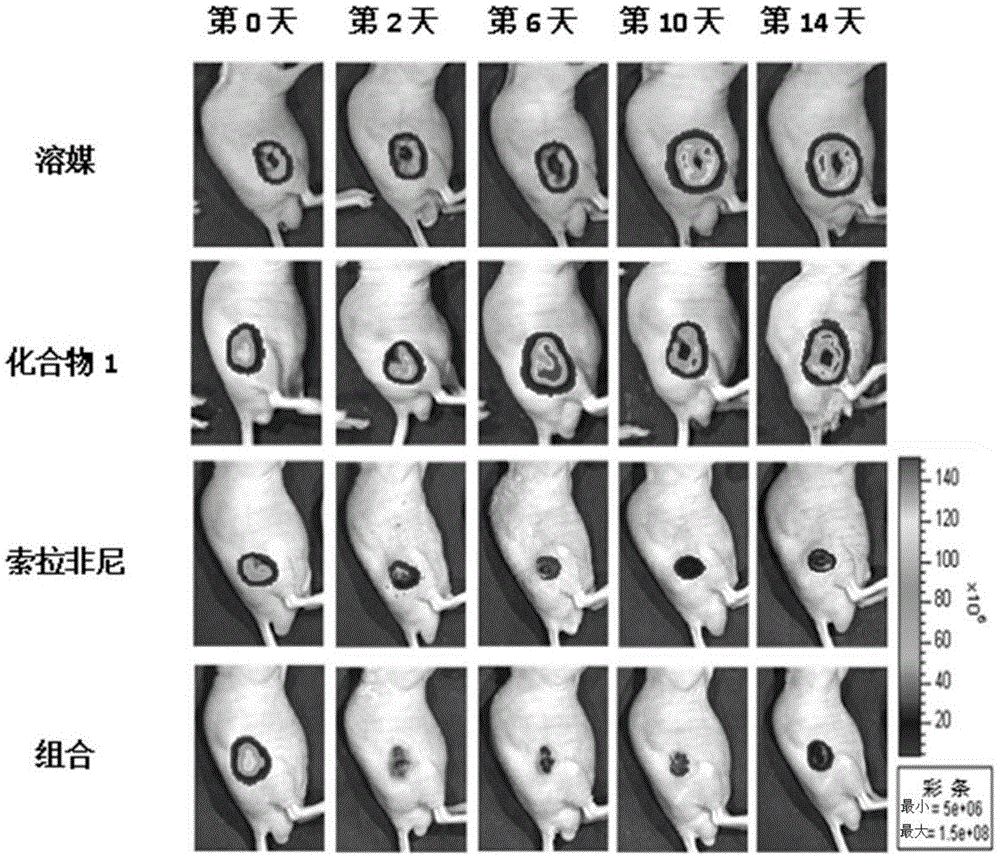
:肝癌是一种预后不良的恶性肿瘤,在癌症死亡原因中,在韩国排名第二,在世界排名第三。70%或更多的肝癌患者不可能进行根治性手术,即使肝癌患者接受根治性切除,5年内其他区域癌症复发的概率也是50%或更多。此外,晚期肝癌对全身抗癌治疗有反应的可能性非常低(10%或更低)。血管破坏剂(vda)旨在通过选择性地破坏血管内皮细胞的细胞骨架中的微管来快速和选择性地破坏已建立的肿瘤血管,并且可以在肿瘤的中心区域诱导缺血性坏死。因此,使用血管破坏剂(vda)破坏血管的方法最近作为新的抗癌策略受到关注。因此,申请人开发了下述式i化合物用作血管破坏剂(国际专利公开号wo2009-119980)。式i式i化合物是具有双重机制的微管蛋白聚合抑制剂:通过微管不稳定作用快速破坏已存在的肿瘤血管;和细胞周期阻滞导致细胞凋亡。然而,当单独使用血管破坏剂(vda)(包括式1化合物)治疗时,存在肿瘤可能从活的边缘迅速再生,从而降低这些药剂的治疗可用性的问题。因此,本发明人进行了广泛的研究以提供能够通过解决肿瘤再生问题同时利用血管破坏剂作为抗癌剂来治疗肝癌(恶性肿瘤)的组合物和治疗方法。公开技术问题本公开的目的是提供用于治疗或辅助治疗肝癌的组合物,其包含血管破坏剂(vda)。本公开的另一个目的是提供用于治疗肝癌的组合,其包含血管破坏剂(vda)和索拉非尼。本公开的另一个目的是提供用于化学栓塞(chemoembolization)的组合物,其包含血管破坏剂(vda)。针对技术问题的技术方案本发明人为实现上述目的做了大量努力,结果开发了用于治疗或辅助治疗肝癌的药物组合物,其包含血管破坏剂(vda)。在本发明中,用作血管破坏剂的化合物是由下式1表示的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5)-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐:式1在本公开中,术语“药学上可接受的盐”意指通常用于制药领域的盐。药学上可接受的盐的实例包括与无机离子如钙、钾、钠或镁形成的盐;与无机酸如盐酸、硝酸、磷酸、氢溴酸、碘酸、高氯酸或硫酸形成的盐;与有机酸如乙酸、三氟乙酸、柠檬酸、马来酸、琥珀酸、草酸、苯甲酸、酒石酸、富马酸、扁桃酸、丙酸、乳酸、乙醇酸、葡萄糖酸、半乳糖醛酸、谷氨酸、戊二酸、葡萄糖醛酸、天冬氨酸、抗坏血酸、碳酸或香草酸形成的盐;与磺酸如甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸或萘磺酸形成的盐;与氨基酸如甘氨酸、精氨酸或赖氨酸形成的盐;与胺如三甲胺、三乙胺、氨、吡啶或甲基吡啶形成的盐,但本发明中使用的盐的范围不限于这些列出的盐。在本发明中,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑的盐-2-基)-2-氨基-3-甲基丁酰胺可以是盐酸盐。在本公开中,本公开的组合物可以应用于肝癌的治疗或辅助治疗。肝癌包括所有原发性肝癌或转移性肝癌,但是本公开的组合物可以应用于原发性肝癌。此外,肝癌包括所有肝细胞癌或胆管癌,但本公开的组合物可以应用于肝细胞癌。另外,肝癌可以是早期肝癌、晚期肝癌或终末期肝癌,并且本公开的组合物可以应用于晚期肝癌。本公开的药物组合物可以通过根据本领域技术人员容易实施的方法与药学上可接受的载体一起配制而制备成单剂型或多剂量容器。药学上可接受的载体包括但不限于:乳糖,葡萄糖,蔗糖,山梨糖醇,甘露醇,淀粉,阿拉伯树胶,磷酸钙,海藻酸钠,明胶,硅酸钙,微晶纤维素,聚乙烯吡咯烷酮,纤维素,水,糖浆,甲基纤维素,羟基苯甲酸甲酯,羟基苯甲酸丙酯,滑石,硬脂酸镁和矿物油。除上述组分外,药物组合物还包括润滑剂、润湿剂、甜味剂、调味剂、乳化剂、悬浮剂、防腐剂等。合适的药学上可接受的载体和制剂在雷明顿药物科学(第19版,1995)中有详细描述。本公开提供了治疗肝癌的方法,其包括将本公开的药物组合物施用于有需要的受试者。如本文所用,术语“受试者”意指包括哺乳动物,特别是人。根据本公开的一个实施方式,本公开提供血管破坏剂和索拉非尼的组合。具体地,本发明提供了一种治疗肝癌的组合,其包括:(1)由下式1表示的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐,和(2)下式2表示的索拉非尼或其药学上可接受的盐。式1式2抗血管生成剂具有抑制肿瘤血管生成的作用,因此已广泛用于抗癌策略。已知拜尔开发的索拉非尼具有两种性质:直接抗癌作用和血管生成抑制作用。索拉非尼的抗癌作用部分是通过抑制b-raf激酶介导的,并且似乎具有通过抑制vegfr-2和pdgfr-β来抑制血管生成的性质。然而,当单独施用索拉非尼等抗血管生成剂时,其仅抑制血管生成的一个方面,因此其抗癌作用不充分。然而,在本公开的组合中,显示当作为具有不同治疗机制的抗癌剂的式1化合物(血管破坏剂)和索拉非尼(抗血管生成剂)组合施用时,由于它们的协同作用,它们表现出优异的肝癌治疗活性。此外,本公开的组合可以解决当单独施用式1化合物或索拉非尼时发生的肿瘤复发问题,因为施用本公开的组合导致活肿瘤细胞很少或没有。在本公开中,索拉非尼的药学上可接受的盐可以是通常用于制药领域的任何盐,并且可以使用索拉非尼对甲苯磺酸盐。本公开的组合可以包含两种单独的制剂,并且还可以构成单一制剂。本公开的组合可以根据预期用途口服或肠胃外(例如,静脉内,皮下,腹膜内或局部)给药。在本发明中,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-氨基-3-甲基丁酰胺或其药学上可接受的盐可以口服或胃肠外给药,优选口服给药。此外,索拉非尼或其药学上可接受的盐可以口服给药。本公开的组合中活性成分的合适剂量可以根据患者的体重、年龄、性别、健康状况、饮食、给药时间、给药方式、排泄率、疾病的严重程度等等而变化。在本发明中,所示(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐的日剂量为约1.0至40.0mg,优选约2.5至25.0mg。此外,索拉非尼或其药学上可接受的盐的日剂量为约50至1500.0mg,优选约100至1000mg。另外,可以根据其剂量确定本公开组合中的活性成分的施用频率。在本发明中,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐可以每天一次至每周一次的频率施用,并且还可以每周施用1至5次,这取决于给药途径。此外,在本公开中,索拉非尼或其药学上可接受的盐可以每天两次至每周一次的频率施用,并且还可以每周施用1至7次或每天施用1至2次。根据本发明的一个实施方式,当本公开的组合中的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐通过胃肠外给药时,可以每周施用一次,在这种情况下,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或者其药学上可接受的盐可以在每周的第1天施用一次,并且索拉非尼或其药学上可接受的盐可以在每周的第1天至第6天每天施用一次。根据本公开的另一个实施方式,当本发明的组合中的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐口服给药时,可以在每周的第1、3和5天每天施用一次(每周三次),索拉非尼或其药学上可接受的盐可以每天施用一次(每周7次)。另外,根据本公开的一个实施方式,索拉非尼或其药学上可接受的盐可以在口服(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐后20分钟至1小时施用。本公开的组合具有非常优异的肝癌治疗活性,并且当施用时,将存在很少或没有活的癌细胞,因此癌症复发的可能性将显著降低。因此,本公开的组合物可以有效地用于治疗肝癌。根据本公开的又一个实施方式,本公开提供用于化学栓塞的组合物,其包含血管破坏剂。具体地,本发明提供了一种化学栓塞用组合物,其包含:(1)由下式1表示的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐,和(2)下式3表示的索拉非尼或其药学上可接受的盐;和(3)碘化油(lipiodol):式1式3在用于治疗肝癌的图像引导疗法中,通常使用两种疗法。在这两种疗法中,一种是经动脉化学栓塞(tace),其包括通过肝动脉局部注射抗癌剂进入肝癌并使肝动脉进行化学栓塞,另一种是射频消融(rfa)。经动脉化学栓塞(tace)采用通过向肝动脉注射抗癌剂并用栓塞剂阻塞肝动脉,从而最大程度提高对肝癌的局部抗癌作用的策略。在许多情况下,经动脉化学栓塞(tace)用作姑息性疗法。正常肝组织通过门静脉接受70%或更多的血液,但肝癌组织通过肝动脉接受90%或更多的血液。因此,当在经动脉化学栓塞(tace)中通过肝动脉注射抗癌剂时,与正常肝组织相比,抗癌剂可以以相对高的浓度施用于肝癌组织。特别地,与全身注射相比,这种通过肝动脉的直接注射可以使肝癌组织中累积的抗癌剂的量增加约100倍。然而,众所周知,由化学栓塞诱发的缺氧反应引起的肝癌组织周围新血管的生长是复发的主要原因之一。然而,在本发明的用于化学栓塞的组合物的情况下,当将式1化合物(血管破坏剂)和多柔比星组合局部施用于肝癌细胞时,由于它们的协同作用,它们将表现出非常高的肝癌治疗活性。另外,由于施用本公开的组合物导致存在很少或没有活肿瘤细胞,因此本公开的组合物可能解决当式1化合物或多柔比星单独施用时发生的肿瘤复发问题。本公开的用于化学栓塞的组合物中的碘化油执行各种功能。具体地,碘化油用于将化学栓塞的组合物中的抗癌剂递送至肝癌细胞。此外,碘化油还可用作造影剂。此外,碘化油具有栓塞肝动脉的作用,因此可以增强栓塞剂如明胶的栓塞效果。因此,本发明的用于化学栓塞的组合物中的碘化油用于将组合物中的抗癌剂递送至肝癌细胞,起到造影剂的作用从而可以进行化学栓塞,并且还表现出栓塞效应以辅助治疗肝癌。本发明的用于化学栓塞的组合物还可包括水性造影剂。多柔比星是一种难以溶解在碘化油中的亲水性物质。因此,本发明的组合物优选还包括水性造影剂。根据本发明的一个实施方式,本发明的组合物中的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐和多柔比星或其药学上可接受的盐可以以5:1至1:5的重量存在,优选2:1至1:2。根据本公开的另一个实施方式,本发明的组合物中的(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐的含量优选为每毫升碘化油5.0至30.0毫克。根据本公开的又一个实施方式,本公开组合物中的多柔比星或其药学上可接受的盐的含量优选为每毫升水性造影剂5.0至50.0毫克。本公开的用于化学栓塞的组合物可以应用于各种化学栓塞疗法。因此,本公开的用于化学栓塞的组合物可以局部施用于待栓塞的区域。最优选地,通过经动脉化学栓塞(tace)将其局部施用于肝脏。此外,本公开的用于化学栓塞的组合物具有非常优异的肝癌治疗活性,并且当施用时,很少或没有活的癌细胞,因此癌症复发的可能性将显著降低。因此,本发明的用于化学栓塞的组合物可以有效地用于治疗肝癌。在注射本发明的用于化学栓塞的组合物之后,可以注射栓塞剂以栓塞所需区域。栓塞剂优选是明胶样物质。在本发明的化学栓塞组合物中,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)-2-氨基-3-甲基丁酰胺或其药学上可接受的盐和多柔比星或其药学上可接受的盐的优选剂量可根据各种因素而变化,包括患者的体重、年龄、性别、健康状况、饮食、施用时间、给药方式、排泄率、疾病的严重程度等。在本发明的用于化学栓塞的组合物中,(s)-n-(4-(3-(1h-1,2,4-三唑-1-基)-4-(3,4,5-三甲氧基苯甲酰基)苯基)噻唑-2-基)2-氨基-3-甲基丁酰胺或其药学上可接受的盐的日剂量为约1.0至40.0mg/kg,优选2.5至25.0mg/kg。此外,本发明的用于化学栓塞的组合物中,多柔比星或其药学上可接受的盐的日剂量为约10.0至1000.0mg/kg,优选20.0至800.0mg/kg。发明的有益效果本公开的组合物表现出优异的肝癌治疗活性,并且起到使癌症复发的可能性低的作用。因此,本公开的组合物可用于肝癌的治疗或辅助治疗。附图简要说明图1显示了在hep3b异种移植小鼠模型中对照组和每个实验组(肠胃外给药)的生物发光成像(bli)的结果。图2是比较hep3b异种移植小鼠模型中对照组和每个实验组(肠胃外给药)之间的相对bli信号强度的图。图3显示对照组和每个实验组(肠胃外给药)的h&e染色和tunel染色图像。图4是比较对照组和每个实验组(肠胃外给药)之间凋亡率的图。图5显示对照组和每个实验组(肠胃外给药)的cd31染色图像。图6是比较对照组和每个实验组(肠胃外给药)之间的微血管密度(mvd)的图。图7显示了在hep3b异种移植小鼠模型中对照组和每个实验组(口服给药)的生物发光成像(bli)的结果。图8是比较hep3b异种移植小鼠模型中对照组和每个实验组(口服给药)之间的相对bli信号强度的图。图9是比较对照组和每个实验组(口服给药)之间凋亡率的图。图10显示对照组和每个实验组(口服给药)的cd31染色图像。图11是比较cd31染色的对照组和每个cd31染色的实验组(口服给药)之间的微血管面积(mva)的图。图12显示meca-32染色的对照组和每个meca-32染色的实验组(口服给药)的图像。图13是比较meca-32染色的对照组和每个meca-32染色的实验组(口服给药)之间的微血管面积(mva)的图。图14是通过经动脉化学栓塞(tace)将测试物质注射到兔vx2肝癌模型中之后比较实验组之间的活肿瘤部分的图(a:施用碘化油的组;b:施用碘化油+vda的组;c:施用碘化油+多柔比星的组;d:施用碘化油+vda+多柔比星的组)。发明模式在下文中,将参考实施例进一步详细描述本发明。然而,应理解,这些实施例仅用于说明目的,并不意图限制本发明的范围。实施例1:胃肠外给药的式1化合物(vda)和索拉非尼的协同作用的评价1.测试方法细胞系的制备及hep3b细胞的感染人肝细胞癌hep3b细胞购自韩国细胞系库(首尔,韩国)。用含有萤火虫荧光素酶报告基因的慢病毒感染hep3b细胞,分离出强烈表达报告基因的克隆,以建立hep3b+luc细胞系。通过生物发光成像(bli)(珀金埃尔默,沃尔瑟姆市,马萨诸塞州,美国)证实荧光素酶的稳定表达。用含有10%胎牛血清(fbs;格兰德岛生物公司,格兰德岛,纽约,美国)和1%抗生素(格兰德岛生物公司,美国)的杜尔贝科改良的伊格尔(eagle)培养基(dmem,威健股份有限公司,首尔,韩国)维持hep3b+luc细胞系。动物肿瘤模型的制备雄性balb/c裸鼠(6周龄)购自东方生物(首尔,韩国)。通过27号针头注射器将1×106hep3b+luc细胞接种到每只动物的右胁腹中。当接种后约14天肿瘤直径超过5mm时,将动物用于试验。活性成分的制备通过国际专利公开号wo2009-119980中公开的制备方法制备式1化合物(vda),并将其溶于0.9%的盐水中。将索拉非尼对甲苯磺酸盐(lc实验室,沃本市,马萨诸塞州,美国)4倍溶解于蓖麻油聚氧乙烯醚(cremophorel)/乙醇(50:50;西格玛蓖麻油聚氧乙烯醚,95%乙醇)中,然后在使用当天用水稀释。体内生物发光成像在药物施用前(第0天)和药物施用后第2天、第6天、第10天和第14天进行生物发光成像(bli)。麻醉小鼠,然后用ivis光谱系统(珀金埃尔默,沃尔瑟姆市,马萨诸塞州,美国)成像。将磷酸盐缓冲盐水(pbs)中的d-荧光素(0.375mg/ml;普洛麦格,麦迪逊市,威斯康星州,美国)腹膜内注射到小鼠体内。将生物发光记录为感兴趣区域(roi)中每秒测量光子的总和。为了测量体内生物发光,使用具有相同大小和形状的roi,其包括每只动物的肿瘤。使用动态图像软件(版本2.50.1,珀金埃尔默,沃尔瑟姆市,马萨诸塞州,美国)进行bli数据的定量。肝癌治疗活性的评价在hep3b异种移植模型中评估式1化合物和索拉非尼的抗癌效果。动物随机分为以下几组:对照组:溶媒(生理盐水,腹膜内给药);单独施用式1化合物(vda)的组:式1化合物(5mg/kg,每周腹膜内给药一次(第1天));单独施用索拉非尼的组:索拉非尼(30mg/kg,每周口服6次(第1天至第6天每天一次));给予组合的组:式1化合物(5mg/kg,每周腹膜内给药一次(第1天))和索拉非尼(30mg/kg,每周口服6次(每周第1至第6天每天一次))。所有实验组施用每种药物2周。通过测量小鼠的体重观察毒性,并且在测试期间使用测径器测量肿瘤生长。使用以下等式计算肿瘤体积:肿瘤体积=(长×宽2)/2组织病理学研究在测试结束时(第14天),处死所有小鼠用于组织处理、免疫组织化学染色和组织学分析。在组织处理中,将试验动物深度麻醉,通过心室导管用盐水经心脏灌注,然后通过重力流动用10%多聚甲醛(pfa)处理10分钟。收集肿瘤组织,在福尔马林中固定,并包埋在石蜡中。将肿瘤的最大部分切成4μm的厚度。为了评估肿瘤细胞的凋亡程度,使用试剂盒(密理博,比尔里卡,马萨诸塞州,美国)进行末端脱氧核苷酸转移酶dutp缺口末端标记(tunel)染色。对收集的肿瘤组织的代表性区域进行苏木紫和曙红(h&e)染色。使用软件(nih-imagej,贝塞斯达,马里兰州,美国)计算细胞凋亡百分比。为了评估肿瘤血管的变化,使用抗小鼠cd31的家兔一抗(艾博抗,剑桥,马萨诸塞州)进行cd31的免疫组织化学染色。使用达科快速染色试剂盒(envision+systemhrp)标记的聚合物抗-家兔(达科)进行目测观察30分钟。用光学显微镜(型号dm2500,徕卡,德国)观察图像。免疫染色区域的测量基于韦德纳法。使用徕卡显微镜(型号dm2500,徕卡,德国),在低放大倍数(40x和100x)下观察三个区域。在200x视野(20x物镜,10x目镜)中测量每个区域中的微血管数量。使用软件(图像分析,徕卡,德国)进行血管数量的图像分析。在该软件中,将染色腔中的一个细胞计算为一个点。将所有测量区域中的微血管的平均数量确定为微血管密度(mvd)。统计学分析定量数据表示为平均值±标准偏差。使用曼-惠特尼的u-检验分析肿瘤体积、生物发光成像、细胞凋亡百分比和微血管密度之间差异的统计学显著性。p<0.05被认为是显著的。使用商业软件(统计产品与服务解决方案(spss),版本21.0用于windows,spss,芝加哥,伊利诺斯州,美国)计算所有数据。2.测试结果hep3b异种移植模型中式1化合物的肝癌治疗活性为了评价单独施用式1化合物(vda)的抗肿瘤效果,给异种移植小鼠施用式1化合物(vda),并且在测试期间观察小鼠的体重和小鼠中的肿瘤生长。未观察到在测试期间变虚弱的小鼠。在施用开始之前和之后14天之间单独施用式1化合物(vda)的组、单独施用索拉非尼的组和施用vda和索拉非尼组合的组的体重差异没有显著性(p>0.05)。施用vda和索拉非尼组合的组的肿瘤生长抑制效果与单独施用索拉非尼的组相似。这表明单独施用式1化合物(vda)的肿瘤生长抑制效果不显著。bli信号观察在测试期间,对照组的相对bli信号继续增加。这表明hep3b-luc肿瘤积极生长。在单独使用索拉非尼的组中,bli信号逐渐下降6天,并在第6天后显著恢复。在单独施用化合物(vda)的组中,在施用式1化合物(第1天和第8天)后相对bli信号降低,并且这种降低的信号持续2天并在2天后显著恢复。这表明式1化合物在施用后的初始阶段表现出快速关闭效应和细胞毒性作用。然而,在施用vda和索拉非尼的组合的组中,相对bli信号在初始阶段显著降低,并且该组合在施用开始后显示肿瘤抑制效果达10天(参见图1)。此外,相对bli信号强度与bli信号结果一致(参见图2)。这些结果表明式1化合物和索拉非尼具有协同作用。具体地,看起来式1化合物诱导快速治疗效果,并且索拉非尼维持肿瘤抑制效果。bli参数与组织学结果的相关性为了验证bli信号结果,进行了一系列组织病理学研究。在开始施用后第14天,使用tunel测定法定量肿瘤细胞的凋亡百分比。结果,细胞凋亡百分比从高到低的顺序为:施用组合的组(43.0±14.0),单独施用式1的化合物的组(38.0±13.7),单独施用索拉非尼的组(18.3±6.0))和对照组(11.1±3.7)。然而,单独施用式1化合物的组的细胞凋亡结果与施用组合的组的细胞凋亡结果没有显著差异。这表明索拉非尼对肿瘤细胞生长的作用是细胞抑制而不是细胞毒性(参见图4)。此外,如在h&e染色和tunel染色的显微图像中可见,在单独施用索拉非尼的组和对照组中,肿瘤细胞看起来像散布的点,而在施用式1化合物的组中,肿瘤细胞主要出现在凋亡的肿瘤细胞中。此外,施用组合的组中的细胞凋亡类似于给予式1化合物的组中的细胞凋亡(参见图3)。这些结果表明式1化合物由于其肿瘤破坏作用而表现出广泛的凋亡作用,并且由于索拉非尼的肿瘤生长抑制性质而表现出肿瘤生长抑制作用。另外,在单次施用每种药物第14天后获得抗-cd31染色的组织学样品和肿瘤微血管密度(mvd)的定量结果。在对照组中,肿瘤血管很大并且分布在整个肿瘤中。然而,在实验组中,出现了与对照组中观察到的血管图像不同的血管图像。具体地,在实验组中,血管的长度短并且分布在肿瘤的边缘。实验组之间的比较基于血管密度(参见图5)。肿瘤mvd的定量表明,微血管密度(mvd)从高到低的顺序为:生理盐水组(3180±465),施用式1化合物的组(2334±60),施用索拉非尼的组(1871±225),和施用组合的组(1122±212)(参见图6)。在所有组中观察到统计学显著性。这些结果还表明式1化合物的血管破坏性质和索拉非尼的抗血管生成性质表现出协同作用。实施例2:口服给药的式1化合物(vda)和索拉非尼的协同作用的评价1.测试方法以与实施例1中所述相同的方式进行细胞系、动物肿瘤模型和活性成分的制备。体内生物发光成像在药物施用前(第0天)和药物施用后第2天、第4天、第8天、第11天、第15天、第18天和第20天进行生物发光成像(bli)。麻醉小鼠,然后用ivis光谱系统(珀金埃尔默,沃尔瑟姆市,马萨诸塞州,美国)成像。将磷酸盐缓冲盐水(pbs)中的d-荧光素(0.375mg/ml;普洛麦格,麦迪逊市,威斯康星州,美国)腹膜内注射到小鼠体内。将生物发光记录为感兴趣区域(roi)中每秒测量光子的总和。为了测量体内生物发光,使用具有相同大小和形状的roi,其包括每只动物的肿瘤。使用动态图像软件(版本2.50.1,珀金埃尔默,沃尔瑟姆市,马萨诸塞州,美国)进行bli数据的定量。肝癌治疗活性的评价在hep3b异种移植模型中评估式1化合物和索拉非尼的抗癌效果。动物随机分为以下几组:对照组:溶媒(生理盐水,口服给药);单独施用式1化合物(vda)的组:式1化合物(4mg/kg,每周口服给药三次(第1天、第3天和第5天));单独施用索拉非尼的组:索拉非尼(15mg/kg,每周口服7次);施用组合的组:式1化合物(4mg/kg,每周口服给药三次(第1天,第3天和第5天))和索拉非尼(15mg/kg,口服给药7次/周(每天一次))。每天下午2点进行口服给药,在施用组合的组中,在施用式1化合物后30分钟施用索拉非尼。所有实验组施用每种药物3周。通过测量小鼠的体重观察毒性,并且在测试期间使用测径器测量肿瘤生长。使用以下等式计算肿瘤体积:肿瘤体积=(长×宽2)/2组织病理学研究在测试结束时(第21天),处死所有小鼠用于组织处理、免疫组织化学染色和组织学分析。根据实施例1中描述的方法进行提取的肿瘤组织的处理。为了评估肿瘤细胞的凋亡程度,使用试剂盒(密理博,比尔里卡,马萨诸塞州,美国)进行末端脱氧核苷酸转移酶dutp缺口末端标记(tunel)染色。对收集的肿瘤组织的代表性区域进行苏木紫和曙红(h&e)染色。使用图像处理软件(nih-imagej,贝塞斯达,马里兰州,美国)计算细胞凋亡百分比。为了评估肿瘤血管的变化,使用抗小鼠cd31的家兔一抗(艾博抗,剑桥,马萨诸塞州)和抗meca-32的大鼠一抗(诺瓦斯生物制品(novusbiologicals),利特尔顿,科罗拉多州,美国)进行免疫组织化学染色。使用达科快速染色试剂盒(envision+systemhrp)标记的聚合物抗-兔(达科)进行cd31的目测观察30分钟。用光学显微镜(型号dm2500,徕卡,德国)观察图像。为了观察meca-32,使用绿色荧光探针(alexafluor488)抗-大鼠抗体。通过cd31和meca-32的免疫化学染色确定微血管区域(mva)。使用徕卡显微镜(型号dm2500,徕卡,德国),在低放大倍数(40x和100x)下观察三个区域。在200x视野(20x物镜,10x目镜)中测量每个区域中的微血管数量。使用软件(图像分析,徕卡,德国)进行血管数量的图像分析。统计学分析定量数据表示为平均值±标准偏差。使用重复测量方差分析测试(rmanovatest)评估每组的肿瘤体积和生物发光成像,并且使用单因素方差分析测试分析组间凋亡百分比和微血管面积差异的统计学显著性。p<0.05被认为是显著的。使用商业软件统计产品与(服务解决方案(spss),版本21.0用于windows,spss,芝加哥,伊利诺斯州,美国)计算所有数据。2.测试结果bli信号的观察在测试期间,对照组的相对bli信号增加至第11天,然后维持在该水平。在给药期的后半段,观察到癌症中心的信号强度降低。这被认为是随着癌症生长而发生的自然坏死(参见图7)。在施用式1化合物的组中,观察到信号强度迅速下降至第2天,然后增加。在施用索拉非尼的组中,观察到信号强度在整个测试期间逐渐降低。在施用组合的组中,信号强度迅速下降,并且在整个给药期间保持降低的信号(参见图8)。这些结果表明,与实施例1的结果一样,式1化合物诱导快速治疗效果并维持索拉非尼的肿瘤抑制效果。此外,可以看出,即使当口服给予式1化合物时,它也表现出优异的效果,与通过注射给药时相似。bli参数与组织学结果的相关性为了验证bli信号结果,进行了一系列组织病理学研究。在开始施用后第21天,使用tunel测定法定量肿瘤细胞的凋亡百分比。结果,细胞凋亡百分比从高到低的顺序为:施用组合的组(34.18±5.99),单独施用式1的化合物的组(16.24±6.16),单独施用索拉非尼的组(10.36±3.62)和对照组(3.74±2.08)(参见图9)。此外,抗-cd31-染色的组织学样品(图10)和其中肿瘤mva的定量结果(图11)和meca-32-染色的组织学样品(图12)和其中肿瘤mva的定量结果(图13)在每种药物施用开始后第21天获得。使用cd31进行的mva分析表明,对照组、单独施用式1化合物的组和单独施用索拉非尼的组显示肿瘤mov分别为16339±3614,14452±2709和14934±3154,而施用组合的组显示肿瘤mva为7161±4522,显著低于其他组(参见图11)。使用meca-32进行的mva分析表明肿瘤mvd从高到低的顺序为对照组(17183±5508),单独施用索拉非尼的组(14897±3556),单独施用式1化合物的组(12871±3433)和施用组合的组(7631±2872)(参见图13)。这些结果还表明式1化合物的血管破坏性质和索拉非尼的抗血管生成性质表现出协同作用。另外,观察到组间血管形态的差异。具体地,在施用式1化合物的组中,血管数量减少,血管腔变窄(参见图10和12)。这是显示式1化合物的血管破坏活性的良好实例。此外,据信将式1化合物与具有抗血管生成活性的索拉非尼联合施用通过改变肝癌中血管的形态和数量而显示出协同作用。实施例3:本公开的用于化学栓塞的组合物的肝癌治疗效果和肝毒性的评估1.测试方法建立作为肝癌的代表性动物模型的家兔vx2癌肝肿瘤模型并使用。切开雄性新西兰白兔(体重3kg)的腹部,并暴露肝脏的左叶。然后,通过注射器针头将制备成具有1mm3大小的vx2癌肿瘤芯片植入肝脏表面。在肿瘤植入后2至3周,进行非造影计算机断层扫描(ct)和灌注磁共振成像以确定是否将产生肿瘤。建立实验组将动物随机分成下列a至d组,每组由10只动物组成:实验组a:施用碘化油(0.2ml碘化油+0.05ml造影剂[通用医疗威视派克270])的组;实验组b:施用碘化油和式1化合物(vda)的组(0.2ml碘化油+1.26mg式1化合物(vda)+0.05ml造影剂[通用医疗威视派克270]));实验组c:施用碘化油和多柔比星的组(0.2ml碘化油+0.05ml造影剂[通用医疗威视派克270])和1mg多柔比星);实验组d:给予碘化油、式1化合物(vda)和多柔比星的组(0.2ml碘化油+1.26mg式1化合物(vda)+0.05ml造影剂[通用医疗威视派克270])+1毫克多柔比星)。药物的施用将微导管通过家兔耳动脉和腹腔干并进入左肝动脉,然后将对应于实验组a至d的药物小心地注射到左肝动脉中。在tace之前和之后,进行非增强ct以确定肿瘤的存在或不存在,并且在tace之后,评估沉积在肿瘤中的碘化油。2.测试结果肝癌治疗效果评价在施用后1周,提取肝组织以制备病理切片。进行h&e染色和tunel染色以确定坏死的肿瘤区域。使用imagej程序计算坏死肿瘤面积,并将活肿瘤部分表示为百分比。结果如下表1和图14所示。表1实验组活肿瘤部分(%)a47.06±11.38b27.48±13.64c14.36±12.51d0.66±1.03(平均值±标准差)从上表1中可以看出,在实验组a单独施用不含抗癌活性的碘化油的情况下,活肿瘤部分为47.06%,而在实验组b给予碘化油与式1化合物(vda)的组合的情况下,活肿瘤部分为27.48%,并且在组c施用碘化油与多柔比星的组合的情况下,活肿瘤部分仅为14.36%。这些结果表明,在实验组a至c中癌症复发的可能性很高。同时,在实验组d施用碘化油、式1化合物(vda)和多柔比星的组合的情况下,活肿瘤部分为0.66%,表明大多数肿瘤被除去。因此,可以看出,实验组d中癌症复发的可能性显著低于实验组a至c中的癌症复发的可能性。另外,实验组a和c的结果具有10%或更高的标准偏差,表明药物的肿瘤治疗效果的重现性差。然而,实验组d的结果具有仅约1%的标准偏差,表明施用实验组d的药物可重现地治疗肝癌。毒性评估在药物注射之前和药物注射后第1、3和7天,取血样,测量其中的ast和alt水平以评估急性肝损伤的程度。结果显示,实验组d中的急性肝损伤程度最高,但在7天后恢复至与其他实验组相似的水平。此外,在实验组中未观察到家兔死亡,表明实验组d中的肝损伤是可接受的。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1