帕金森氏病的治疗的制作方法
本申请要求2016年6月2日提交的印度专利申请号in201621019087以及2016年6月2日提交的in201621019185的权益,其由此通过引用并入。本发明涉及治疗或预防对象中帕金森氏病的方法,其包括施用式i的化合物或其药学上可接受的盐其中,r1是–nhc(o)c3-6环烷基和r2是氢;或r1和r2连同其所附接的碳原子形成六元芳香环,其中该环取代有选自氢、卤素和c1-6烷基的一个或多个基团;r3和r4独立地选自氢、卤素、c1-3烷基、oc1-3烷基、no2、sc1-3烷基、c1-3卤烷基、oc1-3卤烷基和sc1-3卤烷基。
背景技术:
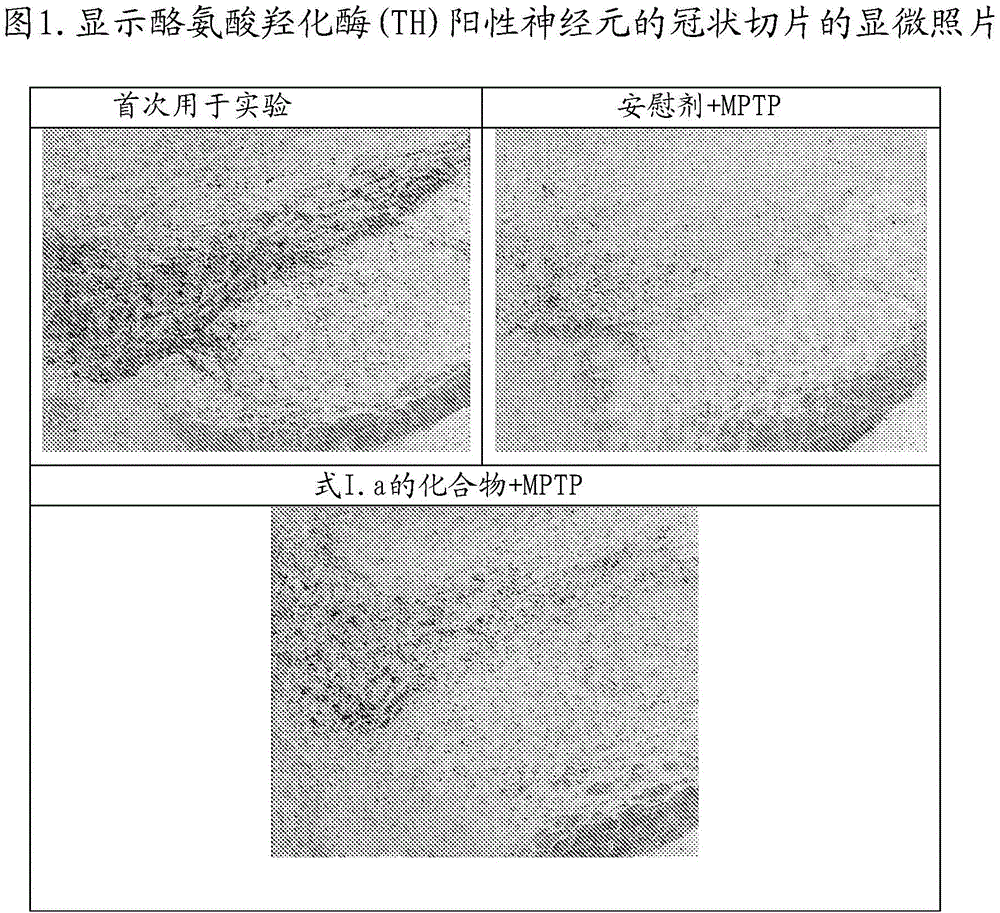
:c-ab1是一种非受体蛋白酪氨酸激酶,其涉及各种细胞过程,包括细胞存活、生长和游动性的调节。c-abl激酶抑制剂,诸如伊马替尼尼罗替尼达沙替尼和普纳替尼已经开发并上市,用于治疗慢性髓细胞性白血病的临床用途。近来的研究已经证明了c-abl在氧化应激诱导的神经细胞死亡中起到重要作用(wu等人,celldeathdiffer.,2016;23:542-552)。已经报道了c-abl参与帕金森氏病(gonfloni等人,int.j.cellbiol.,2012;2012:1-7)。而且,已知c-abl受多巴胺能应激以及受多巴胺能神经毒素——即1-甲基-4-苯基-1,2,3,6-四氢吡啶(mptp,体内酶促转化为其活性形式mpp+)——活化,引起帕金蛋白的酪氨酸磷酸化,导致帕金蛋白的e3连接酶活性丧失。这导致帕金蛋白的各种底物的积累并最终导致多巴胺能神经元的死亡(ko等人,pnas,2010;107:16691-16696)。帕金森病(pd)是一种常见的神经变性疾病,其特征在于称为lewy小体的细胞内内含物中的蛋白积累,和lewy神经炎,以及随后的多巴胺能神经元的丧失。罕见的家族性突变提供了对这种慢性、进行性神经变性疾病的了解,如α-突触核蛋白和lrrk2中的突变引起常染色体显性pd,而dj-1、pink1和帕金蛋白中的突变导致常染色体隐性pd。帕金蛋白是一种e3泛素连接酶,并且家族性突变被认为损害帕金蛋白的e3连接酶活性(ko等人,pnas,2010;107:16691-16696)。已显示c-abl调节涉及pd发病机理的两种蛋白质——即帕金蛋白和α-突触核蛋白——的降解(mahul-mellier等人,hum.mol.genet.,2014;23:2858-2879)。c-abl磷酸化帕金蛋白的酪氨酸143。该磷酸化抑制帕金蛋白的e3泛素连接酶活性,导致aimp2和fbp1(帕金蛋白底物)的积累和帕金蛋白的细胞保护功能的丧失,导致细胞死亡(ko等人,pnas,2010;107:16691-16696;imam等人,j.neurosci.,2011;31:157-163)。进一步,c-abl调节α-突触核蛋白(一种突触蛋白,其强烈涉及pd的发病机理)的清除率。已经描述了α-突触核蛋白和c-abl之间体内双向关系,其中α-突触核蛋白表达的增加促进其磷酸化以及随后的c-abl的活化。相反地,c-abl表达和活化的增加导致α-突触核蛋白积累和聚集,这暗示c-abl的抑制可能构成保护多巴胺能神经元免受pd中积累的α-突触核蛋白毒性的可行策略(hebron等人,hum.mol.genet.,2013;22:3315-3328;hebron等人,autophagy,2013;9:1249-1250)。已知c-abl抑制剂如尼罗替尼跨越血脑屏障并保护1-甲基-4-苯基-1,2,3,6-四氢吡啶(本文称为mptp)诱导的pd小鼠模型中的多巴胺能神经元(karuppagounder等人,sci.rep.2014;4:4874)。已经显示了尼罗替尼经由自噬途径增加α-突触核蛋白清除率并保护免受该pd小鼠模型中多巴胺能神经元的α-突触核蛋白积累诱导的丧失(hebron等人,hum.mol.genet.,2013;22:3315-3328;imam等人,j.neurosci.,2011;31:157-163)。进一步,us20150087653公开了治疗神经变性疾病的方法,其包括施用酪氨酸激酶抑制剂,诸如尼罗替尼。然而,尼罗替尼具有若干主要药物相关的不利作用。usfda已发布胶囊的加框(boxed)警告,因为其治疗潜在地与患者中严重的心脏副作用(qt延长)和猝死相关。已知达沙替尼引起胸腔积液和出血。普纳替尼也与严重的副作用相关,其包括血栓栓塞和血管闭塞。伊马替尼不是abl激酶的强效抑制剂。而且,为p-糖蛋白(p-gp)底物的伊马替尼和达沙替尼二者都显示差的脑浓度。因此,存在对于强效abl激酶抑制剂的需要,其可以跨越血脑屏障并且不会引起心血管副作用。技术实现要素:本发明提供治疗或预防对象中帕金森氏病的方法,其包括施用治疗有效量的式i的化合物或其药学上可接受的盐,其中,r1是–nhc(o)c3-6环烷基和r2是氢;或r1和r2连同其所附接的碳原子形成六元芳香环,其中该环取代有选自氢、卤素和c1-6烷基的一个或多个基团;r3和r4独立地选自氢、卤素、c1-3烷基、oc1-3烷基、no2、sc1-3烷基、c1-3卤烷基、oc1-3卤烷基和sc1-3卤烷基。附图说明图1.显示酪氨酸羟化酶(th)阳性神经元的冠状切片的显微照片,其显示了通过式i.a的化合物预防神经变性。图2.施用式i.a的化合物时th阳性神经元的面积百分比。图3.施用式i.a的化合物时th-免疫反应性的积分密度。图4.显示酪氨酸羟化酶(th)阳性神经元的冠状切片的显微照片,其显示了通过式i.b的化合物预防神经变性。图5.施用式i.b的化合物时th阳性神经元的面积百分比。图6.施用式i.b的化合物时th-免疫反应性的积分密度。具体实施方式一方面,本发明提供了治疗或预防对象中帕金森氏病的方法,其包括施用治疗有效量的式i的化合物或其药学上可接受的盐,其中,r1是–nhc(o)c3-6环烷基和r2是氢;或r1和r2连同其所附接的碳原子形成六元芳香环,其中该环取代有选自氢、卤素和c1-6烷基的一个或多个基团;r3和r4独立地选自氢、卤素、c1-3烷基、oc1-3烷基、no2、sc1-3烷基、c1-3卤烷基、oc1-3卤烷基和sc1-3卤烷基。另一方面,本发明提供了治疗或预防对象中帕金森氏病的方法,其包括选择遭受帕金森氏病或处于发展帕金森氏病的风险下的对象和施用治疗有效量的式i的化合物。本文所使用的短语“治疗有效量的式i的化合物”指如此量的式i的化合物:由于施用其引起治疗效果。术语“烷基”指在主链中仅包括碳和氢原子的饱和烃链自由基,其为直链或支链的并且通过单键与分子的其余部分附接,如,甲基、乙基、正丙基、1-甲基乙基(异丙基)、正丁基和正戊基。短语如“c1-6烷基”中的数字指在烷基链中存在1至6个碳原子。术语“c3-6环烷基”指3至6个碳原子的非芳香单环体系。单环包括环丙基、环丁基、环戊基和环己基。术语“卤烷基”指取代有选自氯根、溴根、碘根和氟根的一个或多个卤素自由基的烷基链。在一个实施方式中,本发明提供了治疗或预防帕金森氏病的方法,其包括施用治疗有效量的式i的化合物,其中式i的化合物中的r1为–nhc(o)环丙基和r2为氢。在另一实施方式中,本发明提供了治疗或预防帕金森氏病的方法,其包括施用治疗有效量的式i的化合物,其中式i的化合物中的r1和r2连同其附接的碳原子形成六元芳香环,其中该环取代有选自氢、卤素和c1-6烷基的一个或多个基团。优选地,芳香环取代有氢,即未取代的。在另一实施方式中,本发明提供了治疗或预防帕金森氏病的方法,其包括施用治疗有效量的式i的化合物,其中式i的化合物中的r1和r2连同其附接的碳原子形成六元芳香环,其中该环取代有氢,即未取代的;和r3是氯和r4是甲基并且作为环中2位和6位处的取代基存在。在另一实施方式中,本发明提供了治疗或预防帕金森氏病的方法,其包括施用治疗有效量的式i的化合物,其中式i的化合物中的r3和r4选自卤素和c1-6烷基。在优选实施方式中,r3和r4是卤素和甲基并且作为环中2位和6位处的取代基存在。下面在表1中提供一些式i的优选化合物的化学名称。表1:本发明化合物的合适的药学上可接受的盐可以是无机酸的盐,诸如盐酸、氢溴酸、磷酸等的盐,或者是有机酸的盐,例如乙酸、苯磺酸、甲磺酸、苯甲酸、柠檬酸、乙醇酸、乳酸、富马酸、琥珀酸、己二酸、庚二酸、辛二酸、壬二酸、苹果酸、酒石酸或氨基酸——诸如谷氨酸或天冬氨酸——等的盐。式i化合物的一个或多个氢原子可以被氘化,即用氘原子取代。wipo公开wo2012098416(‘416公开)公开了起c-abl激酶抑制剂活性的马库什组化合物以及其治疗癌症如慢性骨髓性白血病(cml)的有用性。本发明的式i的化合物可以通过wo2012098416和wo2016185490中描述的过程制备,其通过引用并入本文。发明人已经发现,本发明的式i的化合物是强效的abl激酶抑制剂,并且有利地有效跨越血脑屏障,导致高比率的式i的化合物的脑与血浆浓度和高治疗指数。具体地,发明人已经发现,治疗有效剂量的式i的化合物当测试其对herg通道的体外影响及其对有意识的比格犬和豚鼠中的ecg参数如qt间期、qtc间期、qtcf间期和心率的体内影响时,没有心血管副作用。发现式i的化合物是安全的,因为它们对ecg参数和心率没有显示出任何不适当的影响,如本文实施例中所描述的。式i的化合物可以以合适的剂型的形式口服施用。合适的剂型可包括片剂、丸剂、胶囊、小药囊、小袋中的丸剂、胶囊中的丸剂、粉末、颗粒等。式i的化合物可以配制为口服剂型,其可以包括本领域技术人员公知的药学上可接受的赋形剂。remington'spharmaceuticalsciences,sixteenthedition,e.w.martin(mackpublishingco.,easton,pa.,1980)公开了可用于制备合适剂型的药学上可接受的载体。下面的实施例用于说明本发明而不是将本发明限制在其范围内。实施例1abl激酶抑制在最终25μl的反应体积中,利用8mmmopsph7.0、0.2mmedta、50μmeaiyaapfakkk、10mmmg(oac)2和[γ-33p-atp](比活性约500cpm/pmol,浓度按需)培育abl(人)(5-10mu)。通过添加mgatp混合物开始反应。室温下培育40分钟后,通过添加5μl的3%磷酸溶液停止反应。然后将10μl的反应点加至p30过滤垫(filtermat)上并以75mm磷酸洗涤3次持续5分钟和以甲醇洗涤一次,然后干燥和闪烁计数。在表2中提供式i的代表性化合物的结果。表2:c-abl中酪氨酸激酶抑制剂的比较效力实施例2脑/血浆药代动力学研究为了确定式i.a的化合物是否跨越血脑屏障,给c57bl6小鼠口服施用30mg/kg的式i.a化合物、100mg/kg的尼罗替尼或30mg/kg的达沙替尼。在治疗后的1、4和8小时时间点,用异氟烷麻醉小鼠,并从眼眶后丛中取出0.4ml的血液到含有8μl肝素钠作为抗凝剂(100iu/ml)的微量离心管中,并转移到冰容器。将血液样品在8500rpm、4℃下立即离心7分钟。在预先标记的微量离心管中分离血浆并在-70℃下储存,直到至进一步分析。此后立即处死小鼠,移出全脑并用冰冷的磷酸盐缓冲盐水(pbs)冲洗以除去外来血液并印迹干燥。将脑组织样品称重并使用组织匀浆器在1:2体积的pbs中匀浆,并在-70℃下储存在标记的小瓶中,直至进一步分析。使用lc-ms技术确定脑和血浆中式i.a的化合物的浓度。发现式i.a的化合物的脑与血浆比显著高于尼罗替尼或达沙替尼的脑与血浆比(参见表3)。表3:脑和血浆中化合物的浓度。bql-在量化限以下实施例3在帕金森氏病的动物模型中式i.a的化合物的功效给c57bl/6小鼠(6-8周龄,25-30g体重)口服施用运载体、式i.a的化合物(10或30mg/kg,每天一次),持续7天。在第7天,这些动物以2小时间隔接受四次腹膜内注射盐水中的神经毒素:1-甲基-4-苯基-1,2,3,6-四氢吡啶(mptp-hcl;17mg/kg游离碱;sigma)。在最后一次注射mptp后,每天给予化合物持续另外的7天。简言之,在mptp施用前的6天、mptp施用当天和mptp施用后的7天,施用化合物。在mptp施用后7天处死所有动物并处理脑组织用于免疫组织化学评估。标准的抗生物素蛋白-生物素方法(benno等人,brainres.,1982;246:225-236)用于酪氨酸羟化酶(th)的免疫组织化学检测。最初,使用恒冷切片机在黑质致密部(snpc)水平下将脑切片并安装在载玻片上。将具有脑切片的载玻片在0.1mpbs中冲洗3×5分钟。首先将切片在室温下用具有0.3%tritonx-100的0.1mpbs中的正常封闭血清培育30分钟,并用具有正常封闭血清的0.1mpbs中以1:1000稀释的小鼠酪氨酸羟化酶的兔多克隆抗体(invitrogen)培育另外的2小时。将具有切片的载玻片在0.1mpbs中进一步冲洗3×5分钟,并在室温下用生物素化的抗兔抗体(abc试剂盒)培育1小时。将切片在0.1mpbs中冲洗3×5分钟,并在室温下用0.1mpbs中1:50稀释的a和b溶液(abc试剂盒)培育1小时。1小时后,将切片在0.1mpbs中冲洗3×5分钟,并在3,3'-二氨基联苯胺(dab)/h2o2溶液(sigma)中培育约5-8分钟(在显微镜下检查显影过程以获得最佳信噪比)。首先将切片在0.1mpbs中冲洗2×5分钟,然后在milli-q水中冲洗3×5分钟。将载玻片在甘油-明胶溶液中封片(coverslip),并在室温下保持过夜,进行干燥,之后使用连接至显微镜的照相机拍摄染色的脑切片的图像,并使用nih软件(nih,bethesda,md)进行分析。通过转换为8位分辨率和最佳亮度/对比度来分析图像。去除各个图像上的背景和伪信号,并测量由细胞体和纤维覆盖的面积。计算与总图像面积相比的面积百分比和积分密度。使用graphpad6.0比较所得结果的任何组间或组内差异(variation)和统计学显著性。口服施用式i.a的化合物显著地预防了由mptp诱导的神经变性,如从显示th阳性神经元的冠状切片的显微照片(图1)、th阳性神经元的面积百分比的确定(图2)和th-免疫反应性的积分密度(图3)所见的。实施例4在帕金森氏病的动物模型中式i.b的化合物的功效通过与对上面实施例3中的式i.a的化合物的描述相同的方法测试式i.b化合物治疗帕金森氏病的功效。使用graphpad6.0比较所得结果的任何组间或组内差异和统计学显著性。口服施用式i.b的化合物显著地预防了由mptp诱导的神经变性,如从显示th阳性神经元的冠状切片的显微照片(图4)、th阳性神经元的面积百分比的确定(图5)和th-免疫反应性的积分密度(图6)所见的。实施例5电生理学过程-对hergk+通道的影响在体外测试中测试式i.a和i.b的化合物的心血管安全性,以确定对hergk+通道的抑制。式i.a和i.b的化合物在测试的浓度下未显示出对herg电流的任何显著抑制。检查式i.a和i.b的化合物对稳定表达人ether-à-go-go-相关基因(herg)的电压钳制的人胚胎肾细胞(hek293)中的离子电流的体外影响。在接近生理温度下,测试两种浓度的式i.a和i.b的化合物(1和10μm)。将细胞转移至记录室并用运载体对照溶液(hepes缓冲的生理盐水溶液+1%二甲基亚砜+1%牛血清白蛋白)覆盖(superfuse)。用于全细胞膜片钳记录的微量移液管溶液由以下组成:天冬氨酸钾,130mm;mgcl2,5mm;egta,5mm;atp,4mm;hepes,10mm;用10nkoh将ph调节至7.2。微量移液管溶液分批制备、等分、冷冻保存,并且每天解冻新鲜的等分试样。使用直列式溶液预热器、室加热器和反馈温度控制器的组合,在33至35℃的温度下进行记录。在记录室中使用热敏电阻探针测量温度。使用p-97微量移液管拉出器(sutterinstruments,novato,ca)从玻璃毛细管制备用于膜片钳记录的微量移液管。商用膜片钳放大器用于全细胞记录。在数字化之前,电流记录以采样频率的五分之一进行低通过滤。稳定表达herg的细胞被保持在-80mv。使用具有固定振幅的脉冲模式(调节前脉冲+20mv,1s;以5秒间隔重复复极化试验渐变(ramp)至-80mv(-0.5v))测量由于测试化合物(式i.a和i.b)引起的herg钾电流的起始和稳态抑制。每次记录以最终应用超大浓度的参考物质(e-4031,500nm)结束,以评估内源性电流的贡献。从数据中数字地离线减去剩余的未抑制电流,以确定测试化合物对herg抑制的效力。相比在运载体对照中1.0±0.6%(n=3),式i.a的化合物在1μm(n=3)时抑制herg电流(平均值±sem)2.1±0.4%,在10μm(n=3)时抑制9.7±0.4%(见表4)。由于化合物在运载体对照中的溶解度限制,未测试浓度超过10μm的式i.a的化合物。在相似条件下,阳性对照(特非那定,60nm)抑制herg钾电流(平均值±sd;n=2)82.8±1.2%,这确认了式i.a的化合物在其非常高浓度下在herg+通道中轻微作用。相比之下,尼罗替尼在1μm浓度下显示出近90%的抑制(表5)。表4:式i.a和i.b的化合物在每个浓度下的herg电流的平均抑制百分比。*值与单独运载体在统计学上不同。表5:尼罗替尼对hegr电流的抑制百分比#浓度(μm)0.030.10.31抑制%14.9±243.9±0.770.4±2.589.7±1.6#数据来自nda第22-068号(capsule)pg第31号的cder药理学综述实施例6式i.a的化合物对有意识的比格犬中的qt、qtc间期以及心率的影响使式i.a的化合物经历体内测试,以确定在有意识的比格犬中其对qt、qtcb和qtcf间期以及心率的影响。在有意识的遥测(telemetered)雄性和雌性比格犬中经由口服(p.o.)途径以三种剂量水平(5、15和30mg/kg)施用式i.a的化合物。在给药后14分钟用30mg/kg剂量组的式i.a化合物治疗的两只动物(1雄性和1雌性)发生呕吐。因此,用30mg/kg剂量治疗的组未被考虑用于数据分析。用5和15mg/kg剂量没有观察到呕吐。在药物施用(基线)之前2小时以及在施用后连续上至24小时的期间内进行ecg参数(qt间期、qtcb间期、qtcf间期)和心率的测量并记录。每只动物以拉丁方设计(latinsquaredesign)在不同日子接受运载体(安慰剂)和三种不同剂量的式i.a化合物,清除期最少96小时。在单独的一天,进行一次额外的口服给药式i.a的化合物以评估在不同时间点式i.a的化合物的血浆浓度,以导出药代动力学(pk)参数。ecg参数(qt间期、qtcb间期、qtcf间期)和心率的数据如下进行统计学比较:将0.5、1、1.5、2、3、4、5、6、8、12和24小时时间点处的安慰剂组的数据与同组的基线数据进行比较。将用式i.a的化合物治疗的不同组的数据与安慰剂组的对应数据进行比较。基于雄性和雌性狗的汇总数据的ecg分析显示,与基线相比,安慰剂治疗对任何ecg参数(qt间期、qtcb间期、qtcf间期)和心率没有统计学上显著的影响。与安慰剂相比,5和15mg/kg剂量的式i.a化合物对qt、qtcb、qtcf持续时间没有影响。此外,心率等其他参数保持不变。pk分析显示式i.a的化合物对施用不同剂量的式i.a化合物的动物的剂量依赖性全身性暴露。使用5、15和30mg/kg剂量的式i.a化合物,auc0-inf分别在1925至4824、6776至12756和25927至46749hr×ng/ml的范围内。使用5、15和30mg/kg剂量的式i.a化合物,c最大分别在1218至2033、2723至5323和9825至9967ng/ml的范围内。该研究的结果表明,单次口服施用式i.a的化合物上至15mg/kg剂量对有意识的比格犬的心血管功能没有影响。在任何剂量水平的式i.a的化合物下,ecg形态没有变化。对ecg参数和心率的治疗影响无性别差异。实施例7式i.b的化合物对麻醉的豚鼠中的qt、qtc间期以及心率的影响在麻醉的豚鼠中研究了式i.b的化合物对qt、qtc间期和心率的影响。通过腹膜内注射氨基甲酸乙酯(1.5g/kg)麻醉体重范围为300-400g的雄性dunkin-hartley豚鼠。进行气管切开术并在整个实验过程中允许动物自发呼吸。使用填充有肝素化盐水溶液(100iu/ml)的聚乙烯导管插管左颈静脉用于药物施用。将电极置于导线ii位置并记录基线ecg。对于不同的动物,根据体重,称量式i.b的化合物的剂量并在0.5mldmso中制备,并在10分钟时期内经由缓慢静脉内输注施用。在输注完成期间和之后上至0.5小时记录ecg。使用powerlab软件测量qt和rr间期。在输注期间的1、5和10分钟以及输注后5、10、15和30分钟分析数据,取9次搏动的平均值。施用1mg/kg静脉内剂量的式i.b的化合物,qt间期没有变化。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1