一种基于贝叶斯网络推理的基因间交互关系挖掘方法与流程
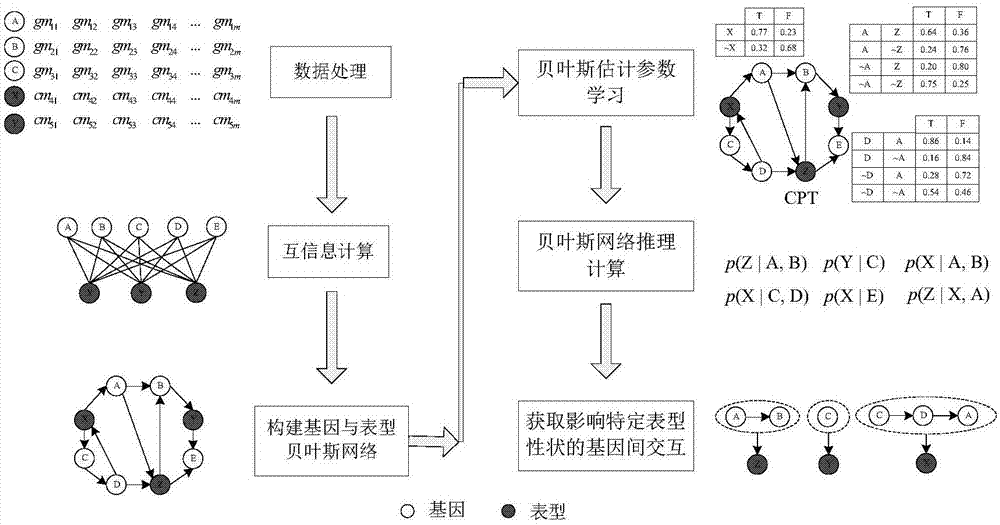
本发明涉及生物信息技术领域,尤其涉及一种基于贝叶斯网络推理的基因间交互关系挖掘方法。
背景技术:
生物信息学是一门通过综合运用生物学(如结构生物学、生物化学、遗传学等),计算机科学(人工智能、机器学习等),数学(概率与统计等)等多门学科而获知海量生物数据背后奥秘的交叉学科。它综合运用各种算法、软件等对大量生物数据进行分析和处理,进而挖掘隐藏在数据背后的生物学规律。疾病是影响人类健康最重要的因素,疾病可以分为简单疾病和复杂疾病。目前大多数疾病都是属于复杂疾病,比如阿兹海默症、哮喘、帕金森氏症、骨质疏松症、结缔组织病等。虽然复杂疾病不遵循孟德尔遗传规律,但疾病易感性在一定程度上可以由基因因素进行解释,拥有某种易感性基因的人更有可能得病。因此,疾病的致病基因挖掘是目前生物信息学领域研究的热点问题之一。
近年来,随着诸多高通量技术的成熟与迅猛发展,产生了海量的生物数据,如基因组、转录组和表型组等。从全基因组数据范围内挖掘影响特定表型性状(如人类疾病、作物产量性状等)的基因位点变得可能,这也成为当今生命科学领域具有挑战的研究课题。全基因组关联研究(genome-wideassociationstudy,gwas)是常用的在全基因组范围内筛检出与表型显著关联的单核苷酸多态性(singlenucleotidepolymorphism,snp)的方法。这种方法在单基因病(即符合孟德尔遗传规律)方面效果良好,能够发现一些致病基因,从而揭示此类疾病的遗传机理。然而,这种方法主要侧重于检测主效基因,并不适用于不符合孟德尔遗传规律的复杂疾病的遗传机理解析。从此,研究者开始转向多基因位点研究,主要包括基因与基因之间的相互作用或基因与环境之间的相互作用对疾病的影响,进行关联分析。目前越来越多的研究表明,基因间的相互作用(即上位性)是影响人类复杂疾病很重要的原因,也被业界认为是遗传性缺失的重要原因,研究者对上位效应的研究兴趣也日益浓厚。然而由于基因-基因、基因-环境相互作用等不符合孟德尔遗传规律,给研究者也带来了很大的困难与挑战。需要研究者以往科学研究的基础上提出一些新的理论和方法,这也给机器学习和数据挖掘方法以用武之地。
技术实现要素:
本发明要解决的技术问题在于针对现有技术中的缺陷,提供一种基于贝叶斯网络推理的基因间交互关系挖掘方法。
本发明解决其技术问题所采用的技术方案是:
本发明提供一种基于贝叶斯网络推理的基因间交互关系挖掘方法,包括以下步骤:
步骤1、获取基因表达量和特定表型数据,对其进行正态化处理,利用高斯核概率密度估计量估测熵的方法,分别计算基因与基因间、基因与表型性状以及表型与表型性状间互信息;
步骤2、在计算节点间互信息和条件互信息的基础上,利用三阶段依赖分析贝叶斯网络结构学习方法,构建包含基因与表型性状节点的贝叶斯网络结构;
步骤3、在步骤2中构建的包含基因与表型性状节点的贝叶斯网络结构网络图的基础上,利用贝叶斯网络参数学习方法学习得到各个节点的条件概率,得到条件概率表;
步骤4、在步骤3所得条件概率表的基础上,利用吉布斯抽样贝叶斯网络近似推理方法计算不同个数的基因与表型性状间条件概率,根据计算结果大小得到影响特定表型性状的基因间交互关系。
进一步地,本发明的步骤2中的三阶段依赖分析贝叶斯网络结构学习方法的具体包括drafting,thickening和thinning三个步骤。
进一步地,本发明的步骤2中的drafting,thickening和thinning三个步骤具体方法为:
步骤2.1、drafting,将基因与表型性状作为网络中节点,利用高斯核概率密度估计量估测熵的方法计算任意两个节点之间的互信息,将互信息大于阈值的节点间的边添加到集合s中,然后根据互信息值的大小对s中节点对进行排序;然后对s中节点对进行循环判断,如果这两个节点之间存在开放路径,则将该节点对加入到集合r中;否则,将该节点对对应的边插入到图中,构造一个初始网络有向图;
步骤2.2、thickening,条件互信息判断,在通过步骤2.1构建初始网络结构图的基础上,对drafting阶段得到的集合r中节点对进行循环,查找能够d-分离该节点对的条件割集cutset,然后利用条件独立性测试判断该节点对是否条件独立;如果条件不独立,将两个节点用有向边相连;否则,对集合r中下一节点对进行循环判断;
步骤2.3、thinning,检查构造的图边集中的每一条边e,暂时移开e,在当前图中查找能够d-分离e连接两个节点的最小割集cutsetmin,利用条件独立性测试判断两个节点在最小割集cutsetmin条件下是否独立;如果条件独立,则删除e;否则,将e重新添加到网络图中,得到基因与表型性状节点的贝叶斯网络。
进一步地,本发明的步骤4的具体方法为:
步骤4.1、利用quantile方法对基因表达和表型数据进行n值离散化处理,将同一基因的表达和特定表型数据划分为n个区间,将这些区间表示n1,n2,n3…nm;
步骤4.2、利用吉布斯抽样贝叶斯网络近似推理方法计算任意多个基因与表型性状间条件概率;然后对这些条件概率进行求和,得到多个基因对表型性状的影响大小。
进一步地,本发明的步骤2.1中利用高斯核概率密度估计量估测熵的方法计算任意两个节点之间的互信息的方法具体为:
用p(x)表示x集合中变量x的概率,对于基因或表型性状变量x,用信息熵h(x)度量x的平均不确定性,其公式为:
计算x集合和y集合的联合熵,其公式为:
基于条件概率熵,计算变量x与y的互信息,其公式为:
mi(x,y)=h(x)+h(y)-h(x,y)
同时计算给定z条件下x与y的条件互信息,其公式为:
mi(x,y|z)=h(x,z)+h(y,z)-h(z)-h(x,y,z)
采用高斯核概率密度估计量估测熵的方法计算节点间互信息,得到:
其中,c表示变量的协方差矩阵,|c|表示矩阵c的行列式。
本发明产生的有益效果是:本发明的基于贝叶斯网络推理的基因间交互关系挖掘方法,利用三阶段依赖分析贝叶斯网络结构学习方法,利用高斯核概率密度估计量估测熵的互信息计算方法,构建包含基因与表型性状节点的贝叶斯网络结构。在构建贝叶斯网络的基础上,利用贝叶斯估计参数学习方法进行参数学习,得到节点间条件概率表。最后,利用吉布斯抽样贝叶斯网络近似推理方法计算不同个数的基因与表型性状间条件概率,根据计算的条件概率大小得到影响特定表型性状的基因间交互关系,进而得到影响特定表型性状的上位性基因位点,辅助基因功能挖掘。该可以帮助生物学研究者获得影响特定表型性状的上位性基因位点,进而辅助基因功能挖掘,以及为不同物种的复杂数量性状的遗传基础解析提供借鉴。
附图说明
下面将结合附图及实施例对本发明作进一步说明,附图中:
图1为本发明实施例的具体实施的原理示意图;
图2为本发明实施例的学习得到的贝叶斯网络条件概率表;
图3为本发明实施例的基因与表型性状网络结构图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
1、对基因表达转录数据和特定表型数据进行正态化处理,将其处理为特定区间(如[-3,3])的连续数据。
2、利用三阶段依赖分析贝叶斯网络结构学习方法,构建包含基因与表型性状节点的贝叶斯网络结构,主要包括drafting,thickening和thinning三个步骤。
(1)初始网络图为空
用p(x)表示x集合中变量x的概率,对于基因或表型性状变量x,用信息熵h(x)度量x的平均不确定性,用式(1)表示。
用式(2)计算x和y的联合熵,如下所示。
基于条件概率熵,进而利用式(3)计算变量x与y的互信息,同时利用式(4)计算给定z条件下x与y的条件互信息。较大的互信息与条件互信息表示变量间存在较强的联系,比如基因间调控关系、基因与表型性状间的影响关系。
mi(x,y)=h(x)+h(y)-h(x,y)(3)
mi(x,y|z)=h(x,z)+h(y,z)-h(z)-h(x,y,z)(4)
为了提高计算的准确性,采用高斯核概率密度估计量估测熵的方法计算节点间互信息。对于连续数据,我们可以利用式(5)计算p(xi),利用式(6)计算p(xi,yi),其中d1,d2为平滑参数,
根据式(1)和式(5),可通过式(7)计算得到h(x)。同理,根据式(2)和式(6),可通过式(8)计算得到h(x,y)。
最后,根据式(3),式(4),式(7)和式(8),计算得到mi(x)与mi(x,y),如式(9)和式(10)所示。
采用式(9)计算基因间、基因与表型间、表型性状间互信息,以及相关节点间的条件互信息。
(2)根据计算的互信息值的大小对s中节点对进行排序,然后对于s中的每一节点对<nodei,nodej>进行循环,判断节点nodei与nodej,之间是否存在开放路径。如果存在开放路径,则执行r=r∪<nodei,nodej>,将该节点对<nodei,nodej>加入到r中。否则,则执行network=network∪<nodei,nodej>,将该节点对对应的边插入到图network中。根据上述方法依次对s中节点对进行判断,构造一个初始网络有向图。
(3)对r中每一节点对<nodem,noden>进行循环,在网络图network中查找能够d-分离节点nodem与noden的条件割集cutset。然后利用式(10)计算mi(nodem,noden|cutset),如果该值大于阈值,则说明节点nodem与noden条件不独立,执行network=network∪<nodem,noden>,将该节点对对应的边插入到network中。根据上述方法依次对r中节点对进行判断,对初始网络有向图进行更新。
(4)对network中每一条边e,假设连接e的两个节点为nodei和nodej,首先从network中移除e,在network中查找能够d-分离节点nodei与nodej的最小条件割集cutsetmin。然后利用式(10)计算mi(nodei,nodej|cutsetmin),如果该值大于阈值,说明节点nodei与nodej条件不独立,则执行network=network∪<nodei,nodej>,将该节点对对应的边重新插入到network中。否则,如果该值小于阈值,则删除边e。依次类推,对网络中每一条边进行判断,进而得到最终的贝叶斯网络结构图。
3、贝叶斯网络参数学习是在已知网络结构的条件下学习每个节点的条件概率分布。在完整数据集下,参数学习主要分为最大似然估计和贝叶斯估计两种方法。鉴于贝叶斯估计方法具有可以综合利用先验知识和后验信息,可以避免主观偏见、盲目搜索和噪音影响等优点,主要采用贝叶斯估计参数学习方法计算得到不同节点的条件概率表。
设由n个基因与表型性状节点x={x1,x2,…,xn}组成的贝叶斯网络network,节点xi共有ri个取值1,2,…,ri,其中基因和表型性状节点的取值为对转录组和表型数据进行离散化处理之后的值。节点xi的父节点π(xi)共有qi个组合的取值,表示为{1,2,…,qi}。样本集记为d。dirichlet分布是一种应用最为广泛的参数先验分布,设参数θ的先验分布p(θ|s)为dirichlet分布,得到θij的先验分布,如式(11)所示。
其中
如图2所示,ac与bc表示两个不同表型性状节点,g1~g5表示基因节点,每个节点都分别有两个取值:t和f。通过参数学习可以学习得到各个节点的条件概率,如p(g1=t),p(g3=t|ac=f),p(ac=t|g1=t,g2=t),p(g5=t|bc=f)等,这些条件概率组成条件概率表。
4、贝叶斯网络推理是指给定网络结构和证据变量集合,利用联合概率分布公式,计算某一事件发生的后验概率。贝叶斯网络推理一般分为精确推理和近似推理两种方式。精确推理的复杂度高且效率比较低,对大规模网络具有不可操作性,是nphard问题。蒙特卡罗方法是最常用的贝叶斯网络近似推理方法,吉布斯抽样算法是一种常用的马尔可夫链蒙特卡罗(mcmc)近似推理方法。该方法采用马尔科夫覆盖,保证了算法返回的结果收敛于真正的后验概率。本发明主要采用该方法计算表型性状节点与具有调控关系的多个基因节点间的条件概率。
设图3为通过三阶段依赖分析贝叶斯网络结构学习方法得到的基因与表型性状节点网络结构,并已通过参数学习得到条件概率表。其中g1~g11表示基因节点,ac与bc表示不同表型性状节点。
图3中,基于参数学习得到的节点条件概率表,利用贝叶斯网络推理方法可以计算任意节点(集)间的条件概率。设m为样本数量,ac与raci分别表示查询变量(表型性状)节点及其取值,g2=r2j,g4=r4k分别表示证据变量(具有调控关系的多个基因)节点及其取值。利用吉布斯抽样贝叶斯网络近似推理算法计算p(ac=raci|g2=r2j,g4=r4k)的主要步骤如下:
(1)设mq=0,随机生成与证据变量节点g2,g4一致的样本,即满足条件g2=r2j,g4=r4k。如果该样本满足ac=raci,将mq加1。
(2)根据拓扑顺序依次对非证据变量节点进行循环,得到该节点的马尔科夫覆盖节点集,然后取马尔科夫覆盖节点在样本中的值valmb。
(3)在valmb的条件下,计算非证据变量节点发生的条件概率,并进行抽样,用抽样结果更新样本中非证据变量节点的取值。根据抽样结果判断样本是否满足ac=raci,如果满足该条件,则将mq加1。
(4)根据上述方法循环执行m次,计算mq/m,得到条件概率p(ac=raci|g2=r2j,g4=r4k)。
在根据上述步骤计算得到p(ac=raci|g2=r2j,g4=r4k)的基础上,利用式(13)计算基因g2,g4对表型性状ac的影响p(ac|g2=r2j,g4=r4k)。
通过上述贝叶斯网络近似推理方法可以计算得到具有调控关系的多个基因与表型性状间的条件概率,如p(ac|g5,g10)、p(ac|g2,g4,g8)、p(bc|g3,g5,g6)等,进而根据计算得到的条件概率大小,灵活高效的挖掘特定表型性状相关的基因集合。
应当理解的是,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,而所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!