法尼酯X受体和可溶性环氧化物水解酶双重调节剂的制作方法
本发明涉及一种新型法尼酯x受体(fxr)和可溶性环氧化物水解酶(seh)双重调节剂。本发明的调节剂旨在提供具有fxr激动剂和seh抑制剂(拮抗剂)双重活性的化合物。本发明还提供了治疗患有fxr和seh有关疾病(如代谢紊乱,特别是非酒精性脂肪肝或非酒精性脂肪性肝炎(nash))的受试者的治疗方法。
背景技术:
营养过剩和久坐不动的生活方式所导致的酒精性脂肪性肝病(nafld)对成年人,特别是在西方国家甚至全世界范围内,的影响越来越大。最近的研究评估认为,世界范围内有三分之一的成年人患有nafld,并且高达15%的成年人发展成为非酒精性脂肪肝(nash)。最令人担忧的是,研究还认为高达11%的青少年受到nafld的影响。nafld和nash疾病综合征——通常被认为是代谢综合征的肝脏表现,因此成为了对健康的严重威胁。然而,目前nafld的治疗十分有限,药理学选择也不充分。因此,迫切需要新型药物干预来治疗nafld和nash(rinella,m.e.“非酒精性脂肪性肝病:系统综述”.《美国医学会杂志(jama)》2015,313(22),2263-2273;gawrieh,s.;chalasani,n.“非酒精性脂肪性肝病的药物治疗”.semin.liverdis.2015,35(3),338-348;chalasani,n.;younossi,z.;lavine,j.e.;diehl,a.m.;brunt,e.m.;cusi,k.;charlton,m.;sanyal,a.j.“非酒精性脂肪性肝病的诊断和治疗:美国肝病研究学会、美国胃肠病学院及美国胃肠病学会实践指南”.《肝脏病学(hepatology)》2012,55(6),2005-2023)。
体内模型和临床试验已经确定了几种nafld/nash的潜在治疗策略。尤其是所述核法尼酯x受体(fxr)(arab,j.p.;karpen,s.j.;dawson,p.a.;arrese,m.;trauner,m.“胆汁酸与非酒精性脂肪性肝病:分子视角和治疗视角”.《肝脏病学》2017,65(1),350-362)似乎是用于nafld/nash治疗的非常有前途的靶标,因为已经证实了由内源性fxr激动剂鹅脱氧胆酸(cdca,1b)开发的奥贝胆酸(6α-乙基-鹅脱氧胆酸、oca,1a)在nash治疗中的临床疗效(adorini,l.等人“法尼酯x受体靶向治疗非酒精性脂肪性肝炎”.《今日药物发现(drugdiscov.today)》2012,17(17-18),988-997;neuschwander-tetri,b.等人“法尼酯x核受体配体奥贝胆酸治疗非肝硬化、非酒精性脂肪性肝炎(flint):一项多中心、随机性、安慰剂对照试验”.《柳叶刀(lancet)》2014,385(9972),956-965)。fxr是配体激活的转录因子,主要存在于肝脏、肠和肾脏中,在生理上被胆汁酸激活(makishima,等人“胆汁酸核受体的鉴定”.《科学(science)》1999,284(5418),1362-1365;parks,d.j.等人“胆汁酸:孤儿核受体的天然配体”.《科学》1999,284(5418),1365-1368;wang,h.等人“内源性胆汁酸是核受体fxr/bar的配体”.《分子细胞(mol.cell)》1999,3(5),543-553)。fxr通过控制几种胆汁酸、脂质和葡萄糖稳态相关基因起到代谢调节的作用,并通过感知累积胆汁酸的毒性水平起到重要的肝脏保护作用(gadaleta,r.m等人“fxr在代谢和癌症中的组织特异性作用”.《生物化学与生物物理学报(biochim.biophys.acta)》2014,1851,30-39;pellicciari,r.等人“法尼酯x受体:从结构到潜在的临床应用”.《药物化学杂志(jmedchem)》2005,48(17),5383-5403)。
临床试验报告了nafld/nash的组织学特性和临床标志物的改善和oca(1a)治疗后代谢参数的改善,这证实了fxr是治疗脂肪性肝紊乱的靶点和其他代谢疾病的潜在靶点(mudaliar,s等人“法尼酯x受体激动剂奥贝胆酸在治疗2型糖尿病和非酒精性脂肪性肝病患者方面的疗效和安全性”.《肠胃病学(gastroenterology)》2013,145(3),574-582.e1)。然而,由于fxr激活可能导致抑制胆固醇7α-羟化酶(cyp7a1),而cyp7a1是胆固醇代谢转化为胆汁酸的限速酶,因此fxr的完全激活可能导致胆固醇的不良累积。fxr部分激动作用低于典型的fxr激动剂的最大功效,例如1c似乎是在不干扰胆固醇稳态的前提下发挥fxr激活的理想效果的一种潜在方法(merk,d等人“邻氨基苯甲酸衍生物作为法尼酯x受体(fxr)的新型配体”.《生物有机化学与医药化学通讯(bioorg.med.chem.2014,22(8),2447-2460)》;merk,等人“邻氨基苯甲酸衍生物作为法尼酯x受体调节剂的构效关系扩展:一种高效法尼酯x受体部分激动剂的研发”.《药物化学杂志(j.med.chem.)》2014,57(19),8035-8055;merk,d.等人“法尼酯x受体配体的药物化学:从激动剂和拮抗剂到调节剂”.《未来医药化学(futuremed.chem.)》2012,4(8),1015-1036)。
所述可溶性环氧化物水解酶(seh)是花生四烯酸代谢的cyp途径的下游酶,在nafld/nash和其他代谢疾病(如2型糖尿病)的治疗中也具有潜在功效(shen,h.c;hammock,b.d.“可溶性环氧化物水解酶的发现:一个具有多种潜在治疗指征的靶点。《药物化学杂志》2012,55(5),1789-1808;newman,j.w.等人“环氧化物水解酶:他们的作用和与脂代谢的相互作用”.《脂类研究进展(prog.lipidres.)》2005,44(1),1-51;imig,j.d.“心血管生理学中的环氧化物和可溶性环氧化物水解酶”.《生理学评论(physiol.rev.)》2012,92(1),101-130;huang,h.;weng,j.;wang,m.h.“eets/seh在糖尿病和肥胖引起的心血管疾病中的作用”.《前列腺素和其他脂质介(prostaglandinsotherlipidmediat.)》2016,125,80-89.)。seh将由cyp酶形成的环氧二十碳三烯酸(eets)从花生四烯酸转化为相应的二羟二十碳三烯酸(dhets)。由于eets具有强大的抗炎活性,因此seh抑制是一种抗炎策略。seh在全身都有表达,尤其是在心脏、肝脏和肾脏具有高表达。小鼠nash模型的最新结果证实,seh基因敲除或抑制减少了高脂饮食下的肝脏脂肪堆积并改善了肝脏炎症。
nash与2型糖尿病或肥胖等多种危险因素有关,并且具脂肪变性、肝脏炎症和纤维化等多种表现形式。因此,一些实验研究已经揭示了其对nash的治疗效果(sanyal,a.j.“脂肪性肝炎的新型治疗靶点”.《肝脏病学和胃肠病学的临床和研究(clin.res.hepatol.gastroenterol.)》2015,39增刊1,s46-50;milic,s,等人“非酒精性脂肪性肝炎:优化治疗方案的新兴靶向疗法”.《药物设计开发和治疗(drugdes.devel.ther.)》2015,9,4835-4845.),对于这种多因素疾病,似乎更应该用多于一种治疗机制针对不同的病理因素进行治疗。然而,使用多种药物的复方用药是不利的,例如来自复杂且不清楚的药物-药物相互作用和额外的副作用的危害。能够解决多种治疗机制需要的多靶点药物能够避免多重药理学的诸多弊端。
技术实现要素:
鉴于fxr激活在减缓nash和脂质积累进展方面的临床效果以及seh抑制在肝脏抗炎和抗脂肪变性方面的作用,fxr和seh的双重调节似乎是一种可能产生协同效应的治疗nafld/nash的有效方法。因此,本发明旨在提供对fxr具有部分激动活性并对seh具有抑制效力的双重调节剂。
上述问题由具有化学式i的化合物解决:
其中,r1、r2、r3以及r4各自独立地选自h,非取代的、单取代的或多取代的c1-c18烷基或杂环烷基,其中所述烷基为直链、支链或环状,非取代的、单取代的或多取代的c1-c18烯基或杂环烯基,其中所述烯基为直链、支链或环状,非取代的、单取代的或多取代的芳基和杂芳基,非取代的、单取代的或多取代的苄基,酰基,如甲酰基、乙酰基、三氯乙酰基、反丁烯二酰基、顺丁烯二酰基、琥珀酰基、苯甲酰基或支链、杂原子取代的或芳基取代的酰基,糖或其他缩醛基以及磺酰基,和/或其中r2、r3和/或r4一起形成非取代的、单取代的或多取代的环,优选芳香环,
z是含有或不含任何取代的c,优选地用h或烷基取代;
或其异构体、外消旋体、前体药或衍生物或这些化合物药学上可接受的盐或溶剂化物。
在一些优选的实施例中,r2是c1-c10烷基,优选支链烷基,更优选-c(ch3)3基团,r3优选h、-oh或ome,r4优选h、-oh或-ome。
在另一优选实施例中,所述化合物是上述定义的化合物组,不包括本文公开的化合物4a、4b、6、7、9、14、16、19、29、33、36、42和45。在这一方面,进一步优选的是本申请的实例部分的表1到8中公开的所述其它化合物中的任意一种(不包括上述化合物)。
在一个优选实施例中,上述化合物中的r3和r4是h。
在一些实施例中,r1是具有化学式ii的基团:
其中,r5、r6和r7各自独立地选自h,非取代的、单取代的或多取代的c1-c18烷基或杂环烷基,其中所述烷基为直链、支链或环状,非取代的、单取代的或多取代的c1-c18烯基或杂环烯基,其中所述烯基为直链、支链或环状,非取代的、单取代的或多取代的芳基和杂芳基,非取代的、单取代的或多取代的苄基,酰基,如甲酰基、乙酰基、三氯乙酰基、反丁烯二酰基、顺丁烯二酰基、琥珀酰基、苯甲酰基或支链、杂原子取代或芳基取代的酰基,糖或其他缩醛基以及未取代的、单取代的或多取代的酰胺或磺酰基。
优选地,r6和r7是h或卤素,优选地,卤素选自f或cl。最优选地,r6是h,r7是h、f或cl。
优选地,r5是任何长度的支链,其包含羧酸或适当的羧酸替代物,例如典型的生物电子等排体,其包括但不限于四唑、磺胺、酰胺(例如有机酰胺、磺胺或磷酰胺)等。
本文所使用的术语“生物电子等排体”涉及一种化学成分,该成分取代活性化合物分子中的另一部分且对其生物活性没有显著影响。活性化合物的其他特性,例如其作为药物的稳定性,会受此影响。
由于羧基(cooh)的生物电子等排体部分值得注意,特别是具有从氮、氧和硫中选择的1至4个杂原子的5元杂环基团,例如1,3,4-恶二唑基、1,2,3-恶二唑基、1,2,5-恶二唑基、1,2,4-恶二唑基、1,3,4-噻二唑基、1,2,4-噻二唑基、1,2,3-噻二唑基、1,2,5-噻二唑基、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、异恶唑基、异噻唑基以及n-取代四唑基。5元杂环基团可以任选地被1或2个取代基取代,所述取代基选自包括以下的群组:苯基、吡啶基、直链或支链烷基、氨基、羟基、氟、氯、溴、碘、三氟甲基、三氟甲氧基、三氟硫代甲氧基、烷氧基和硫代烷氧基。
由于羧基(cooh)的生物电子等排体部分也是值得注意,特别是具有从氮、氧和硫中选择的1至4个杂原子的6元杂环基团,例如吡啶基、吡嗪基、哒嗪基、嘧啶基、三嗪基、四嗪基等。苯基和6元杂环基团可以任选地被1或2个取代基取代,所述取代基选自包括以下的群组:苯基、吡啶基、直链或支链烷基、氨基、羟基、氟、氯、溴、碘、三氟甲基、三氟甲氧基、三氟硫代甲氧基、烷氧基和硫代烷氧基。
在本发明的一些实施例中,r1选自以下取代基中的任一个:
最优选地,本发明的化合物具有上述化学式i,其中r1选自由以下组成的群组:
其中,z是c,r2是-c(ch3)3,r4、r3是h。
在另一个实施例中,进一步优选地是具有化学式i的上述化合物,其中r1选自由以下组成的群组:
其中z是c,r3是h或oh,r4是h或oh,特别地r3和r4都不是oh;并且其中r2选自-c(ch3)3、-n(ch3)2,或所述r2是以下中的任一个
根据权利要求1至5中任一项的化合物,该化合物为法尼酯x受体(fxr)激动剂和可溶性环氧化物水解酶(seh)抑制剂。
具有药学上不可接受的阴离子的盐同样构成本发明要求范围的一部分,该盐可作为制备或纯化药学上可接受的盐的有利中间体和/或用于非治疗性应用(如体外应用)。本发明所述化合物也可以以各种多态形式存在,例如不规则和晶体多态形式。本发明化合物的所有多态形式都在本发明要求的范围内,并且属于本发明的另一方面。
如本文所使用,术语“法尼酯x受体”或“fxr”是指该类受体的所有哺乳动物形式,包括例如选择性剪接的亚型和天然存在的亚型(参见例如r.m.huber等人,《基因(gene)》2002,290,35)。典型的fxr类包括但不限于所述受体的大鼠(genbank登录号nm_21745)、小鼠(genbank登录号nm_09108)以及人类(genbank登录号no.nm_05123)形式。
术语“可溶性环氧化物水解酶(seh)”是指存在于人类体内、由ephx2基因(hgnc:3402)编码的双功能酶。seh是环氧化物水解酶家族的成员。该酶存在于细胞溶质和过氧化物酶体,与特定的环氧化物结合并将其转化为相应的二醇。
在另一方面,本发明还提供了一种合成本文所公开的新型化合物的方法。
本发明的化合物在治疗受试者疾病的方法中特别有效。优选地,根据本发明,需要治疗的疾病是与fxr和seh有关的紊乱。
因此,本发明还提供了一种同时调节fxr和seh的方法,所述方法包括向受试者或细胞施用如前所述的fxr和seh双向调节剂的步骤。
另一方面,涉及治疗受试者疾病的方法,所述方法包括向受试者施用治疗有效量的本发明的化合物或本发明的药物组合物的步骤。
在本发明公开的上下文中,术语“受试者”优选地指哺乳动物,优选地是小鼠、大鼠、驴、马、猫、狗、豚鼠、猴子、猿,或者优选地指人类患者,例如患有本文所述紊乱的患者。
在优选的实施例中,所述疾病或紊乱是代谢紊乱,优选地是由高脂肪饮食引起的或与高脂肪饮食相关的代谢紊乱。
肝病是一种肝脏部位的典型损伤或疾病。肝病的种类超过一百种。最广泛传播有:肝片吸虫病;肝炎;酒精性肝病;脂肪性肝病;肝硬化;肝脏;胆汁;硬化性胆管炎;小叶中心坏死;布加综合征;遗传性肝病(血色素沉着症,包括体内铁的积聚和威尔逊病);甲状腺素运载蛋白相关遗传性淀粉样变性以及gilbert综合征。
如本文所用,术语“肝病”是指影响肝脏的任何疾病或紊乱。肝病的实例包括但不限于alagille综合征;酒精性肝病;α-1抗胰蛋白酶缺乏症;自身免疫性肝炎;良性肝肿瘤;胆道闭锁;肝硬化;半乳糖血症;gilbert综合征;血色素沉着症;甲型肝炎;乙型肝炎;丙型肝炎;肝细胞癌;肝性脑病;肝囊肿;肝癌;新生儿黄疸;非酒精性脂肪性肝病(包括非酒精性脂肪肝和非酒精性脂肪性肝炎);原发性胆汁性肝硬化(pbc);原发性硬化性胆管炎(psc);reye综合征;i型糖原贮积症和威尔逊病。
术语“非酒精性脂肪肝”或“非酒精性脂肪性肝病”(nafld)是指导致脂肪肝的一种疾病,由脂肪沉积于肝脏而非过度饮酒导致。nafld与胰岛素抗性和代谢综合征有关,并且可能对最初针对其他胰岛素抗性状态(例如2型糖尿病)开发的治疗方法有反应,如减重、二甲双胍和噻唑烷二酮类药。nafld可以进一步分为非酒精性脂肪性肝炎(nash)和非酒精性脂肪肝(nafl)。nash是nafld的最极端形式,并且被认为是不明原因的肝硬化的主要病因。
大多数nafld患者只有一些症状或没有症状。患者可能会诉疲劳、莫名不适和右上腹部不适。可能会出现轻度黄疸,虽然这比较罕见。更常见的nafld是在血常规检查中出现肝功能异常后诊断出来的。nafld与胰岛素抗性和代谢综合征(肥胖、混合型高脂血症,ii型糖尿病以及高血压)有关。常见的症状是肝酶升高和肝超声显示脂肪变性。超声也可以用来排除胆结石问题(胆石症)。肝活检(组织检查)是唯一被广泛接受的、明确区分nash和其他形式肝疾病的检查方式,可以用于评估炎症的严重程度和由此引起的纤维化。
非酒精性脂肪性肝炎(nash)是一种常见的、“沉默的”(通常情况下)肝病。nash的主要特征是肝脏脂肪以及炎症和损伤。大多数患有nash的人感觉良好,并没有意识到他们患有肝脏问题。然而,nash是很严重的,并且可能导致肝硬化,肝脏可能受到永久性的损害和创伤并且不能再正常工作。
当在一个人的血常规检测中发现肝脏检查结果(如丙氨酸氨基转移酶(alt)或天冬氨酸氨基转移酶(ast))升高时,通常首先怀疑nash。当进一步的评估显示没有明显的肝病原因且肝部x光或影像学检查显示肥胖时,即怀疑nash。唯一能明确诊断nash并将其与单纯性脂肪肝区分的方法是肝脏活检。如果活检中发现肥胖伴随炎症或损伤,则诊断为nash。如果组织显示肥胖但没有炎症或损伤,则诊断为nafl或nafld。目前,没有血液检查或扫描能够可靠地提供这一信息。
因此,本发明的化合物治疗的优选疾病是肝病,如非酒精性脂肪性肝病或非酒精性脂肪性肝炎(nash)。
如本文所用,术语“治疗有效量”是指能引起组织、系统、动物或人类的生物或医学反应的化合物的量,所述组织、系统、动物或人类由研究人员或临床医生寻找。此外,术语“治疗有效量”是指与未接受该量的相应受试者相比,能够改善治疗、治愈、预防或改善疾病、紊乱或副作用或减缓疾病或紊乱进展速度的量。
在另一方面,还提供了一种药物组合物,其包含本发明的化合物和药学上可接受的载体和/或赋形剂。
本发明的化合物还能与其他活性成分结合施用。为达到预期的生物学效果,具有化学式i的化合物的量取决于诸多因素,例如所选择的特定化合物、预期用途、使用方式以及患者的临床条件。
每日剂量范围一般为0.3mg至100mg/天/公斤体重(通常为3mg至50mg/天/公斤体重),例如3至10mg/kg/天。例如,静脉注射剂量范围可以为0.3mg至1.0mg/kg,其可以适当地以10ng至100ng/公斤体重/分钟以输液方式给药。用于这些目的的恰当输注溶液可以含有,例如0.1ng至100mg,通常为1ng至100mg/毫升。单剂量可以含有,例如1mg至10g活性成分。因此,注射用安瓿可以含有,例如1mg至100mg,口服单剂量制剂(如片剂或胶囊)可以含有,例如1.0至1000mg,通常为10至600mg。对于上述条件的处理,可以将具有化学式i的化合物本身用作所述化合物,但是它们优选地以药物组合物的形式与兼容载体一起存在。当然,所述载体必须是可接受的,即它与所述化合物的其他成分相容,并且对患者的健康无害。所述载体可以是固体或液体或两者都有,并且优选地将所述化合物以单一剂量(例如片剂)配制,其可能含有0.05%至95%的活性成分重量。其他药学的活性物质也可以存在,包括具有化学式i的其他化合物。本发明的药物组合物可以由已知的药学方法中的一种生产,其基本上涉及将成分与药学上可接受的载体和/或赋形剂相混合。
本发明的药物组合物为适用于口服给药、直肠给药、局部给药、经口给药(如舌下给药)以及肠外给药(如皮下给药、肌肉给药、皮内给药或静脉给药),最合适的给药方式取决于每种个体情况下待治疗疾病的性质和严重程度和每种情况下使用的具有化学式i的化合物的性质。包衣制剂或药物形式也在本发明要求的范围之内。糖衣制剂和糖衣缓释制剂也在本发明的保护范围之内。优先选择耐酸和耐胃液的制剂。适宜的耐胃液包衣包括醋酸邻苯二甲酸纤维素、聚醋酸乙烯酞酸酯、羟丙基甲基纤维素邻苯二甲酸酯以及甲基丙烯酸和甲基丙烯酸甲酯的阴离子聚合物。适于口服给药的药物化合物可以是单独的形式,即单剂量、单元,例如胶囊、药膏、含片、薄膜衣片、糖衣片、可溶性片、舌下片、口服片或片剂,每种形式都含有一定量的具有化合式i的化合物;可以作为粉末或颗粒;作为水或非水溶液或悬液;或作为水包油或油包水乳液。如前所述,这些组合物可能通过任何适当的药学方法进行制备,该方法包括使活性成分和载体(其可以由一个或多个附加成分组成)进行接触的步骤。所述组合物通常是由所述活性成分与液体和/或细碎固体载体均匀且一致地混匀而形成,然后在必要时形成产品。例如,片剂可以通过对化合物的粉末或颗粒进行压缩或定型而形成,在适当情况下添加一种或多种额外成分。压缩片剂可以通过将化合物(如粉末或颗粒)以自由流动的形式进行压片来生产,在适当的情况下在合适的机器中与粘合剂、油膏、惰性稀释剂(填料)和/或一种(或多种)表面活性剂/分散剂进行混合。模制片剂或颗粒可以在适当的机器中对用惰性液体稀释剂润湿的所述粉状化合物进行模压来生产。
适用于经口(舌下)给药的药物组合物包括含片,该含片包括具有化学式i的化合物和调味料(通常为蔗糖、阿拉伯树胶或黄芪胶),以及在惰性基中含有所述化合物的软锭剂,例如明胶和甘油或蔗糖和阿拉伯树胶。
适用于肠外给药的药物组合物优选地包括具有化学式i的化合物的无菌水溶液制剂,其优选地与所述预期受试者的血液等渗。这些制剂优选地经静脉给药,尽管也可以通过皮下、肌肉或皮内注射给药。这些制剂可以优选地通过以下步骤制备:将化合物与水混合,然后将所得溶液通过适当的灭菌过程(例如蒸气灭菌、无菌过滤)进行灭菌,然后与血液等渗。本发明的可注射组合物通常含有0.1至5重量%的活性化合物。适用于直肠给药的药物组合物优选地为单剂量栓剂的形式。这些可以通过将具有化学式i的化合物与一种或多种常规固体载体(例如可可脂)混合并将所得混合物成型来制备。
适用于皮肤上局部使用的药物组合物优选地为软膏、霜、散剂、乳液、糊剂、喷剂、气雾或油的形式。可使用的载体有矿脂、羊毛脂、聚乙二醇、醇以及两种或多种前述物质的混合物。所述活性成分通常以0.1至15重量%组合物的浓度存在,例如0.5至2重量%。
经皮给药也是可能的。适用于经皮使用的药物组合物可以是适合于长期密切接触患者表皮的单个贴片的形式。这些贴片适当地含有水溶液中的活性成分,该水溶液在适当情况下被缓冲、溶解和/或分散在粘合剂中或分散在聚合物中。适宜的活性成分浓度为约1%至35%,优选地为3%至15%。一种特殊的选择是,通过电转运或离子电渗疗法对活性成分进行释放,例如《药学研究(pharmaceuticalresearch)》,2(6):318(1986)所述。
附图说明
现在将参照附图和序列在以下实例中对本发明进行进一步说明,但是并不限于此。为了本发明的目的,本文引用的所有参考文献通过引入并入其实体中。图中:
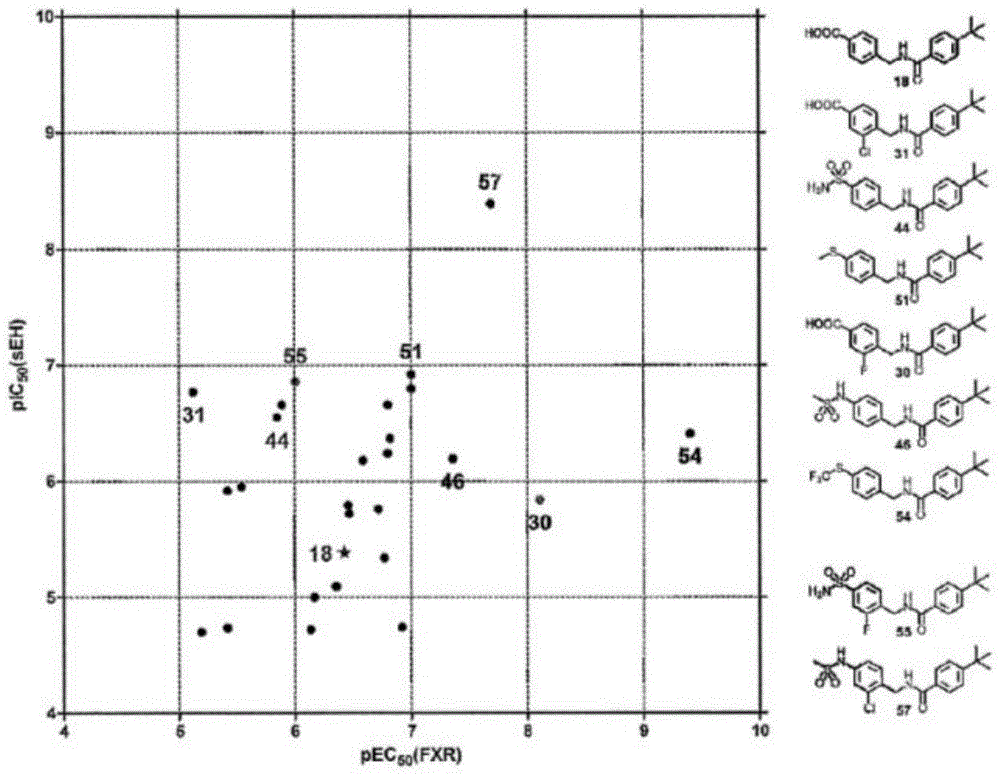
图1双重调节剂的效力图表明30、46和54(红色)是最有效的fxr部分激动剂,而31、44和51(蓝色)出现了最高的seh抑制效力。因此,将这些化合物的结构特征结合起来以提高双重活性,从而产生了55和57(绿色,表7)。
图2:57的分子对接:(a)化合物57在fxr-lbd(pdb代码4qe8)中的结合模式。(b)化合物57在seh(pdb代码3i28)中的结合模式。分子表面的颜色是亲油性的(绿色:亲油的;紫红色:亲水的)。选定的侧链显示为线,化合物57显示为棒。
图3:57的体外表征:(a)选择性概括:除对ppary的适当活性外(ec50=14·7±0.9μμ,30±1%max.),57在相关的核受体上无活性,因此fxr具有高度选择性(数值是平均值±sem;n=3)。(b)在常规的wst-1试验中,57在hepg2细胞高达100μμ时无细胞毒性(数值是平均值±sem;n=4)。(c)与wistar大鼠肝微粒体孵育60分钟后,57显示出可接受的体外微粒体稳定性,剩余仍超过50%(数值是平均值±sem;n=3)。
图4:57的细胞活性测定:(a)与cdca(50μμ,1b)相比,57(0.1μμ和1μm)与hepg2细胞孵育8或16小时后的fxr靶基因mrna定量:57引起bsep、shp、pparα、glut4、fgf19、pdk4和fabp1非浓度依赖的部分诱导,以及cyp7a1、srebp1c和fas非浓度依赖的部分抑制(载体处理的对照细胞定义为100%,数值为平均值±sem;n=4)。(b)57不引起ppary靶基因cd36的显著调节(数值为平均值±sem;n=3),其中fam3a(吡咯列酮,pio)作为阳性对照。(c)hepg2细胞匀浆中的可溶性环氧化物水解酶活性:双重调节剂57通过胞内seh(ic50约为10nm)抑制14.15-eet-d11向14.15-dhet-d11的转化并且在低至0.1nm的浓度下即有显著的统计学抑制作用(数值为平均值±sem;n=3)。(*p<0.05;**p<0.01;***p<0.00)
图5:57的体内特征:(a)57的体内药代动力学评估显示,双重调节剂吸收迅速、生物利用度高且半衰期适度、可接受。在单次口服10mg/kg体重剂量后,57的有效血浆浓度维持在ic50值(seh)和ec50值(fxr)以上约3.5小时。(b)给药8小时后,小鼠血浆的eet/dhet比值提高约2倍,表明57在体内抑制了seh。(c)与载体处理的小鼠(100%)相比,给药8小时后,fxr靶基因bsep、shp、cyp7a1、srebp1c和fgf15和pparγ靶基因fatp的mrna水平定量。57表现出诱导bsep和shp的趋势,并且轻度抑制了cyp7a1。此外,57显著抑制了srebp1c并显著诱导了fgf15,表明fxr的体内调节效果和在nash中的潜在作用。ppary靶基因fatp在体内不受调节。(n(载体)=3;n(57)=6;*p<0.05;**p<0.01;***p<0.001)。
图6:(或方案10)57的体外代谢。双重调节剂57的磺酰胺部分水解产生代谢物69a(由lc-ms-ms确认),该代谢产物显示出高效的双重调节效力并且有助于促进药学活性。lc-ms-ms分析还表明存在叔丁基苯甲酰胺残基羟基化产生的代谢产物,在三种可能的异构体77、78和79中,77和78均未检测到,从而确定了叔丁基的羟基化导致了79。此外,57在苄基取代基的芳香环上被羟基化。
图7:(或方案1)展示了重要的fxr配体:奥贝胆酸(1a)、生理激动剂cdca(1b)以及合成的参考fxr激动剂(1c)。
图8:(或方案2)展示了将seh抑制剂gsk2188931b(2)和部分fxr激动剂3的药效团合并制成先导化合物5的发现过程。
具体实施方式
实例
为了开发对fxr和seh具有强效双重活性的试剂,发明人最初筛选了fxr调节剂内部库的代表性化合物,但未能从中确定对seh具有抑制效力的先导化合物。因此,发明人从已知的部分fxr激动剂和已知的seh抑制剂中寻找合并的药效团。一些seh抑制剂含有酰胺或尿素残基,模拟酶裂解的环氧化物部分。包括作为药效团存在的n-苄基酰胺残基的seh抑制剂gsk2188931b(2)与最近报道的部分fxr激动剂(例如3)具有一些结构相似性。因此,发明人提取了这两种化合物的相似特征结构并把它们结合在最小双效药效团4a中,该药效团含有用于seh抑制的n-苄基酰胺残基和用于fxr激活的羧酸基(图8,方案2)。化合物4a在50μμ浓度时的seh抑制作用适中(37±1%),但是对fxr无激活作用。结合模板2的哌啶部分的4b展示出更低的活性并且对seh和fxr无活性。然而,当发明人用5中的芳香族部分替代4a和4b中的饱和环时,发明人在50μμ浓度条件下达到了预期的双重活性(12±1%的fxr激活作用和16±2%的seh抑制作用)。对于5的低片段样大小(mw255da),发明人认为该中等效力足够并且系统地分析了5作为seh和fxr双重调节剂的结构活性关系(sar)(图2)。
实例1:合成
按照方案3-9制备了n-苄基苯甲酰胺4-57和77-78。氨基甲苯前体58a-j的合成,首先用nbs和aibn与各自的甲苯衍生物59a-j进行自由基溴化反应,生成溴甲基苯60a-j。然后将溴甲基苯60a-j应用于叠氮化钠两步staudinger反应,在水溶液中生成叠氮化物61a-j和三苯基膦进行还原。用liaih44还原4-氨基-2-氯苯甲腈62,制备氨基甲苯衍生物58k。氨基甲苯衍生物58i-w可购买。然后,将58a-w与羰基氯化物63a-o在吡啶存在下进行反应,或者将58a-w与羧酸64a-f在edc及4-dmap存在下进行反应,得到化合物18、19、22、35、36、44、47-51、54、55、68及69a-c或其酯65a-h(方案3)。化合物68经brch2cooch3处理生成酯65i。所有的酯类65a-i在碱性条件下水解为终产物16、20及23-32(方案4)。尿素21由4-氨基苯甲酸(66)和4-叔丁基苯基异氰酸酯(67)与net3反应制备,然后由氢氧化锂水解(方案5)。游离的羧酸18和氯化铵、甲基氯化铵或二甲基氯化铵以及edc/dmap用于制备酰胺37-39。lialh4还原18得到乙醇衍生物33、33进一步通过pcc转化成醛34(方案6)。转化的酰胺40/41和56和转化的磺胺46和57以及n-酰基磺酰胺45经过edc/dmap由42、44和69a进行羧酸活化生成。在cu2o的催化下,通过nan3的环加成从腈36中得到四唑43(方案7)。用合适当量的间氯过氧苯甲酸(mcpba,方案8)将甲硫醇51氧化为亚砜52和砜53,并最终按照所述标准规程制备甲氧基衍生物76a-b(方案3),然后与bbr3脱甲基化生成酚衍生物77和78(方案9)。
方案3:试剂和条件(a)nbs、aibn、chcl3、回流4h;(b)nan3、dmf、80℃、16h;(c)pph3、h2o、thf、室温(rt)、12h;(d)lialh4、thf、回流、18h;(e)吡啶、thf、rt、2h;(f)edc-hcl、dmap、chcl3、50℃、6h;(g)socl2、dcm、dmf、回流、4h。
方案4:试剂和条件(a)brch2cooch3、dmf、k2co3、rt、18h;(b)koh、h2o、meoh、μw、15min。
方案5:试剂和条件(a)dcm、net3、rt、24h;(b)lioh、thf、meoh、h2o、rt、15h。
方案6:试剂和条件(a)lialh4、thf、rt、18h;(b)pcc、dcm、rt、2h;(c)r2nh2·cl、edc·hcl、dmap、chcl3、50℃、4h。
方案7:试剂和条件(a)r-oh、edc-hcl、dmap、chcl3、50℃、4h或甲磺酰氯、thf、rt、2h;(b)cu2o、nan3、dmf、meoh、90℃、24h;(c)bbr3、dcm、0℃至rt、2h。
方案8:试剂和条件(a)mcpba、chcl3、0℃、2h或rt、18h。
方案9:试剂和条件(a)甲磺酰氯、thf、rt、2h;(b)bbr3、dcm、0℃至rt、2h。
实例2:生物学评估
为了测定fxr激动剂活性,在hela细胞中用全长(fl)fxr受体基因试验对受试化合物4-57进行表征。本试验基于含有虫荧光素酶的报告基因,受胆盐输出蛋白(bsep)中的fxr反应原件调控。将受cmv启动子调控的fxr和其异源二聚体伴侣类视黄醇x受体(rxr)表达载体及用于标准化和毒性控制的结构性表达肾荧光素酶(sv40启动子)共转染。合成的fxr激动剂gw4064(1c)作为参比激动剂,其3μm浓度下的转录激活活性定义为100%激活。该试验由多种已知的fxr激动剂进行验证,该激动剂效力与文献相符(gw4064(1c):ec50=0.51±0.16μμ,oca(1a):ec50=0.16±o.02μμ,cdca(1b):ec50=18±1μμ)。在10μμ浓度时,本试验中,特征良好的seh抑制剂ciu36没有活性,排除了seh抑制剂的非特异性。使用重组酶和荧光性seh底物phome37(由seh水解为荧光性6-甲氧基萘醛),采用荧光方法测定了受试化合物的seh抑制效力。
实例3:结构活性关系与结构优化
作为sar研究(表1)的第一个步骤,发明人在5的苯甲酰胺部分结构中的每个位置引入了额外的甲基(6-8)。虽然所有甲基化衍生物6-8都表现出微弱的seh抑制剂活性,但fxr只耐受8中的4-甲基,这表明fxr结合袋提供了额外的空间,尤其是在这一方向上。3-甲基(7)在甲基化衍生物中产生最高的seh抑制性,因此,发明人制备了包含两个甲基取代基的化合物(9、10),以结合两个标靶的改进。2,4-二甲基衍生物9几乎无活性,而3,4-二甲基衍生物10虽然对fxr的最大相对活性较低,但具有改进的双重活性。
由于额外的甲基基团没有显著地提高任何一个标靶的效力,发明人还研究了在苯甲酰胺部分上引入较大残基,并对联苯衍生物11-13进行了表征。三种联苯11-13的效力均显著高于相应的甲基苯甲酰胺6-8,而效力的等级次序保持不变。fxr耐受3-取代(12),但偏爱4-取代(13),seh同样受3-(12)和4-联苯衍生物(13)的抑制。
表1:4-13对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
初步sar结果表明,两种靶标均耐受5中本甲酰胺残基的3-和4-取代,最终改善了双重效力,因此,发明人研究了各种3,4-双取代(14-16)和2-萘衍生物17(表2)。然而,14-17未能显著改善对fxr和seh的双重效力。3,4-二氯(14)和3,4-双(三氟甲基)取代(16)只能被seh耐受,2-萘衍生物17对fxr的活性低于10。只有3,4-二甲氧基苯甲酰胺15是有利的,因为其提高了对fxr的最大相对活性而不影响ec50值,并且微弱地抑制了seh活性。
表2:14-17对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
目前为止,4-联苯衍生物13显示出最高的双重效力,表明两种靶标均耐受苯甲酰胺残基处的大体积的4-取代基(表3)。在18中引入4-叔丁基部分可进一步提高双重活性,而更具极性的4-二甲氨基苯甲酰胺19作为叔丁基模拟物的活性则大大降低。在20中,有利的大体积18的叔丁基残基和15的3-甲氧基的组合未产生额外的效应,因此,发明人选择了4-叔丁基衍生物18进行进一步优化。
表3:18-20对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
以18为新导向,发明人随后致力于优化n-苄基取代基(表4)。在21中,对于用二苯脲交换18的n-苄基苯甲酰胺结构作为经典的seh药效基团,两种靶标对其的耐受性均较差。类似地,将羧酸部分从18的4位转移到22的3位导致效力显著下降。苯甲酸(18)到苯乙酸(23)侧链长度的变化降低了两个靶标的效力,而苯丙酸24的活性与18相同。正如先前的研究所观察到的那样,该sar可能最终水介导的18的相互作用和由24介导的水置换得到解释。15苯氧乙酸25对fxr的活性类似于苯丙酸24,但seh对乙醚残基的耐受性差,乙醚残基显著降低了抑制效力,约10倍。
表4:21-25对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
由于改变分子几何结构和药效特征间的距离没有显著提升双重效力,发明人随后探索了在18的苯甲酸芳香环处引入额外取代基的可能性(表5)。2位(26-28)的取代基显著提高了2-氯衍生物28作为高效部分fxr激动剂对fxr的效力。但同时,seh的抑制活性显著下降。相比之下,3-取代(29-31)随着取代基大小的增加有利于3-氯苯甲酸31作为高效seh抑制剂对seh的抑制效力。在fxr上,3-甲基取代(29)活性完全消失,而3-氯衍生物31仍具有活性,但效力大大较低。3-氟苯甲酸30对fxr有很强的效力。苄基位置(32)的甲基化显著增强了fxr的激动活性,但不奇怪的是,seh对其不耐受。由于所述酰胺部分模拟eets的环氧化物和酶的活性位点中的攻击性水分子34,苄基位空间位阻显著降低了对seh的抑制效力。总之,苯甲酸残基上的一些额外残基提高了fxr或seh的活性,但是无法确定能够产生增强和平衡的双重效力的进一步取代的位置。
表5:26-32对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
因此,发明人研究了18中羧酸的sar,并引入了几种替代的极性残基和生物电子等排体(表6)。醇33对seh的效力与18相同,但对fxr无效,而醛34仍然激活了fxr,其效力比18低10倍,但对seh的效力明显更强。甲基酮35对两种标靶都表现出明显提高的活性,是第一个对fxr和seh具有纳摩尔效力的双调节剂。该sar表明,羧酸的羰基部分,而不是醇部分,足以激活fxr,seh在该位置上优选低极性基团。醛34的效力降低可能是由于flfxr试验的细胞环境的稳定性低。腈36对seh有纳摩尔级抑制效力,但对fxr无活性。酰胺37-39只是中等效力的seh抑制剂,这再次表明这一位置上较多的极性残基是不利的。在fxr上,随着氮原子取代的增加,酰胺37-39的效力显著提高。伯酰胺37的活性明显低于羧酸18,而n-甲基酰胺38的效力与18相当。n,n-二甲基酰胺39中的另一个甲基进一步提高了效力。酰胺在n-乙酰苯胺40中的转化降低了fxr的激动活性,但在n-三氟乙酰苯胺41中引入三个氟原子是非常有利的,能够产生平衡的纳摩尔级的双重调节剂。游离苯胺42对seh仍有抑制作用,但对fxr无活性。
与18相比,四唑43作为羧酸的典型生物电子等排体,对fxr具有稍微增强的效力,并且降低了seh的抑制活性。酸性低得多的磺胺44在一定程度上能够反转18的活性曲线,与fxr相比,其对seh更有效。为了提高44的酸度,发明人制备了n-乙酰磺胺45,但与44相比,其对fxr无活性并且对seh的效力显著降低。46中的磺胺残基的转化与40中的酰胺的转化具有相似效果,能够得到有效的双重调节剂。
最后,发明人在用47中的所述甲氧基交换了18的所述羧酸,出其不意地在两个靶标上都产生了相当大的效果。乙氧基衍生物48在fxr和seh上表现出同样的效力,而异丙氧基类似物49在fxr上表现出同样的活性,但由于其不溶性而不能在seh上进行表征。三氟甲氧基衍生物50对seh的效力略有提高,显示出高且平衡的双重效力。在甲基硫醇51中用硫代替氧,产生了更有效的双重调节剂,其在两个靶标上的半最大效应浓度约为0.1μm。当硫醇51被氧化成亚砜52或硫酸基53时,fxr上的效力显著下降而52在seh上仍有活性,只有53显示出明显的抑制效力降低。在硫醇作为羧酸替代物的情况下,在54中引入三氟甲基导致fxr上的效力显著增加,并生成亚纳摩尔级部分激动剂。然而,与甲基硫醇51相比,三氟甲基硫醇54对seh的活性略有降低,因此其具有高但略不平衡的双重效力。
表6:33-54对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
由于优化的先导化合物18的单一改性都不能将两个靶标的效力提高到较低的纳摩尔值,因此发明人评估了将每个靶标的最有利结构改变组合在一个分子中的可能性。为了确定对其中一个靶标具有突出活性的衍生物,发明人绘制了最有效化合物的pic50(seh)与pec50(fxr)值的对比图(图1)。该效力图显示,苄基部分(30)的3-氟取代、三氟甲基硫醇残基(54)和转化的磺胺46对fxr非常有利。由于三氟乙酰胺41在结构上类似于46,并且对fxr也具有很高的效力,因此它被另外选择用于组合。关于seh抑制,苄基部分(31)的3-氯取代、甲硫醇残基(51)和磺胺44较其它衍生物更为突出。由于51和54溶解度差,并且由于这两个部分不能合并,所以在重组时忽略了它们。相反,选择30、31、41、44和46进行结构重组(表7)。
在磺胺44中引入30的3-氟原子产生了双重调节剂55,揭示了fxr效力的预期增加。然而,这种改进仅仅是适度的,并没有产生期望的低纳摩尔效力的双重调节剂。相比之下,在n-(3-氯苯基)三氟乙酰胺56中31和41的合并以及在n-(3-氯苯基)甲磺酸酰胺57中31和46的合并伴随着对两个靶标效力的显著提高。部分fxr激活的ec50值分别为14±1nm和20.4±4.2nm,seh抑制的ic50分别为8.9±1.6nm和4.1±0.4nm,双重调节剂56和57最终对两个靶标都构成了期望的低纳摩尔效力。在这两种双调节剂中,57显示出明显高于56(<0.1μg/ml(lloq))的水溶性(1.5μg/ml),因此被选择用于进一步的体外评价。等温滴定量热法(itc,支持图s7)揭示了0.13μm57的kd并且揭示了焓结合(δη=-19.5kcal/mol,δs=-34.0cal/mol·k)。细胞ec50值与kd之间的差异似乎是由于itc实验中缺少可能显著影响结合平衡的共激活剂。高结合能可能部分来自fxr-lbd结合时潜在的显著构象变化。
表7:55-57对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
此外,还生产了以下额外化合物并测试了其对fxr激活和seh抑制的活性(见表8)。
表8:额外化合物对fxr和seh的体外活性(数据表示平均值±sem;n=3-6)
采用以下步骤合成化合物:
1-(4-氨基-2-氯苯基)甲胺(58k):将lialh4(1m溶于thf,16.4ml,16.4mmol,2.5eq)冷却至0℃。将4-氨基-2-氯苯甲腈62(1.0g,6.6mmol,1.0eq)溶于3mlthf,然后缓慢加入到混合物中。在h2析出停止后,将混合物加热至室温,然后回流16h。冷却至室温后,用10mlthf稀释混合物,然后冷却至0℃。逐滴加入1ml10%naoh溶液和1.8ml水。用硅藻土对无色沉淀物进行过滤并用15ml乙醚洗涤。滤液中有机溶剂的蒸发产生58k(黄色油)(0.77g,75%)。1hnmr(500mhz,dmso-d6)δ=7.11(d,j=8.2hz,1h),6.58(d,j=2.2hz,1h),6.48(dd,j=8.2,2.2hz,1h),5.19(s,2h),3.59(s,2h).13cnmr(126mhz,dmso-d6)δ=148.56,132.33,129.67,127.74,113.63,112.72,42.88。
n-(4-氨基-2-氯苄基)-4-(叔丁基)苯甲酰胺(69a):1-(4-氨基-2-氯苯基)甲胺58k(0.31g,2.0mmol,1.1eq)溶解于10mlchcl3,加入5mlnet3并将混合物冷却至0℃。在10min内,将4-叔丁基苯甲酰氯630(0.35ml,1.8mmol,1.0eq)缓慢加入,并在室温下将混合物搅拌两小时。然后,加入50ml10%盐酸水溶液,分离相,用30mletoac清洗水相。用na2co3将水相ph调至10,并用80mletoac一次性提取3次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。以石油醚/etoac(90:10)作为流动相,采用层析柱进行进一步纯化得到69a,黄色固体(0.57g,97%)。rf{石油醚/etoac=67:33)=0.26.1hnmr(500mhz,meoh-d4)δ=7.81(dt,j=8.6,2.3hz,2h),7.53(dt,j=8.6,2.3hz,2h),7.45(d,j=8.3hz,1h),7.31(d,j=2.2hz,1h),7.15(dd,j=8.3,2.2hz,1h),4.64(s,2h),1.36(s,9h).13cnmr(126mhz,meoh-d4)δ=170.32,156.67,140.89,135.23,132.33,131.39,128.29,126.59,122.96,120.82,119.16,42.13,35.36,31.54.hrms(maldi):c18h21cln2o的m/z计算为317.14152,发现为317.14130[m+h]+。
n-(4-氨基-2-氯苄基)-4-(叔丁基-2-甲氧基)苯甲酰胺(69b):将4-氨基-2-氯苄胺(58k,0.23g,0.15mmol,1.3eq))溶解于30ml无水chcl3中。加入4-dmap(0.12g,1.15mmol,1.00eq)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(0.24g、1.27mmol、1.10eq)和4-叔丁基-2-甲氧基苯甲酸(64b,0.24g,1.15mmol,1.0eq)。将混合物在室温下搅拌16小时。然后加入25ml5%盐酸水溶液,用15mletoac萃取水相三次。用na2so4干燥结合的有机层,并在真空中除去溶剂。进一步用己烷/乙酸乙酯结晶纯化得到69b,淡黄色固体(0.324g,81%)。1hnmr(250mhz,dmso-d6):δ=8.62(t,j=6.0hz,1h),7.73(d,j=8.0hz,1h),7.27(d,j=8.3hz,1h),7.07(dd,j=10.1,2.0hz,3h),6.97(t,j=7.2hz,1h),4.47(d,j=6.0hz,2h),3.94(s,3h),1.31(s,9h).13cnmr(75mhz,dmso-d6)δ=164.89,156.98,155.90,150.38,150.31,132.32,130.41,129.61,119.72,117.52,109.08,106.97,55.89,34.91,30.90。
4-(叔丁基-3-甲氧基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(76a):将n-(4-氨基-2-氯苄基)-4-(重丁基-2-甲氧基)苯甲酰胺69b(0.32g,0.93mmol,1.00eq)溶解于20mlchcl3,并加入5ml吡啶。小心添加甲磺酰氯(70,0.09ml,1.12mmol,1.20eq)。将混合物在室温下搅拌两小时。然后加入15ml10%盐酸水溶液,用30ml乙酸乙酯萃取三次。用na2so4干燥结合的有机层,并在真空中去除溶剂。通过己烷/乙酸乙酯结晶进一步纯化得到76a,淡棕色固体(0.172g,35%)。1hnmr(250mhz,dmso-d6)δ=9-97(s,1h),8.72(t,j=6.0hz,1h),7.79(d,j=8.0hz,1h),7.36(dd,j=14.9,5.2hz,2h),7-25-7.13(m,3h),4.56(d,j=6.0hz,2h),4.00(s,3h),3.08(s,3h),1.37(s,9h).13cnmr(126mhz,dmso-d6)δ=165.47,157.45,156.36,138.79,132.71,132.32,130.85,129.87,120.27,117.99,109.54,60.22,56.36,35.39,31.37,21.24,14.56。
4-(叔丁基-2-羟基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(77):将4-(叔丁基-2-甲氧基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(76a,0.17g,0.40mmol,1.0eq)溶解于30mldcm并在0℃条件下加入bbr3(4.05ml,4.05mmol溶解于dcm,10.0eq))。将混合物在室温下搅拌16小时,然后稀释于30ml冰水。用nahco3将ph调整到6,并用30ml乙酸乙酯一次性萃取3次。将结合的有机层在真空中浓缩。用己烷/乙酸乙酯结晶进一步纯化得到77,无色固体(0.098g,60%)。1hnmr(500mhz,dmso-d6)δ=12.30(s,1h),9.95(s,1h),9.21(t,j=5.5hz,1h),7.84(d,j=8.4hz,1η),7.35-7-24(m,2h),7.15(d,j=8.5hz,1h),6.98-6.93(m,1h),6.89(s,1h),4.51(d,j=5.6hz,2h),3.02(s,3h),1.26(s,9h).13cnmr(126mhz,dmso-d6)δ=168.84,159.78,157.29,138.57,132.42,131.05,129.75,127.69,119.71,118.24,116.17,113.95,112.50,34.65,30.71,30.49.hrms(maldi):c19h24cln2o4s的m/z计算为411.11398,发现为411.11373[m+h]+。
n-(4-氨基-2-氯苄基)-4-(叔丁基-3-甲氧基)苯甲酰胺(69c):将4-氨基-2-氯苄胺(58k,0.23g,0.15mmol,1.3eq)溶解于30ml干燥chcl3。加入4-dmap(0.12g,1.15mmol,1.00eq)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(0.24g,1.27mmol,1.10eq)和4-叔丁基-3-甲氧基苯甲酸(64a,0.24g,1.15mmol,1.0eq)。将混合物在室温下搅拌16小时。然后加入25ml5%盐酸水溶液,用15mletoac一次性提取3次。用na2so4干燥结合的有机层并在真空中去除有机溶剂。用己烷/乙酸乙酯结晶进一步纯化得到淡69c,黄色固体(0.312g,78%)。1hnmr(250mhz,dmso-d6):δ=8.84(t,j=5.7hz,1h),7.45(d,j=9.7hz,2h),7.29(d,j=7.9hz,1h),7.13(d,j=8.4hz,1h),6.86(s,1h),6.72(d,j=8.1hz,1h),4.42(d,j=5.7hz,2h),3.86(s,3h),1.34(s,9h).ms(esi+):m/z369.10([m+na]+,100)
4-(叔丁基-3-甲氧基)-n-(2-氯-4-(甲磺胺基)苄基)苯甲酰胺(76b):将n-(4-氨基-2-氯苄基)-4-(叔丁基-3-甲氧基)苯甲酰胺69c(0.31g,0.89mmol,1.00eq)溶解于20mlchcl3并加入5ml吡啶。小心加入甲磺酰氯70(0.07ml,1.07mmol,1.20eq)。将混合物在室温下搅拌两小时。然后加入15ml10%盐酸水溶液,用30ml乙酸乙酯萃取三次。用na2so4干燥结合的有机层,并在真空中去除溶剂。用己烷/乙酸乙酯结晶进一步纯化得到76b,淡棕色固体76b(0.315g,83%)。1hnmr(250mhz,dmso-d6)δ=9.92(s,1h),8.95(t,j=5-8hz,1h),7.46(dd,j=9.6,1.6hz,2h),7.34-7.26(m,3h),7.15(dd,j=8.4,2.2hz,1h),4.49(d,j=5.7hz,2h),3.87(s,3h),3.01(s,3h),1.34(s,9h).13cnmr(126mhz,dmso-d6)δ=172.05,170.40,166.16,158.02,140.79,138.38,133.14,132.33,131.72,129.59,126.16,119.82,119.39,118.34,110.73,59.79,55.42,34.68,29.43。
4-(叔丁基-3-羟基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(78):4-(叔丁基-3-甲氧基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(76b,0.31g,0.74mmol,1.0eq)溶解于50mldcm并在0℃条件下加入bbr3(7.40ml,7.40mmol溶于dcm,10.0eq)。将混合物在室温下搅拌16小时,然后稀释于50ml冰水中。用nahco3调节ph至6,然后用30ml乙酸乙酯提取混合物三次。将结合的有机层在真空中浓缩。用己烷/乙酸乙酯结晶进一步纯化得到78,无色固体(0.145g,48%)。1hnmr(500mhz,dmso-d6)δ=9.92(s,1h),9.62(s,1h),8.81(t,j=5.8hz,1h),7.31-7.27(m,2h),7.27-7.25(m,2h),7.21(d,j=8.1hz,1h),7.15(dd,j=8.4,2.2hz,1h),4.45(d,j=5.8hz,2h),3.01(s,3h),1.35(s,9h).13cnmr(126mhz,dmso-d6)δ=170.82,166.91,156.27,139.24,138.78,133.49,132.72,132.23,129.83,126.61,120.22,118.72,117.77,115.88,34.93,29.61.hrms(maldi):c19h24cln2o4s的m/z计算为411.11398,发现为411.11383[m+h]+。
n-(4-氨基-2-(三氟甲基)苄基)-4-(叔丁基)苯甲酰胺(69d):将1-(4-氨基-2-(三氟甲基)苯基)甲胺58z(0.25g,1.3mmol,1.1eq)溶解于7mlchcl3,加入7mlnet3并将混合物冷却至0℃。在10min内,将4-叔丁基苯甲酰氯630(0.28ml,1.4mmol,1.1eq)缓慢加入,并在室温下将混合物搅拌两小时。然后加入10ml10%盐酸水溶液,分离相,用10mletoac清洗水相。用na2co3将水相ph调整到10,并用10mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用己烷/etoac(90:10)作为流动相,采用柱层析法进一步纯化得到69d,黄色固体(0.35g,84%)。rf(己烷/etoac=90:10)=0.27.1hnmr(250mhz,dmso-d6)δ=8.77(t,j=5-7hz,1h),7.87-7.79(m,2h),7.48(d,j=8.5hz,2h),7.13(d,j=8.4hz,1h),6.90(d,j=2.3hz,1h),6.74(dd,j=8.3,2.1hz,1h),5.44(s,2h),4.47(d,j=5.3hz,2h),1.30(s,9h).13cnmr(75mhz,dmso-d6)δ=166.14,154.04,147.81,131·50,129.71,127.15,125.05,123.08,116.80,110.55,110.47,34.60,30.94。
4-(叔丁基)-n-(2-三氟甲基-4-(甲基磺胺基)苄基)苯甲酰胺(80):n-(4-氨基-2-(三氟甲基)苄基)-4-(叔丁基)苯甲酰胺69d(0.35g,1.0mmol,1.00eq)溶解于50mlthf并加入5ml吡啶。小心加入甲磺酰氯(70,0.23ml,3.0mmol,3.0eq)。将混合物在室温下搅拌过夜。然后加入50ml10%盐酸水溶液,并用20ml乙酸乙酯萃取三次。用na2so4干燥所结合的有机层并在真空中除去溶剂。用己烷/etoac(67:33)作为流动相,采用柱层析法进一步纯化得到80,淡粉色固体(0.09g,22%)。rf(己烷/etoac=67:33)=0.19.1hnmr(500mhz,dmso-d6)δ=10.06(s,1h),9.00(t,j=5.8hz,1h),7.89-7.82(m,2h),7.54-7.43(m,5h),4-59(d,j=5.5hz,2h),3.03(s,3h),1.30(s,9h).13cnmr(126mhz,dmso-d6)δ=166.42,154.33,137.52,132.71,131.25,129.78,127.24,125.20,123.13,116.72,116.67,34.69,30.98.hrms(maldi):c20h24f3n2o3s的m/z计算为429.14542,发现为429.14523[m+h]+。
4-(叔丁基)-n-(2-氯-4-(异丙基磺胺基)苄基)-苯甲酰胺(81):将n-(4-氨基-2-氯苄基)-4-(叔丁基)苯甲酰胺69a(0.154g,0.49mmol,1.0eq)溶解于20mlthf并加入2ml吡啶。小心加入异丙基磺酰氯86a(0.57ml,4.89mmol,10eq),并在室温下将混合物搅拌24小时。然后加入15ml盐酸水溶液,并用30mletoac一次性萃取混合物三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用己烷/etoac()67:33)作为流动相,采用柱层析法进一步纯化得到81,黄色固体(0.041g,20%)。rf(己烷/etoac=67:33)=0.20.1hnmr(500mhz,dmso-d6)δ=9.95(s,1h),8.92(t,j=5.7hz,1h),7.84(d,j=8.4hz,2h),7.49(d,j=8.4hz,2h),7.36-7.24(m,2h),7.17(dd,j=8.5,2.1hz,1h),4.46(d,j=5.7hz,2h),1.30(s,9h).13cnmr(126mhz,dmso-d6)δ=166.28,154.21,138.65,132.33,131.37,131.34,129.56,127.20,125.13,119.19,117.78,51.59,34.65,30.96,16.10.hrms(maldi):c21h28cin2o3s的m/z计算为423.15037,发现为423.14967[m+h]+。
4-(叔丁基-n-(2-氯-4-(乙基磺胺基)苄基)-苯甲酰胺(82):将n-(4-氨基-2-氯苄基)-4-(叔丁基)苯甲酰胺69a(0.16g,0.51mmol,1.0eq)溶解于20mlthf并加入2ml吡啶。小心加入乙磺酰氯86b(0.5ml,5.1mmol,10eq),并在室温下将混合物搅拌24小时。然后加入15ml10%盐酸水溶液,并用30mletoac一次性萃取混合物三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用己烷/etoac(50:50)作为流动相,采用柱层析法进一步纯化得到82,黄色固体(0.125g,60%)。rf(己烷/etoac=50:50)=0.51.1hnmr(500mhz,dmso-d6)δ=9.98(s,1h),8.93(t,j=5.8hz,1h),7.84(d,j=8.4hz,2h),7.49(d,j=8.4hz,2h),7.29(d,j=8.4hz,1h),7.27(d,j=2.1hz,1h),7.15(dd,j=8.4,2.1hz,1h),4.47(d,j=5.7hz,2h),3.11(q,j=7.3hz,2h),1.30(s,9h),1.18(t,j=3.6hz,3h).13cnmr(126mhz,dmso-d6)δ=166.27,154.20,138.42,132.34,131.51,131.34,129.57,127.19,125.13,119.35,117.89,59.78,45.29,39.52,34.65,30.96,8.02.hrms(maldi):c20h26cin2o3s的m/z计算为409.13472,发现为409.13444[m+h]+。
n-(4-氨基-2-氯苄基)-4-(二甲氨基)苯甲酰胺(69e):将1-(4-氨基-1-氯苯基)甲胺58k(0.30g,1.92mmol,1.1eq)溶解于10mldmf。加入10mlnet3并将混合物冷却至0℃。在10min内,将4-二甲氨基苯甲酰氯63p(0.32ml,1.73mmol,1.0eq)缓慢加入,并在室温下将混合物搅拌5h。然后加入50ml10%盐酸水溶液,分离相,并用na2co3溶液将水相ph调整至10,用80mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用etoac/己烷/net3进行柱层析进一步纯化得到69e,黄色固体(0.205g,39%)。rf(etoac/己烷/net3=65:33:2)=0.47.1hnmr(250mhz,dmso-d6)δ=8.42(t,j=5.7hz,1h),7.76(d,j=9.0hz,2h),6.98(d,j=8.3hz,1h),6.70(d,j=9.0hz,2h),6.61(d,j=2.2hz,1h),6.47(dd,j=8.3,2.3hz,1h),5.24(s,2h),4.34(d,j=5.7hz,2h).13cnmr(75mhz,dmso-d6)δ=166.25,152.23,148.96,132.34,129.62,128.71,123.13,121.02,113.65,112.70,110.88,39.87。
4-(二甲胺基)-n-(2-氯-4-(甲基磺胺基)苄基)苯甲酰胺(83):将n-(4-氨基-2-氯苄基)-4-(二甲氨基)苯甲酰胺69e(0.204g,0.67mmol,1.0eq)溶解于30mlthf并加入3ml吡啶。小心加入甲磺酰氯70(0.27ml,3.36mmol,5,0eq),并在室温下将混合物搅拌2h。然后加入15ml10%盐酸水溶液,并用30mletoac一次性萃取混合物三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用etoac作为流动相,采用柱层析法进一步纯化。在dcm/己烷和丙酮/水中进一步结晶得到83,黄色固体(0.064g,25%)。rf(etoac)=0.64.1hnmr(500mhz,meod-d4)δ=7.76(d,j=9.0hz,2h),7.33(t,j=5.5hz,2h),7.14(dd,j=8.4,2.2hz,1h),6.73(d,j=9.0hz,2h),5.49(s,1h),4.59(s,2h),3.02(s,6h),2.96(s,3h).13cnmr(126mhz,meod-d4)δ=170.50,154.38,139.71,134.72,133.51,130.77,129.89,121.74,121.54,119.63,112.13,41.90,40.22,39.32.hrms(maldi):c17h19cln3o3s的m/z计算为380.08302,发现为380.08274[m-h]-。
4-((1-三氟甲基)环丙-1-基)苯甲酸(64i):pd(oac)2(0.045mmol,0.01g,3mol%),xantphos(0.045mmol,0.01g,3mol%)溶解于10mldmf。然后逐滴加入甲酸(10.6mmol,0.4ml,7.0eq)和1-溴-4-(1-三氟甲基)环丙-1-基)苯87(1.5mmol,0.4g,1.0eq)。然后在混合物中加入1-(3-二甲基氨基丙基)-3-乙基碳二亚胺和三乙胺。将混合物在50℃条件下搅拌20h。将混合物冷却至室温后,添加10ml10%盐酸水溶液,并用30mletoac一次性萃取混合物三次。用30ml饱和na2co3水溶液一次性萃取结合的有机层。然后用浓盐酸将有机水相的ph调整至1,并用30mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂获得64i,米色固体(0.177g,51%),无需进一步纯化。1hnmr(300mhz,dmso-d6)δ=13.04(s,1h),8.00-7.93(m,2h),7.60(d,j=8.1hz,2h),1.40(dd,j=7.0,5.0hz,2h),1.23-1.11(m,2h).13cnmr(75mhz,dmso-d6)δ=166.85,140.01,131.17,130.85,129.40,128.15,27.75-27.32,9.78-9.75。
n-(4-氨基-2-氯苄基)-4-((1-三氟甲基)环丙-1-基)-苯甲酰胺(69f):将1-(4-氨基-1-氯苯基)甲胺58k(0.31g,2.0mmol,3,0eq)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(0.31g,2.0mmol,3,0eq)和4-(二甲氨基)吡啶(0.66mmol,0.08g,1,0eq)溶解于10mlchcl3和1mldmf。然后将4-((1-三氟甲基)环丙-1-基)苯甲酸64i(0.15g,0.66mmol,1.0eq)溶解于5mlchcl3,并在10min内,将0.5mldmf缓慢加入,将混合物在60℃条件下搅拌5h。将混合物冷却至室温后,添加10ml10%盐酸水溶液,分离相,用na2co3溶液将水相ph调整至10,并用20mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用己烷/etoac(57:43)作为流动相,采用柱层析法进一步纯化得到69f,黄色固体(0.123g,50%)。rf(己烷/etoac=57:43)=0.45.1hnmr(300mhz,丙酮-d6)δ=7.95-7.90(m,2h),7.57(d,j=8.1hz,2h),7.15(d,j=8.3hz,1h),6.72(d,j=2.3hz,1h),6.58(dd,j=8.3,2.3hz,1h),4.85(s,1h),4-54(d,j=5-6hz,2h),1.39(dd,j=6.9,5.0hz,2h),1.18-1.12(m,2h).13cnmr(75mhz,meod-d4)δ=δ=169.60,149-99,140.91,135.73,134.95,132.42,131.42,128.50,124.97,116.42,114.75,42.29,29.28,28.83,10.41
4-((1-三氟甲基)环丙-1-基)-n-(2-氯-4-(甲磺胺基)苄基)苯甲酰胺(84):将n-(4-氨基-2-氯苄基)-4-((1-三氟甲基)环丙-1-基)-苯甲酰胺69f(0.1g,0.028mmol,1.0eq)溶解于15mlthf并加入1.5ml吡啶。小心加入甲磺酰氯70(0.22ml,2.8mmol,10.0eq.),并在室温下将混合物搅拌24h。然后加入15ml10%盐酸水溶液,用30mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用己烷/etoac(50:50)作为流动相,采用柱层析法进一步纯化得到84,白色固体(0.052g,41%)。rf(己烷/etoac=50:50)=0.36.1hnmr(500mhz,dmso-d6)δ=9-93(s,1h),9.04(s,1h),7.91(d,j=8.3hz,2h),7-57(d,j=8.1hz,2h),7.32(d,j=8.4hz,1h),7.26(d,j=2.1hz,1h),7.15(dd,j=8.4,2.1hz,1h),4.48(d,j=5.7hz,2h),3.02(s,3h),1.38(s,2h),1.17(s,2h).13cnmr(126mhz,meod-d4)δ=169.74,141.13,139.98,135.45,134.93,132.85,132.52,131.10,128.53,121.72,119.59,42.16,39.37,29.21-28.95,10.44-10.43.hrms(maldi):c19h19clf3n2o3s的m/z计算为447.07515,发现为447.07455[m+h]+。
n-(4-氨基-2-氯苄基)-4-(吡咯烷)-苯甲酰胺(69g):将1-(4-氨基-1-氯苯基)甲胺58k(0.49g,3.1mmol,3.0eq)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺(0.49g,3.1mmol,3.0eq)和4-(二甲氨基)吡啶(1.1mmol,0.13g,1.0eq)溶解于20mlchcl3和2mldmf。然后在10分钟内,将溶解于5mlchcl3和0.5mldmf的4-吡咯烷苯甲酸(0.2g,1.1mmol,1.0eq)缓慢加入,将混合物在60℃下搅拌5h。将混合物冷却至室温后,加入10ml10%盐酸水溶液,分离相,用na2co3溶液将水相的ph调整至10,并用20mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用etoac/己烷(86:14)为流动相,采用柱层析法进一步纯化得到69g,黄色固体(0.100g,29%)。rf(etoac/己烷=86:14)=0.45.1hnmr(250mhz,dmso-d6)δ=8.39(s,1h),7.76(d,j=8.8hz,2h),6.98(d,j=8.3hz,1h),6.61(d,j=2.2hz,1h),6.53(d,j=8.9hz,2h),6.47(dd,j=8.3,2.3hz,1h),5.24(s,2h),4.35(d,j=5.7hz,2h),3.27(d,j=7.2hz,4h),1.96(t,j=4.8hz,4h).13cnmr(75mhz,dmso-d6)δ=162.32,149.50,148.89,132.23,129.53,128.75,123.11,120.31,113.53,112.59,110.54,47.21,35.78,24.96。
4-(吡咯烷)-n-(2-氯-4-(甲磺胺基)苄基)苯甲酰胺(85):将n-(4-氨基-2-氯苄基)-4-(吡咯烷)-苯甲酰胺69g(0.1g,0.03mmol,1.0eq)溶解于15mlthf并加入2ml吡啶。小心加入甲磺酰氯70(0.12ml,1.55mmol,5,0eq),并在室温下将混合物搅拌2h。然后加入15ml10%盐酸水溶液,并用30mletoac一次性萃取混合物三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。用etoac/己烷(67:33)为流动相,采用柱层析法进一步纯化得到85,白色固体(0.033g,26%)。rf(etoac/己烷=67:33)=0.37.1hnmr(500mhz,dmso-d6)δ=9.91(s,1h),8.60(s,1h),7.77(d,j=8.8hz,2h),7.28(d,j=8.4hz,1h),7.25(d,j=2.1hz,1h),7.14(dd,j=8.4,2.2hz,1h),6.54(d,j=8.9hz,2h),4.44(d,j=5.5hz,2h),3.28(s,4h),3.01(s,3h),1.98-1.94(m,4h).13cnmr(126mhz,dmso-d6)166.38,149.61,138.20,132.30,132.19,129.43,128.81,119.97,119.76,118.29,110.59,47.23,42.98,36.56,24.98.hrms(maldi):c19h23cln3o3s的m/z计算为408.11432,发现为408.11361[m+h]+。
实例4:实质性检查
利用seh和fxr的x-射线结构,通过分子对接分析化合物57在硅中的结合,x射线结构含有构建先导化合物5的配体(fxr:化合物3/pdb-id:4qe8;seh:化合物2/pdb-id:3i28)。得到的结合模式(如图2所示)与n-苄基苯甲酰胺5-57在两个靶标上的所述sar一致。在fxr中57的结合模式下(图2a),叔丁基部分紧密结合在结合袋中,并通过螺旋12的稳定化介导受体激活。相邻的苯环被很好地放置在亲脂袋中,不允许在2-或3-位发生变化。磺酰胺占据亲水区域,并且不表现出特定的相互作用,这解释了苄基部分这一位置的亲水部分的广泛耐受性。酰胺部分不形成定向h键,这也适用于fxr的x射线结构4qe8中的参考配体3。亚甲基桥结合在leu287附近,说明了化合物32的效力增强,该化合物在这一位置携带另外的甲基。氯原子指向由ile352定义的紧固子袋和耐受氯或氟的tyr369的酚基部分(30)(支持图s1),但没有如29的甲基取代基的纯亲脂残基。
提出的57与seh的结合模式(图2b)表明,其酰胺基与催化残基tyr383、tyr466和asp335相互作用。亚甲基桥位于不允许任何结构修改的狭窄通道中。苄基部分的氯取代基指向亲脂袋,对结合至关重要。与fxr结合模式类似,磺胺基部分结合在更亲水的子袋中,不会形成特定的相互作用。4-叔丁基苯基残基位于紧固疏水袋中,为芳香环的4或3位,而不是2位的取代基提供空间。
为了研究57在相关核受体中的选择性分布,发明人在10μm浓度条件下,确定了它们在相应受体的gal4-杂合报告基因分析中对palps、lxrs、rxrs、rars、pxr和vdr的活性(图3a)。57对pparα和pparδ无活性,对lxr亚型和rxrα均无活性。57仅对pparγ表现出微弱的部分激动活性,ec50值为14.7±0.9μμ,因此其在核受体中对fxr具有高度选择性(选择性≥720)。此外,在水溶性四唑(wst-1)测定中,高达100μm浓度的57仍无细胞毒性活性(图3b)。为了评估代谢稳定性,57与wistar大鼠肝微粒体进行孵育,60分钟后,仍有超过50%的化合物残留,证明了可接受的稳定性。尽管如此,发明人更详细地研究了57的体外代谢转化并鉴定了其代谢产物(图6,方案10)。根据lc-ms-ms分析(支持图s2-s6),57通过以下代谢:通过磺胺部分水解产生苯胺69a、通过叔丁基苯甲酰胺部分的羟基化可产生三种异构体77、78和79(图6,方案10),以及通过对在苄基取代基的芳香环的羟基化。发明人合成了77和78,其在苯甲酰胺芳香环上带有羟基,但在代谢残留物中未检测到这两种异构体,因此认为79为57的代谢物。代谢物69a具有相当大的激活fxr的效力(ec50值为0.046±0.006μμ),和抑制seh的效力(ic50值为0.040±0.006μμ)。因此,代谢物69a可能有助于体内双重调节的药效学活性,并且能够延长原始化合物57的药理作用。
为了评估57在比报告基因分析更少的人工条件下的fxr激动作用,发明人还量化了化合物对hepg2肝癌细胞中fxr靶基因表达的影响(图4a)。为此目的,将细胞与内源性fxr激动剂cdca(1b)(50μm)、部分激动剂57(0.1μm和1μm)或dmso(0.1%,作为对照)孵育8或16小时,然后对fxr靶基因mrna进行定量。根据该方法对数据进行分析,并将所有结果用看家基因甘油醛3-磷酸脱氢酶(gapdh)进行归一化。载体处理的对照细胞的基因表达被定义为100%。57显示出部分fxr激动分布,并调节了与内源激动剂cdca(1b)相似但幅值较小的的所有9个研究基因。57仅适度地增加了胆酸盐输出蛋白(bsep)的表达,而微小异源二聚体伙伴(shp)、过氧化物酶体增殖物激活受体α(pparα)、成纤维细胞生长因子19(fgf19)、丙酮酸脱氢酶激酶4(pdk4)和肝型脂肪酸结合蛋白(脂肪酸结合蛋白、fabp1)的引入使幅值达到cdca(1b)的50-80%。此外,57还抑制了胆固醇7α-羟化酶(cyp7a1)、固醇调节元件结合蛋白1c(srebp1c)和脂肪酸合成酶(fas),这些酶由fxr通过诱导shp进行间接调节。在cyp7a1中,57表现为部分激动剂,其抑制作用低于1b,而57和1b对srebp1c和fas的抑制作用幅值相同。对于所有基因,57在0.1μμ和1μm浓度下产生相同的效果,证实了其在这些浓度下已经饱和。相反地,即使在10μm的高浓度条件下,57也不能显著调节pparγ靶基因清道夫受体3b(cd36)和细胞因子样蛋白2-19(fam3a),进一步证明了所述化合物的选择性(图4b)。
同时,通过对氘化seh底物14.15-eet-d11和不同浓度的抑制剂17进行转化进行定量,在更接近自然的背景下对hepg2细胞匀浆中的可溶性环氧化物水解酶的抑制作用进行了研究(图4c)。即使在1nm浓度条件下,化合物对胞内seh也有较强的抑制作用,eet/dhet比值在统计学上有有显著的增大。在细胞环境中,57的seh抑制作用的ic50值约为10nm。
实例5:体内表征
受到57的高效力和良好的体外特性鼓舞,发明人在雄性野生型c57bl6/j小鼠体内进行了化合物的试验性研究(图5)。为了记录体内的药代动力学曲线并评估有关fxr激活和seh抑制的药效学数据,给予6只小鼠单一剂量的57(口服,10mg/kg体重)。另外三只动物作为载体对照。
双重调节剂57显示了良好的口服生物利用度(cmax1182ng/ml)和快速摄取度(tmax=0.5h),同时具有适中的半衰期(t1/2=0.7h)。总之,单一剂量的57在约四到五小时内的有效浓度高于ec50(fxr)和ic50(seh)值(图5a)。为了评估57的药效学作用,对小鼠血浆进行eet/dhet比值分析,并在用药8小时后测定小鼠肝脏中fxr靶基因的表达(图5b)。8.9-和11.12-同分异构体的eet/dhet比值在57处理时增加约2倍,表示双重调节剂在体内抑制了seh活性。此外,fxr靶基因在给予57的小鼠的肝脏中的表达发生了改变,以下的表达增加:bsep(约3倍)、shp(约4倍)和fgf15(约2.5倍),srebp1c的表达降低(约5倍),这也证明了fxr在体内的激活(图5c)。cyp7a1mrna水平显示出轻微的抑制趋势。pparγ靶基因脂肪酸转运蛋白(fatp)在体内的表达不受57影响。因此,实验动物研究证明其药代动力学可接受,并明确指出了57在体内的双重靶标作用。
肝细胞癌和肝硬化是nash最严重的潜在后果,其发病率的上升代表了全球健康问题的迅速增长。虽然肝移植是目前唯一有效的治疗方法,但对药物选择的研究非常深入。fxr激动剂oca(1a)正处于研发的前沿,有望成为第一种有效的nash治疗药物。它在临床试验中显示出抗脂肪变性和抗纤维化作用,同时伴有多种代谢改善。6,12双重pparα/δ激动剂elafibranor已经成功地完成了ii期临床试验,并可能作为一种治疗选择推进oca的研究进程。38然而,nash的多因素性质,涉及脂肪变性、纤维化和炎症(重要的)可能需要更广泛的治疗策略来处理所有相关因素。鉴于fxr激活对脂肪变性的显著效果6,12和seh抑制的肝脏抗脂肪变性和抗炎作用的相关报道23,39,发明人认为这些策略的组合可能是协同的。因此,发明人研制了一种高效且具有良好平衡活性的fxr和seh双重调节剂。
发明人成功地将部分fxr激动剂和seh抑制剂的已知药效团合并,生成了先导结构5,其在两个靶标上显示出微弱但具有统计学意义的活性。该先导化合物的低碎片样特性和结构柔韧性能容忍较大的结构变化以实现优化,因此,适度的活性似乎就足够了。在连续的四个步骤中,发明人系统地研究了所述化合物类对fxr和seh的sar,并强化了双重效力。然而,尽管发明人发现了几种单靶标的高效调节剂,但在系统性sar研究中,没有发现对两个靶标均具有低纳摩尔效力的化合物。因此,在最后的决定性优化步骤中,我们将对单靶标具有最大活性的剂的结构元素进行组合,开发出了高效的双重调节剂57,其对部分fxr激活的ec50值是20.4±4.2nm,对seh抑制的ic50值是4.1±0.4nm。57的更广泛的体外表征揭示,其在相关核受体上具有非常好的选择性分布,并且浓度为100μm时仍没有细胞毒性活性。在体外代谢的评估中,57表现出中等的稳定性,由体内适当半衰期所证实。然而,对主要代谢物的进一步评价和表征表明,由57的磺酰胺部分水解形成的苯胺69a具有几乎相等的效力,并且可能具有延长57的双重调节作用的药理活性。
与内源性激动剂cdca(1b)相比,在hepg2细胞中,57对fxr靶基因有部分诱导作用。在未转染的肝细胞中进行的观察以及在1μm和0.1μm浓度条件下部分激动剂调节作用相等的事实证实了,双重调节剂57的部分fxr真实激动特性。奥贝胆酸(1a)6的临床研究表明,用全部fxr激动剂治疗后,由于fxr靶基因胆固醇7α羟化酶(cyp7a1)的强烈抑制,胆固醇稳态受到干扰。该酶是胆固醇代谢转化为胆汁酸的第一酶并且是限速酶,因此,fxr的完全激活阻断了胆固醇消除的主要途径之一。为了避免这种副作用,部分fxr激活似乎更可取。
用57刺激后,hepg2细胞中的fxr靶基因表达谱显示,57对nafld和nash有有益影响。最近的研究报道nafld和nash患者40的成纤维细胞因子19(fgf19)的血清水平降低,并且fdf19改善了小鼠的胰岛素敏感性、减轻了体重并降低了肝脂肪含量。在oca(1a)治疗下观察到fgf19水平的升高,并认为其具有重要的有益药效作用。12通过β-氧化41完成的pparα诱导(pparα被认为是肝脏脂肪酸的主要调节因子)联合肝脏fas的抑制(该抑制能够导致脂肪酸从头合成减少)有利于减少肝脂肪变性。值得注意的是,nafld和nash中的肝脏脂肪主要是游离脂肪酸。4诱导pdk4导致糖酵解减少,从而进一步增强该效应,最终完成脂肪酸的能源利用。42肝型脂肪酸结合蛋白(fabp1)参与多种生理过程,并在全身范围内影响脂质稳态。在肝脏中,fabp1具有保护细胞的功能,并对抗氧化性细胞损伤。43由于肝细胞氧化应激是nafld/nash发展和表现的主要因素,fabp1在57作用下的增强表达似乎对nash是有利的。
而且,通过在hepg2细胞裂解物中定量氘化的seh底物14.15-eet-d11的转化,在更接近自然的设置下,研究了57对可溶性环氧化物水解酶的抑制效力。在该体系中,57的ic50值是1.6±0.5nm,与重组蛋白活细胞荧光分析结果完全一致。因此,在存在来自肝细胞的其他蛋白质和细胞成分的情况下,双重调节剂57在抑制人类seh方面同样有效。
受体外研究前景的鼓舞,发明人将57应用于雄性野生型c57bl6/j小鼠的体内试验研究,以评估双重调节剂的药代动力学和药效学作用。57显示出良好的快速摄取率和口服生物利用度,尽管分子的半衰期相当短,但发明者仍在单次口服10mg/kg体重后约3.5-4小时内观察到活性浓度。给予57后8h,小鼠肝脏fxr靶基因mrna定量结果显示shp和bsep由明显的上调趋势(没有达到统计学意义的显著性)并且显著影响fgf15和srebp1c的表达。cyp7a1表现出轻微的下调趋势。特别是bsep的诱导表明57在体内激活了fxr,因为该基因几乎完全由fxr调节。44-46此外,如上所述,srebp1c的抑制和fgf15的诱导在nafld/nash的治疗中显示出良好的效果。对于seh的体内抑制作用,发明人评估了57对血浆中seh底物(eets)与seh产物(dhets)的比值的影响,在给予双重调节剂的小鼠中,该比值显著升高(倾向eets)。eets的这种积累表明57在体内抑制了seh,并且可能表现出抗炎活性,这将对nash十分有益。
本文报道的双重调节剂57是第一个具有低纳摩尔级效力的、能够部分激活fxr并且抑制seh的化合物。其调节fxr靶基因表达和eet/dhet比值的药学作用表明,57在体内能够靶向两个靶标。由于这种独特的活性,双重调节剂完全符合大规模动物模型的要求,用以研究其治疗功效和对nash及相关代谢或心血管紊乱的双重fxr/seh调节的概念。
材料和方法
化学
常规。所有化学品和溶剂均为试剂级,除非另有规定,否则无需进一步纯化即可使用。所有的反应均于氩气条件下烘干的玻璃器皿和绝对溶剂中进行的。nmr光谱用brukerav400、brukerav300、brukeram250xp或brukerav500(bruker公司,billerica,马萨诸塞州,美国)分光光度计进行记录。以四甲基硅烷(tms)为参照,报道了在ppm中的化学位移(δ);多重性:s,单峰;d,双峰;dd,两个双峰;t,三重峰;dt,两个三重峰;m,多重峰;近似耦合常数(j),赫兹(hz)。在vg平台ii(thermofischerscientific公司,waltham,马萨诸塞州,美国)上使用电喷雾电离(esi)方法进行质谱。高分辨率质谱用maldiltqorbitrapxl仪器(thermofisherscientific)进行记录。化合物纯度由varianprostarhplc(spectralabscientific公司,markham,on,加拿大)进行分析,该仪器配有multohigh100phenyl-5μ240+4mm柱(cs-chromatographieservicegmbh,langerwehe,德国),以1ml/min的流速进行梯度洗脱(h2o/meoh80:20+0.1%等比例甲酸5min,meoh+0.1%等比例甲酸45min,meoh+0.1%等比例甲酸10min),并在245nm和280nm处进行紫外检测。对化合物16进行元素分析,因为在ms中未发现分子离子。所有用于生物评价的最终化合物纯度均≥95%。
双重调节剂57的制备:
1-(4-氨基-1-氯苯基)甲胺(58k):将lialh4(1m溶于thf,16.4ml,16.4mmol,2.5eq)冷却至0℃。4-氨基-2-氯苯甲腈62(1.0g,6.6mmol,1.0eq)溶于3mlthf并缓慢加入到混合物中。停止生成h2后,将混合物加热至室温,然后回流16h。冷却至室温后,用10mlthf稀释混合物,然后冷却至0℃。逐滴加入1ml10%naoh溶液和1.8ml水。用硅藻土过滤无色沉淀物,并用15ml乙醚进行洗涤。滤液中有机溶剂蒸发产生58k,黄色油(0.77g,75%)。1hnmr(500mhz,dmso-d6)δ=7.11(d,j=8.2hz,1h),6.58(d,j=2.2hz,1h),6.48(dd,j=8.2,2.2hz,1h),5.19(s,2h),3.59(s,2h).13cnmr(126mhz,dmso-d6)δ=148.56,132.33,129.67,127.74,113.63,112.72,42.88。
n-(4-氨基-2-氯苄基)-4-(叔丁基)苯甲酰胺(69a):将1-(4-氨基-1-氯苯基)甲胺58k(0.31g,2.0mmol,1.1eq)溶解于10mlchcl3,加入5mlnet3并将混合物冷却至0℃。在10min内,将4-叔丁基苯甲酰氯630(0.35ml,1.8mmol,1.0eq)缓慢加入,并在室温下将混合物搅拌两小时。然后,加入50ml10%盐酸水溶液,分离相,用30mletoac清洗水相。用na2co3溶液将水相ph调整至10,并用80mletoac一次性萃取三次。用na2so4干燥结合的有机层并在真空中蒸发溶剂。以石油醚/etoac(9:1)为流动相,采用柱层析法进一步纯化得到69a,黄色固体。rf(石油醚/etoac=2:1)=0.26.1hnmr(500mhz,meoh-d4)δ=7.81(dt,j=8.6,2.3hz,2h),7.53(dt,j=8.6,2.3hz,2h),7.45(d,j=8.3hz,1h),7.31(d,j=2.2hz,1h),7.15(dd,j=8.3,2.2hz,1h),4.64(s,2h),1.36(s,9h).13cnmr(126mhz,meoh-d4)δ=170.32,156.67,140.89,135.23,132.33,131.39,128.29,126.59,122.96,120.82,119.16,42.13,35.36,31.54.hrms(maldi):c18h21cln2o的m/z计算为317.14152,发现为317.14130[m+h]+。
4-(叔丁基)-n-(2-氯-4-(甲磺胺基)苄基)苯甲酰胺(57):将n-(4-氨基-2-氯苄基)-4-(叔丁基)苯甲酰胺69a(0.04g,0.12mmol,1.0eq)溶解于5mlchcl3并加入0.5ml吡啶。小心加入甲酰氯70(0.02ml,0.14mmol,1.2eq),并在室温下将混合物搅拌两小时。然后,加入15ml10%盐酸水溶液,用30mletoac一次性萃取三次。用na2so4干燥结合的有机层,并在真空中蒸发溶剂。以石油醚/etoac(4:1)为流动相,采用柱层析法进一步纯化得到57,无色固体(0.047g,66%)。rf(石油醚/etoac=2:1)=0.13.1hnmr(500mhz,meoh-d4)δ=7-80(dt,j=8.6,2.0hz,2h),7.52(dt,j=8.6,2.0hz,2h),7.36(d,j=8.4hz,1h),7-33(d,j=2.2hz,1h),7.16(dd,j=8.4,2.3hz,1h),4.62(s,2h),2.97(s,3h),1.35(s,9h).13cnmr(126mhz,meoh-d4)δ=170.30,156.55,139.90,134.88,133.07,132.46,130.99,128.29,126.56,121.75,119.62,42.06,39.36,35.81,31.55.ms(esi-):m/z393.1(100,[m-h]-).hrms(maldi):c19h24cin2o3s的m/z计算为395.11907,发现为395.11892[m+h]+。
有关化合物4-56和77-78以及相应中间体的制备和表征,请参考支持信息。
生物学评估
全长fxr反式激活分析
质粒:pcdna3-hfxr含有人fxr序列,已在其他地方发表。47pgl3basic(promegacorporation公司,fitchburg,威斯康星州,美国)用作报告基因质粒,是将胆盐输出蛋白(bsep)的启动子的短链结构克隆到荧光素酶基因前的sacl/nhel酶切位点。48以prl-sv40(promega)为对照,用于转染过程中转染效率和细胞生长的归一化。psg5-hrxr也已在其他地方发表。49
分析方法:用高糖dmem培养基(补充以10%fcs,1mm丙酮酸钠,100u/ml青霉素)在37℃、5%co2条件下培养hela细胞。转染前24小时,将hela细胞接种于96孔板中,密度为8000个/孔。转染前3.5h,将培养基改为高糖dmem(补充以1mm丙酮酸钠,100u/ml青霉素,100μg/ml链霉素,0.5%去固醇fcs)。采用磷酸钙转染法,用bsep-pgl3、prl-sv40和表达质粒pcdna3-hfxr和psgs-hrxr瞬时转染hela细胞。转染16h后,将培养基改为高糖dmem培养基(补充以1mm丙酮酸钠,100u/ml青霉素,100μg/ml链霉素,0.5%去固醇fcs)。转染24h后,将培养基改为不含酚红的dmem培养基(补充以1mm丙酮酸钠,100u/ml青霉素,100μg/ml链霉素,2mm左旋谷氨酰胺,0.5%去固醇fcs),现在,另外含有0.1%dmso和相应的受试化合物或仅含有0.1%dmso作为未处理对照。对每种浓度进行测试,一式三份,且每个实验至少独立重复三次。与所述受试化合物孵育24h后,根据生产商的方案,使dual-glotm荧光素酶测定系统(promega)测定细胞的荧光素酶活性。使用tecaninfinitem200光度计(tecandeutschlandgmbh,crailsheim,德国)检测发光值。用虫荧光素酶数据除以海肾萤光素酶数据,再乘以1000得到相对光单位(rlu),实现转染效率和细胞生长的归一化。用所测试化合物的平均rlu除以相应浓度下的未处理对照的平均rlu,得到激活倍数。用相应浓度下的所测试化合物的激活倍数除以fxr全激动剂gw4064(1c)在3μμ的激活倍数,得到相对激活。用sigmaplot10.0(systatsoftwaregmbh,erkrath,德国)采用四参数逻辑回归分析计算至少三个独立试验的平均相对激活值,得到ec50和平均值的标准误差。用fxr激动剂1b对该分析进行验证验证(ec50=18±1μμ,88±3%相对最大激活),1a(ec50=0.16±0.02μμ,87±3%相对最大激活),1c(ec50=0.51±0.16μm,3μμ定义为100%)。15
seh活性分析
在基于荧光的96孔seh活性检测中,用重组人酶测定了化合物的seh抑制效力。50,51非荧光phome(3-苯基氰基-(6-甲氧基-2-萘基)甲酯2-环氧丙烷乙酸;cayman化学)可被seh水解为荧光6-甲氧基萘甲醛作为底物。重组人seh(溶解于bis-tris缓冲液,ph7,0.1mg/mlbsa,triton-x100终浓度0.01%)与测试化合物(溶解于dmso,dmso终浓度1%)在室温条件下预孵育30min。然后加入底物(终浓度50μμ),用tecaninfinitef200pro(λem=330nm,λex=465nm)测量荧光产物的形成持续30min(每分钟一个点),确定底物的水解程度。设置空白对照组(不含蛋白质和化合物)和阳性对照组(不含化合物)。所有的实验一式三份,并在至少三个独立的实验中进行重复。记录升高化合物浓度的剂量-反应曲线,计算ic50。
pparα/γ/δ、lxrα/β、rxrα/β/γ、rarα/β/γ、vdr和pxr质粒的杂交报告基因检测:gal4-融合受体质粒pfa-cmv-hppara-lbd52、pfa-cmv-hpparγ-lbd52、pfa-cmv-hpparδ-lbd52、pfa-cmvhlxrα-lbd32和pfa-cmv-hlxrβ-lbd32以前均有报道。对商业化cdna(sourcebiosience,nottingham,英国)进行pcr扩增得到cdna片段,并将该片段通过bamhi酶切切割位点和前面插入的kpnl酶切切割位点整合到pfa-cmv载体(stratagene,lajoll,加利福尼亚州,美国)的gal4-bamhi切割位点,以此构建编码各自核受体典型亚型的铰链区和配体结合区(lbd)(uniprot目录号:hrxrα-p19793,225-462残基;hrxrβ-p28702-1,294-533残基;hrxrγ-p48443-1,229-463残基;hrarα-p10276-1,177-462残基;hrarβ-p10826-1,177-455残基;hrarγ-p13631-1,179-454残基;hvdr-p11473-1,119-427残基;hpxr-o75469-1,138-434残基)的融合受体质粒pfa-cmv-hrxrα-lbd、pfa-cmv-hrxrβ-lbd、pfa-cmv-hrxrγ-lbd、pfa-cmv-hrarα-lbd、pfa-cmv-hrarβ-lbd、pfa-cmv-hrarγ-lbd、pfa-cmv-hvdr-lbd和pfa-cmv-hpxr-lbd。测序验证了所有融合受体的结构和序列。pfr-luc(stratagene)用作报告基因质粒,prl-sv40(promega)用于转染效率和细胞生长归一化。
分析方法:用高糖dmem(补充以10%fcs,1mm丙酮酸钠,100u/ml青霉素,100μg/ml链霉素)在37℃和5%co2条件下培养hek293t细胞。转染前一天,将hek293t细胞接种于96孔板(2.5·104细胞/孔)。转染前,将培养基改为opti-mem(无补充物)。根据生产商的方案,用lipofectamineltx试剂(invitrogen)瞬时转染pfr-luc(stratagene)、prl-sv40(promega)和pfa-cmv-hrxrα-lbd。转染5h后,将培养基换为opti-mem(补充以100u/ml青霉素,100μg/ml链霉素),现在,另外含有0.1%dmso和相应的测试化合物或仅含有0.1%dmso作为未处理对照。对每种浓度进行测试,一式三份,每个实验至少独立重复三次。根据生产商的方案,与测试化合物孵育过夜(12-14h)后,使用dual-glotm荧光素酶分析系统(promega)对细胞进行荧光素酶活性分析。使用tecaninfinitem200光度计(tecandeutschlandgmbh)检测发光值。用虫荧光素酶数据除以海肾萤光素酶数据,再乘以1000得到相对光单位(rlu),实现转染效率和细胞生长的归一化。用测试化合物的平均rlu除以相应浓度下的未处理对照的平均rlu,得到激活倍数。用相应浓度下的测试化合物的激活倍数除以各自的对照激动剂(pparα:gw7647;ppary:吡格列酮;pparδ:l165,041;lxrα/β:t0901317;rxrs:视黄醛;rars:维甲酸;vdr:骨化三醇;pxr:sr12813)在1μμ下的激活倍数,得到相对激活。所有的杂交分析都用上述参比激动剂进行了验证,得出的ec50值与文献相符。
fxr靶基因定量(实时荧光定量pcr)
如前所述,对fxr靶基因进行定量。15简言之,用hepg2细胞与测试化合物57(0.1μμ和1μμ)或1b(50μμ)或仅0.1%dmso(作为未处理对照)孵育培养8或16h,收获,用冷磷酸盐缓冲液(pbs)洗涤,然后直接用于rna提取。用总rna小型试剂盒(r6834-02,omegabio-tek公司,norcross,佐治亚州,美国)从hepg2细胞中提取到两微克总rna。根据生产商的方案,用高容量cdna反转录试剂盒(4368814,thermofischerscientific公司)将rna反转录成cdna。使用steponeplustmsystem(lifetechnologies,carlsbad,加利福尼亚州,美国),通过实时荧光定量pc分析r采用powersybrgreen(lifetechnologies;12.5μl每孔)对fxr靶基因表达进行评估。引物列于支持信息中。每个样品一式两份,并在至少三个独立的实验中进行重复。以甘油醛3-磷酸脱氢酶(gapdh)为参比基因,用对比δδct法定量表达水平。结果(以载体组表达水平为100%,表示为变化的平均值±sem%;n≥4):bsep:dmso:100;1b(50μm):557±28;57(0.1μμ):216±18;57(1μμ):222±20.shp:dmso:100;1b(50μμ):368±35;57(0.1μμ):242±61;57(1μm):317±78。cyp7a1:dmso:100;1b(50μμ):34±12;57(0.1μμ):50±7;57(1μμ):52±8。pparα:dmso:100;1b(50μm):289±59;57(0.1μμ):170±11;57(1μm):211±10。srebp1c:dmso:100;1b(50μμ):45±7;57(0.1μμ):49±17;57(1μm):36±12。fas:dmso:100;1b(50μμ):34±14;57(0.1μμ):22±8;57(1μμ):38±15。fgf19:dmso:100;1b(50μμ):407±42;57(0.1μμ):309±101;57(1μμ):325±77。pdk4:dmso:100;1b(50μμ):284±50;57(0.1μμ):255±54;57(1μμ):226±57。fabp1:dmso:100;1b(50μμ):249±17;57(0.1μμ):183±34;57(1μμ):194±42。cd36:dmso:100;吡格列酮(1μμ):353±43;57(1μμ):119±33;57(10μμ):129±42。fam3a:dmso:100;吡格列酮(1μμ):310±66;57(1μμ):112±12;57(10μμ):142±7。
细胞seh分析
根据zha等人所述进行细胞seh代谢活性的量化。53据此,将1μghepg2全细胞匀浆(稀释于含有0.1mg/mlbsa的100μlpbs)与不同浓度的57在37℃条件下孵育15min,n-环己基-n'-(4-碘苯基)尿素(ciu,10μm)36作为阳性对照或载体(最终浓度为1%的dmso)作为阴性对照。加入25ng(±)14(15)-eet-d11(caymanchemical,annarbor,美国)并在37℃条件下继续孵育10min。用同样处理过的pbs(含0.1mg/mlbsa)进行空白实验。加入100μl冰冷甲醇终止反应。在2000rpm,4℃,离心5min,用lc-ms/ms分析上清液并测定(±)14(15)-eet-d11和相应的(±)14(15)-dhet-d11的量。根据定量小鼠血浆中eet和dhet的程序(见下文),采用lc-ms对上清液中的(±)14(15)-eet-d11和(±)14(15)-dhet-d11进行定量。结果((±)14(15)-eet-d11/(±)14(15)-dhet-d11比值的平均值±sem;n=3):空白(无细胞):499±123;dmso(1%):63±16;ciu(10μμ):519±55;57:0.001μμ:191±7;0.01μμ:396±65;0.1μμ:442±7;1μμ:602±56;10μμ:614±96。
动物研究
动物和化合物应用:本研究选用9只雄性c57bl/6jrj小鼠(体重23-26g,购自法国janvierlabs)。动物被安置在温度控制室(20-24℃),并保持12小时光照/12小时黑暗循环。食物和水随意可用。生命期由委托研究机构pharmacelsus(saarbrücken,德国)完成。所有实验程序均由当地动物福利机构(landesamtfürgesundheitundverbraucherschutz,abteilunglebensmittel-undveter-
血液和肝脏取样:在六个时间点(57给药后15min、30min、60min、120min、240min和480min),从尾静脉对六只被束缚且清醒的小鼠取血(20μl)。在最后一个时间点(480min),用异氟醚麻醉小鼠,眶后穿刺取血(约500μl)。将一部分血液离心(6000rpm,10min,+4℃),获得用于定量eet/dhet比值的血浆,并在进一步评估前在-80℃条件下保存。在最后一次采血(给药后8h)后,用颈椎脱臼法处死小鼠,收集肝脏。整个肝脏立即快速冷冻并在进一步评估前在-80℃条件下进行保存。三只对照小鼠口服给予载体(1%hpmc/tween80(99:1))。用同样的方法获得血浆和肝脏。
血液样本中57的定量:校准样本:将测试品的储备溶液(1mg/ml,溶于dmso)用dmso稀释至终浓度00μg/ml(起始溶液)。用dmso稀释工作溶液得到进一步的工作溶液。
使用单个储备溶液制备校准标准品和qcs。用2.4μl工作液加20μl无药的空白血液制备校准标准品和qc。相应地,未知样本、零浓度样本和空白样本加入2.4μldmso。校准标准品和质控品一式两份。将含有内标物(灰黄霉素,600ng/ml)的40μl乙腈加入到22.4μl未知样本、零浓度样本、校准标准品和qc样本中。将无内标物的乙腈加入到空白样本中。所有样本(6000g)在室温条件下剧烈摇动并离心10分钟。用等体积水稀释无颗粒的上清液(50μl)。将试样转移到200μl样本管中,然后取15μl进行lcms分析。lc-ms分析:将hplc泵流速设置为600μl/min,用带有预置柱(kinetexphenyl-hexyl,securityguardultra,2.1mm)的kinetexphenyl-hexyl,2.6μm,50x2.1mm(phenomenex,aschaffenburg,德国)分析柱分离化合物。以水合0.1%甲酸为水相(a),含0.1%甲酸的乙腈为结合的有机层(b)进行梯度洗脱:%b(t(min)),0(0-0.1)-97(0.4-1.7)-0(1.8-3.0)。在正离子模式下,通过注射泵注入获得全扫描质谱,以鉴定质子化的准分子离子[m+h]+。自动调整最大化离子丰度,然后进行特征碎片离子鉴定,通用参数设置:离子迁移毛细管温度350℃,毛细管电压3.8kv,碰撞气体0.8毫巴氩气,保护气体、离子扫描气体和辅助气体压力分别为20、2和8(任意单位)。使用kinetica5.0软件(thermoscientific,waltham,美国)应用非隔室模型进行药代动力学分析。
小鼠肝脏中fxr靶基因mrna的定量:分离小鼠肝组织的肝细胞进行rt-qpcr:为了使肝脏样本均一化,将每个肝脏的三分之一放置在一个孔径为40μm的falcontm细胞过滤器(bdbioscience,erembodegem,比利时)上,并将该过滤器放置于50mlfalcon管中。用含10%fcs和1%链霉素的pbs缓冲液冲洗每个组织,并按压通过细胞过滤器,直到收集5ml细胞悬液。将悬液在4℃,1200rpm条件下离心10min。弃掉上清,用1mlpbs重悬得到的细胞颗粒,然后用e.z.a.总rna提取试剂盒(omegabio-tek公司,narcoss,佐治亚州,美国)按照动物组织协议提取总rna。提取的rna用于qrt-pcr,并按照上述从hepg2细胞定量mrna的方法(见上文)进行同等处理。小鼠基因的pcr引物列于支持信息。结果(表示为与载体对照100%相比,表达变化的平均值±sem%;57:n=6,载体:n=3):bsep:载体:105±24;57(10mg/kg):312±103。shp:载体:108±29;57(10mg/kg):410±147。cyp7a1:载体:106±25;57(10mg/kg):70±n。srebp1c:载体:82±14;57(10mg/kg):17±10。fgf15:载体:119±15;57(10mg/kg):254±g。fatp:载体:101±1;57(10mg/kg):105±5。
小鼠血浆样本的eet/dhet比值分析:(lc-ms/ms测定环氧二十碳三烯酸(eets)及其代谢物二羟环氧二十碳三烯酸(dhets)):使用液相色谱-串联质谱(lc-ms/ms)对提取样品中的8.9-eet,11.12-eet及其脱氢酶代谢物含量进行分析。lc-ms/ms系统包含api5500qtrap(absciex,darmstadt,德国),配备在负esi模式下工作的涡轮-v离子源,agilent1200二元hplc泵和脱气装置(agilent,waldbronn,德国)和配备了25μlleap注射器(axelsemraugmbh,sprockhovel,德国)的htcpal自动进样器(chromtech,idstein,德国)。质谱仪用高纯氮气由ngm22-lc/ms氮气发生器(cmcinstruments,eschborn,德国)生产。所有化合物均来自cayman化学(annarbor,密歇根州,美国)。在甲醇中制备具有2500ng/ml所有分析物的储备溶液。通过进一步稀释环氧二十碳三烯酸及其脱氢代谢物,得到浓度范围为0.1-250ng/ml的工作标准品。采用液-液萃取法提取小鼠血浆样本。将150μl样本/基质匀浆与20μl内标物(200ng/ml8.9-eet-d8和11.12-eet-d8甲醇溶液)轻轻混合,并用600μl乙酸乙酯萃取两次。制备标准曲线和质量控制样本,用150μlpbs代替150μl基质匀浆,再加入20μl甲醇,20μl工作标准品和20μl内标物。有机溶剂在45℃的温度下,在温和的氮气流下蒸发。残余物用50μl甲醇/水(50:50,v/v)重悬,在10000g下离心2分钟,在注入lc-ms/ms系统之前转移到玻璃瓶(macherey-nagel,düren,德国)。色谱分离采用gemininxc18柱和预柱(150mm*2mm内直径,粒径5μm,孔径
- 还没有人留言评论。精彩留言会获得点赞!