口服的卡利拉嗪固体制剂的制作方法
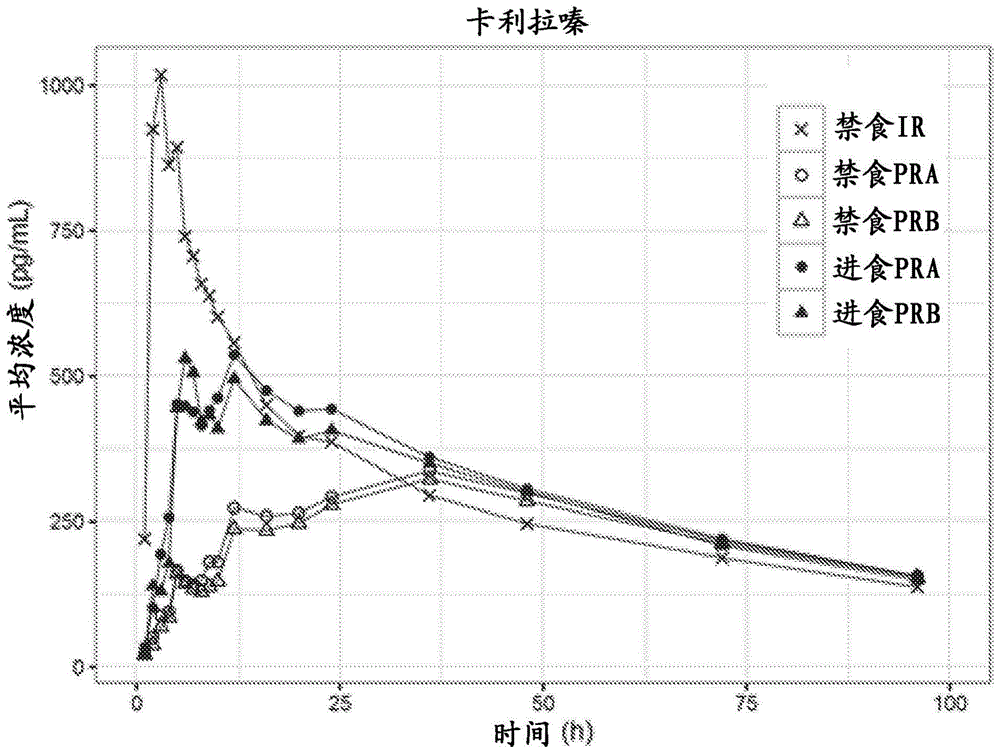
发明领域本发明提供了用于低于每日给药剂量的调释递送卡利拉嗪(cariprazine)(反式-n-{4-[2-[4-(2,3-二氯苯基)-哌嗪-1-基]-乙基]-环己基}-n’,n’-二甲基脲)或其药学上可接受的盐的口服药物组合物和方法。发明背景卡利拉嗪是多巴胺d3优先性d3/d2受体部分激动剂。wo2005/012266a1公开了卡利拉嗪及其药学上可接受的盐。该文件还公开了包含卡利拉嗪的盐酸盐或其他药学上可接受的盐的药物组合物,以及它们治疗和/或预防需要调节多巴胺受体的病理状况的用途,所述的病理状况例如精神病(例如精神分裂症、分裂情感障碍等)、药物滥用(例如酒精、可卡因、尼古丁、阿片类药物等)、伴有认知缺损的精神分裂症(包括阳性症状,例如妄想和幻觉、以及阴性症状,例如缺乏动力和回避社交以及认知症状,例如注意力和记忆的问题)、轻度至中度认知缺陷、痴呆、与痴呆相关的精神病状态、进食障碍(例如贪食症等)、注意缺陷障碍、儿童多动症、精神病性抑郁症、躁狂症、偏执和妄想障碍、运动障碍(例如帕金森病、精神安定药诱发的帕金森综合征、迟发性运动障碍)、焦虑症、性功能障碍、睡眠障碍、呕吐、攻击行为、孤独症。卡利拉嗪产生两种临床上相关的代谢物:脱甲基-卡利拉嗪(dcar)和双脱甲基-卡利拉嗪(ddcar)。由于该代谢物的体外受体特性和效能与母体化合物的相似,并且卡哌嗪、dcar和ddcar的血浆蛋白结合以及脑渗透也相似,因此这些部分的血浆暴露直接反映了它们对药物产品的体内药理作用的贡献。所有这些化合物都应一起被视为药物产品的活性药物成分。目前,只有卡利拉嗪盐酸盐的即释(ir)制剂可以作为药剂使用。wo2010/009309a1公开了该药物的稳定和可生物利用的即释药物组合物。根据wo2009/104739a1,已将卡利拉嗪盐酸盐的口服固体制剂开发为新的即释片剂剂型。卡利拉嗪盐酸盐的另外即释剂型,特别是具有优良特性的颗粒、细颗粒或粉末描述在ep16165247a1中。由于卡利拉嗪及其药学上可接受的盐的固体剂型的即释特性,它们的目前可利用的用途限于每日施用。患者必须施用该药剂的时间越长,对频率较低的给药方案的需求就越高,因为有效的长期疗法与患者的依从性密切相关,尤其是对于用不同中枢神经系统(cns)疾病(包括精神分裂症)的患者。几项研究直接将不依从性与较高的复发率、再次住院的次数增加和对家庭和医护系统的依赖性增加以及长期预后和功能恶化联系起来。根据现有技术,存在几种一般控制药物释放的机制,包括溶解,分配,扩散,渗透,溶胀,浸蚀和靶向。[j.siepmann等人(eds.),fundamentals和applicationsofcontrolledreleasedrugdelivery,advancesindeliveryscience和technology,doi10.1007/978-1-4614-0881-9_2,#controlledreleasesociety2012]。受控递药的方式取决于特定的应用,且它们中的一些可以组合并一起参与,或在最终控制机制的不同阶段参与。现有技术公开了几种不同的减少抗精神病药的给药频率的机制,例如调释口服制剂和长效可注射组合物。wo2008/038003a1公开了包含阿立哌唑的控释口服药物组合物。可以将所述组合物配制成扩散受控的制剂,溶出受控的制剂,易于施用的制剂,肠溶衣制剂,渗透泵技术制剂,防篡改制剂,侵蚀受控的制剂,离子交换树脂或上述的组合。us5910319b1专利公开了肠溶小丸形式的氟西汀肠溶制剂,其包含由氟西汀和一种或多种药学上可接受的赋形剂组成的芯;任选的分离层,其包含非还原糖和一种或多种药学上可接受的赋形剂;肠溶层,其包含乙酸琥珀酸羟丙基甲基纤维素和一种或多种药学上可接受的赋形剂;以及一个任选的修饰层。长效可注射(lai)抗精神病药的开发是治疗因不依从抗精神病药物而复发的精神分裂症患者的药理学策略,因为lai抗精神病药通过间隔两到四周注射施用,与每日口服抗精神病药不同。这些抗精神病药以长效形式上市销售:阿立哌唑(abilifymaintena);阿立哌唑lauroxil(aristada);氟奋乃静(prolixin);氟哌啶醇(haldol);双羟萘酸奥氮平(zyprexarelprevv);帕利哌酮(invegasustenna,invegatrinza)和利培酮(risperdalconsta)。除了长效可注射抗精神病药的已知优点外,还存在许多需要考量的关于临床实践的缺陷。其中一些更相关,例如特征感觉,注射部位的疼痛以及渗入皮下组织和/或皮肤引起刺激和损害(尤其是油性长效注射剂),以及更高的制造成本。因此,需要开发卡利拉嗪及其药学上可接受的盐的可经口施用的非即释(调释)药物制剂,该制剂能够降低给药频率,具有使得该组合物的使用频率低于每日施用频率的生物利用度值,这是一种用于终身治疗和/或预防上述病理状况的有效率、成本有效和便利的工具。需要开发一种新的组合物,与目前每日一次的常规即释(ir)剂量方案相比,该组合物允许以一个剂量施用更大量的卡利拉嗪,而不显著增加不良反应。通过减少间接的人力治疗费用(例如,减少监督药物施用所需的医务人员的时间),降低给药频率相对于目前的剂量方案提供了显著的药物经济学优势。另一方面,需要开发调释药物制剂,该制剂满足“剂量倾泻”的法规要求,改善患者依从性并通过更一致的血浆水平减少副作用,产生更有效的疗法。“剂量倾泻”是指全部剂量或其很大部分在短时间期限内快即释放。与药物施用适时相关的酒精饮料消耗导致的剂量倾泻被称作“酒精诱导的剂量倾泻”。特定的患者群体,例如患有以异常的社会行为为特征的精神障碍的人具有转向饮酒的趋势,作为应对他们的病情的方式。患有精神分裂症的人通常还有其他心理健康问题,例如焦虑症,重度抑郁症或药物滥用症。当改良药物释放时,如果通过控制性药剂在水-醇液体中的溶解破坏了释放控制,则可能发生剂量倾泻。[regulatoryconsiderationsforalcohol-induceddosedumpingoforalmodified-releaseformulations,pharmaceuticaltechnology,第38卷,第10期,pp40-46]因此,调释组合物必须为在治疗期间服用水-醇液体的患者提供安全的应用。另外,还需要提供简单的制备方法,该方法可以按比例放大到工业水平,并且该生产必须长期经济上可行。特别而言,调释产品能够以精确受控速率维持有效剂量,该精确受控速率与对应于所需要的血浆中药物治疗浓度的药物消除速率处于质量平衡,而没有任何不良作用;并且它还能够迅速在体内达到卡利拉嗪的治疗浓度,然后将该浓度维持给定的时间期限。我们的目标在于在长期疗法中以成本有效的方式获得令人满意的耐受性和便利的给药。熟知调释组合物作为口服贮库制剂确保较不频繁的给药方案,并且它适合提供有利的药物动力学特性。为了获得它,必须全面研究药物的药物动力学特性。药物动力学描述给药后人体如何通过吸收和分布机制影响药物以及该物质在体内的代谢变化,以及药物代谢产物的效应和排泄途径。化学品的药物动力学特性受施用途径和施用药物剂量影响。这些可能影响吸收速率。[inmosby'sdictionaryofmedicine,nursing&healthprofessions.philadelphia,pa:elsevierhealthsciences.retrieveddecember11,2008,fromhttp://www.credoreference.com/entry/6686418;jumpup^kathleenknights;bronwenbryant(2002).pharmacologyforhealthprofessionals.amsterdam:elsevier.isbn0-7295-3664-5]。为了开发调释口服药物组合物,必需考虑胃肠道的生理学、活性物质的理化性质、剂型的设计、药物释放机理和药物的生物学性质。为了使药物被吸收,首先它需要溶出,其次,它必须穿过膜;在口服药物的情况下,该膜为胃肠道上皮。在开发过程中必须考虑药物或其他成分在胃肠液中的溶出速率。已知胃肠道腔内的环境对药物溶出和吸收的速率和程度有重大影响。调释递送系统在胃肠道中的停留时间是旨在达到期望的生物利用度的关键因素,并且它受胃排空时间和肠运输时间二者影响。溶解是分子或离子从固态到溶液中的转移。在给定的一组实验条件下溶解进行的程度被称为溶质在溶剂中的溶解度。因此,物质的溶解度是在溶液与过量(未溶解)物质之间建立平衡时该物质进入溶液的量。[pharmaceutics,thescienceofdosageformdesign(2002);chapter1/p16]。吸收是药物运动进入血流。在人胃肠道的几个特征中,运输时间可能是非常可变的。因此,有必要选择适合的赋形剂以便提供期望的药物释放和吸收。许多生理因素,例如胃肠ph,酶活性,胃和肠运输速率,食物或任何种类的胃肠疾病(其通常影响常规口服剂型的药物生物利用度)也可能干扰口服调释形式的药物的溶出和吸收。此外,调释口服产品沿胃肠道的运输速率将单剂量给药后维持治疗反应的最大期限限制为约12小时。此外,应考虑吸收的药物持续发挥其治疗活性的时间长度。[pharmaceutics,thescienceofdosageformdesign(2002);chapter20/p294]另外,还必须考虑活性化合物通过胃肠道的溶解度特性。特别地,流体的ph值沿着胃肠道的长度变化很大。从胃的酸性通过弱酸性的十二指肠至小肠的实质上中性环境(其中ph值在5-8的范围内)存在自然的ph梯度。胃液是高酸性的;在禁食状态下的健康人中它被指定为在1-3.5范围内,并且在摄食后,胃液被缓冲至较不酸的ph。餐后典型的胃ph值在3-7范围内。由于胃酸被胰腺分泌到小肠的碳酸氢根离子中和,因此肠的ph值高于胃的ph值。从十二指肠到回肠,沿着小肠的长度,ph值逐渐升高[pharmaceutics,thescienceofdosageformdesign(2002);16章/p224–p225]。为了治疗功效,所有药物至少表现出有限的水溶性。因此,相对不溶的化合物可能表现出不稳定吸收或不完全吸收,因此使用更可溶的盐或其他化学衍生物可能是合适的。溶解度,尤其是在介质中的饱和度,在已经溶解在液体剂型中的药物的吸收中也可能是重要的,因为可能在胃肠道发生沉淀,并且可以改变生物利用度。酸性或碱性化合物的溶解度为ph依赖性的,可以通过形成盐形式来改变,不同的盐表现出不同的平衡溶解度。但是,与弱酸盐的溶解度相比,强酸盐的溶解度受ph变化的影响较小。在后一种情况下,当ph较低时,在一定程度上盐水解取决于ph和pka,导致溶解度降低。药物的微溶盐通过共同离子效应也可能发生溶解度降低。如果所涉及的离子之一以不同的更水溶性盐的形式加入,则可能超出溶解度积并且一部分药物沉淀[pharmaceutics,thescienceofdosageformdesign;(2002)第1章/p7]。如果弱酸性药物或该药物的盐的溶液的ph降低,则溶液中未离子化酸分子的比例增加。由于未离子化物质的溶解度小于离子化形式的溶解度,因此可能发生沉淀。相反,在弱碱性药物或其盐的溶液的情况下,ph值升高有利于沉淀。对于弱酸性和碱性药物的离子化,ph与离子化溶质的溶解度之间的这种关系极为重要,因为它们通过胃肠道并经历约1至8的ph变化。这将影响药物分子的离子化程度,进而影响它们的溶解度和它们吸收的能力。[pharmaceutics,thescienceofdosageformdesign(2002);第1章/p27]。当在溶液中电离的化合物与带相反电荷的抗衡离子形成强离子相互作用时,形成盐,导致盐形式结晶。所有酸性和碱性化合物均可参与成盐。盐形成为药物产品提供许多优势,因为它可以改善药物的溶解度,溶出速率,通透性和功效。形成盐的主要目的在于增加溶液中药物的量。药物的盐形式对药物的理化性质具有重大影响,影响其质量,安全性和性能。重要的是,不同的盐形式几乎不会改变药物的药理特性。对于弱碱,总浓度随ph的降低而增加,而对于弱酸,总浓度随ph的增加而升高。卡利拉嗪盐在酸性环境中非常易溶。但是,可溶于酸性环境的药物实际上可能不溶于中性或碱性环境。这与卡利拉嗪盐酸盐在ph1时溶解度为3.258mg/ml和在ph7时溶解度为0.001mg/ml的事实相符。根据溶解度研究,卡利拉嗪盐酸盐在ph值为3附近时显示出最佳的溶解度。在37℃下测得的值证明了卡利拉嗪盐酸盐的ph依赖性溶解度特征。ph溶解度[mg/ml]ph溶解度[mg/ml]13.257950.351028.93365.50.1488311.032160.018843.230370.0013表1:卡利拉嗪盐酸盐在37℃的ph-溶解度即释组合物(实施例4中所述)的溶出特性与卡利拉嗪盐酸盐的溶解度相对应,因为在ph5.5以上,药物的溶出显著降低。此外,生物相关溶出介质中存在表面活性剂—模拟进食前(禁食状态模拟肠液(fassif))和进食后(进食状态模拟肠液(fessif))的肠液—在较高的ph值下不会提高卡利拉嗪的溶出度(见表2和3)。表2:卡利拉嗪2.5毫克ir胶囊的溶出特性表3:卡利拉嗪25毫克ir胶囊的溶出特性因此,生产能够适当控制药物通过胃肠道释放的组合物显然不可行。溶解度是主要决定卡利拉嗪的生物利用度的因素,因为根据caco-2研究,它显示出高渗透性。在caco-2药物吸收模型中,卡利拉嗪在向内和向外方向的渗透系数分别被计算为26.4·10-6厘米/秒和51.2·10-6厘米/秒(渗透方向比(pdr):1.9)(arturssonp&karlssonj(1991)."correlationbetweenoraldrugabsorptioninhumansandapparentdrugpermeabilitycoefficientsinhumanintestinalepithelial(caco-2)cells".biochembiophysrescomm175(3):880–5和内部数据)。因此,明显的是该组合物可以改变释放特性,并且它高度依赖于活性物质的溶解度特性。因此,与在小肠和结肠中较慢甚或不完全药物释放相比,预期在胃中从仅含ph依赖性溶胀聚合物的常规系统卡利拉嗪盐酸盐的药物释放更快得多。为了找到卡利拉嗪及其药学上可接受的盐的适合递送系统,已经制备和评估了许多制剂。因此,本发明的目的在于提供包含卡利拉嗪盐与至少一种释放调节剂的口服药物组合物,所述的释放调节剂适合于降低cmax并使auc值维持在有效和可耐受的每日治疗剂量范围内,目的在于在所期望的施用频率下延长作用,该施用频率独立于胃肠道中药物释放的位置。附图简述附图中举例说明了本发明的示例性实施方案,其中相同的标记数字表示相同或相似的要素,并且其中:图1说明一次口服根据实施例13的ir、pra和prb组合物后的平均卡利拉嗪血浆浓度(pg/ml)。图2说明每4天口服6毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图3示例每7天口服10.5毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图4示例每4天口服12毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图5示例每4天口服18毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图6示例每14天口服21毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图7示例每4天口服24毫克根据实施例14的prb后的卡利拉嗪稳态模拟。图8示例每7天口服42毫克根据实施例14的prb后的卡利拉嗪稳态模拟。发明概述卡利拉嗪盐在酸性环境中非常易溶,并且现有技术教导,微环境ph调节或溶解度提高对实现从调释药物组合物中以ph依赖性溶解度为特征的活性成分的完全溶出是必需的。然而,在开发过程中令人惊讶地发现,这些复杂的方法是完全不必要的,并且简单的基质片制剂提供有利的药物动力学特性,因为它们能够降低cmax并将auc值保持在有效且可耐受的治疗每日剂量的范围内。本发明涉及一种用于调释卡利拉嗪或其药学上可接受的盐的可经口递送的固体药物组合物,其中该组合物包含治疗有效量的卡利拉嗪或其药学上可接受的盐和至少一种释放调节剂。本发明还涉及用于治疗和/或预防需要调节多巴胺受体的病理学状况的如上所定义的药物组合物,其中所述的治疗和/或预防包括以少于每日1次的频率施用所述的药物组合物。本发明还涉及如上所定义的药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括以少于每日1次的频率施用所述的药物组合物。本发明还涉及用于制备不同剂型的如上所定义的调释药物组合物的方法,其中该组合物通过本领域中已知的常规方法得到,包括将所述成分直接压制成片剂,任选地将其包衣;流化制粒,此后压制;以及挤出和滚圆所述成分,此后将得到的小球填入胶囊。本发明还涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括施用如上所定义的药物组合物。发明详述本发明提供一种用于调释卡利拉嗪或其药学上可接受的盐的可经口递送的固体药物组合物,该组合物用于治疗和/或预防需要调节多巴胺受体的病理学状况,其包含治疗有效量的卡利拉嗪或其药学上可接受的盐和至少一种释放调节剂。特别地,本发明涉及用于调释卡利拉嗪或其药学上可接受的盐的可经口递送的固体药物组合物,其中该组合物包含治疗有效量的卡利拉嗪或其药学上可接受的盐和至少一种释放调节剂,该释放调节剂适合于降低cmax并且将auc值维持在有效和可耐受的治疗每日剂量的范围内,剂目的在于以所期望的施用频率延长作用,该施用频率独立于胃肠道中药物释放的位置。在优选的实施方案中,本发明提供固体药物组合物,其包含约1.5毫克至约84毫克,包括约1.5毫克、约3毫克、约4.5毫克、约6毫克、约9毫克、约10.5毫克、约12毫克、约15毫克、约18毫克、约21毫克、约24毫克、约27毫克、约30毫克、约31.5毫克、约42毫克、约60毫克、约63或约84毫克药学上可接受的盐形式的卡利拉嗪。在更优选的实施方案中,本发明提供固体药物组合物,其包含约1.5毫克至约31.5毫克,包括约1.5毫克、约3毫克、约4.5毫克、约6毫克、约9毫克、约10.5毫克、约12毫克、约15毫克、约18毫克、约21毫克、约24毫克、约27毫克、约30毫克或约31.5毫克药学上可接受的盐形式的卡利拉嗪。在特别优选的实施方案中,本发明提供固体药物组合物,其包含约1.5毫克至约24毫克,包括约1.5毫克、约3毫克、约4.5毫克、约6毫克、约9毫克、约10.5毫克、约12毫克、约15毫克、约18毫克、约21毫克或约24毫克药学上可接受的盐形式的卡利拉嗪。在最优选的实施方案中,本发明提供固体药物组合物,其包含约1.5毫克至约12毫克,包括约1.5毫克、约3毫克、约4.5毫克、约6毫克、约9毫克、约10.5毫克或约12毫克药学上可接受的盐形式的卡利拉嗪。在本发明的另一个优选的实施方案中,所述的固体药物组合物包含大于1.5毫克药学上可接受的盐形式的卡利拉嗪。在本发明的另一个优选的实施方案中,所述的固体药物组合物包含至多84毫克药学上可接受的盐形式的卡利拉嗪。在另一个优选的实施方案中,本发明提供固体药物组合物,其包含约6毫克至约30毫克药学上可接受的盐形式的卡利拉嗪。在一个优选的实施方案中,本发明提供固体药物组合物,其包含约6毫克至约24毫克药学上可接受的盐形式的卡利拉嗪。在一个优选的实施方案中,本发明提供固体药物组合物,其包含约1.5毫克至约84毫克盐酸盐形式的卡利拉嗪。在一个更优选的实施方案中,本发明提供固体药物组合物,其包含约6毫克至约30毫克盐酸盐形式的卡利拉嗪。在最优选的实施方案中,本发明提供固体药物组合物,其包含约6毫克至约24毫克盐酸盐形式的卡利拉嗪。在本发明的优选的实施方案中,所述的固体药物组合物包含卡利拉嗪的药学上可接受的盐,其选自盐酸、硫酸、磷酸、甲磺酸、樟脑磺酸、草酸、马来酸、琥珀酸、柠檬酸、甲酸、氢溴酸、苯甲酸、酒石酸、富马酸、水杨酸、扁桃酸和碳酸的盐。在本发明的更优选的实施方案中,所述的固体药物组合物包含卡利拉嗪的药学上可接受的盐,其选自盐酸、氢溴酸和甲磺酸的盐。在本发明的最优选的实施方案中,所述的固体药物组合物包含卡利拉嗪盐酸盐。在本发明的优选的实施方案中,所述的固体药物组合物包含至少一种释放调节剂,其选自亲水性聚合物和疏水性聚合物。在本发明的更优选的实施方案中,所述的固体药物组合物包含至少一种亲水性聚合物作为释放调节剂。在本发明的更优选的实施方案中,所述的固体药物组合物包含至少一种基于纤维素的聚合物作为释放调节剂。在本发明的更优选的实施方案中,所述的固体药物组合物包含至少一种基于纤维素的聚合物作为释放调节剂,例如羟烷基纤维素,其选自羟丙基纤维素(hpc)、羟乙基纤维素(hec)、羟甲基纤维素和羟丙基甲基纤维素(hpmc)、羧甲基纤维素、羧甲基纤维素钠、甲基纤维素和羟乙基甲基纤维素。在本发明的最优选的实施方案中,所述的固体药物组合物包含至少一种基于纤维素的聚合物作为释放调节剂,例如羟烷基纤维素,其选自羟丙基纤维素(hpc)、羟乙基纤维素(hec)、羟甲基纤维素和羟丙基甲基纤维素(hpmc)。在本发明的更优选的实施方案中,所述的固体药物组合物包含至少一种疏水性作为释放调节剂。在本发明的优选的实施方案中,所述的固体药物组合物包含约15至约75%重量的至少一种释放调节剂。在本发明的更优选的实施方案中,所述的固体药物组合物包含约25至约65%重量的至少一种释放调节剂。在本发明的优选的实施方案中,如上所定义的固体药物组合物还包含单独或任何组合形式的其他赋形剂,其选自稀释剂,润滑剂,泡腾成分,粘合剂,制粒助剂,成膜剂和助流剂。在本发明的优选的实施方案中,所述的固体药物组合物被设计用于口服,包括但不限于片剂,胶囊,颗粒剂,散剂,微球,微丸和小丸。在优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供溶出特性,其中在4小时,卡利拉嗪总量的约25%至约70%在溶液中,在8小时,卡利拉嗪总量的约45%至约100%在溶液中,在12小时,卡利拉嗪总量的约65%至约100%在溶液中。在更优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供溶出特性,其中在4小时,卡利拉嗪总量的约30%至约65%在溶液中,在8小时,溶液中卡利拉嗪总量的约50%至约95%,在12小时,溶液中卡利拉嗪总量的约70%至约100%。在最优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供溶出特性,其中在4小时,卡利拉嗪总量的约35%至约60%在溶液中,在8小时,卡利拉嗪总量的约55%至约90%在溶液中,在12小时,卡利拉嗪总量的约75%至约100%在溶液中。在本发明的另一个优选的实施方案中,如上所定义的药物组合物表现出的口服后卡利拉嗪auc值为以相同剂量口服时使用卡利拉嗪的即释(ir)剂型实现的卡利拉嗪auc值的约60%至约145%。在本发明的更优选的实施方案中,如上所定义的药物组合物表现出的口服后卡利拉嗪auc值为以相同剂量口服时使用卡利拉嗪的即释(ir)剂型实现的卡利拉嗪auc值的约70%至约135%。在本发明的更优选的实施方案中,如上所定义的药物组合物表现出的口服后卡利拉嗪auc值为以相同剂量口服时使用卡利拉嗪的即释(ir)剂型实现的卡利拉嗪auc值的约80%至约125%。在本发明的更优选的实施方案中,如上所定义的药物组合物表现出的口服后卡利拉嗪auc值为以相同剂量口服时使用卡利拉嗪的即释(ir)剂型实现的卡利拉嗪auc值的约90%至约115%。在本发明的最优选的实施方案中,如上所定义的药物组合物表现出的口服后卡利拉嗪auc值为以相同剂量口服时使用卡利拉嗪的即释(ir)剂型实现的卡利拉嗪auc值的约95%至约105%。在另一个优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约40%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于卡利拉嗪母体以及脱甲基卡利拉嗪和双脱甲基卡利拉嗪总和的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在更优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约35%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在更优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约30%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在另一个更优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约25%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在另一个更优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约20%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在一个最优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供人口服后的pk特性,其中cmax为通过包含与所述调释药物组合物相同量的卡利拉嗪的ir制剂得到的cmax的约8%至约15%;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。在另一个优选的实施方案中,本发明涉及包含卡利拉嗪的药物组合物,其提供pk特性,其中cmax/auc0-∞在0.05-0.20h-1,例如0.08-0.17或0.10-0.15h-1的范围内;此时所述pk特性产生于在禁食过夜给药前至少8小时的人中进行的pk实验;其中所述pk特性基于总卡利拉嗪的血浆浓度;且其中所述的药物组合物包含治疗有效量的卡利拉嗪。本发明涉及如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括以低于每日1次的频率施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括以2-14天期限内施用1次所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每2天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每3天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每4天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每7天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每10天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中所述的治疗和/或预防包括每14天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中将所述药物组合物分成2-15个月剂量。在另一个优选的实施方案中,本发明提供如上所定义的药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理学状况,其中将所述药物组合物分成2、3、4、5、6、7、8、9、10、11、12、13、14或15个月剂量。在优选的实施方案中,本发明提供如上所定义的固体药物组合物,其用于治疗和/或预防需要调节多巴胺受体的病理性状况,例如精神病(例如精神分裂症,分裂情感障碍等),药物滥用(例如,酒精,可卡因,尼古丁,阿片类药物等),伴有认知缺损的精神分裂症(包括阳性症状,例如妄想和幻觉,以及阴性症状,例如缺乏动力和回避社交,以及认知症状,例如具有注意和记忆的问题),轻度至中度认知缺陷,痴呆,与痴呆相关的精神病状态,进食障碍(例如贪食症等),注意缺陷障碍,儿童多动症,精神病性抑郁症,躁狂症,偏执和妄想障碍,运动障碍(例如帕金森病,精神安定药诱发的帕金森综合征,迟发性运动障碍),焦虑症,性功能障碍,睡眠障碍,呕吐,攻击行为和孤独症。在更优选的实施方案中,本发明提供如上所定义的固体药物组合物,其用于治疗和/或预防精神分裂症和/或躁狂症。本发明还涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括以低于每日1次的频率施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括以2-14天期限内施用1次所述的药物组合物。在另一个优选的实施方案中,本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每2天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每3天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每4天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每7天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每10天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包括每14天1次施用所述的药物组合物。在另一个优选的实施方案中,本发明涉及本发明涉及如上所定义的固体药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中将所述药剂分成2-15个月剂量。在另一个优选的实施方案中,本发明涉及如上所定义的药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,其中所述的治疗和/或预防包含施用,其中将所述药剂分成2、3、4、5、6、7、8、9、10、11、12、13、14或15个月剂量。在优选的实施方案中,本发明涉及如上所定义的药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的病理学状况的药剂中的用途,所述的病理学状况例如精神病(例如精神分裂症,分裂情感障碍等),药物滥用(例如,酒精,可卡因,尼古丁,阿片类药物等),伴有认知缺损的精神分裂症(包括阳性症状,例如妄想和幻觉,以及阴性症状,例如缺乏动力和回避社交,以及认知症状,例如具有注意和记忆的问题),轻度至中度认知缺陷,痴呆,与痴呆相关的精神病状态,进食障碍(例如贪食症等),注意缺陷障碍,儿童多动症,精神病性抑郁症,躁狂症,偏执和妄想障碍,运动障碍(例如帕金森病,精神安定药诱发的帕金森综合征,迟发性运动障碍),焦虑症,性功能障碍,睡眠障碍,呕吐,攻击行为和孤独症。在更优选的实施方案中,本发明涉及如上所定义的药物组合物在制备用于治疗和/或预防需要调节多巴胺受体的精神分裂症和/或躁狂症的药剂中的用途。本发明还涉及用于制备不同剂型的如上所定义的调释药物组合物的方法,其中该组合物通过本领域公知的常规方法得到,包括将成分直接压制成片剂,任选地对它们进行包衣;流化制粒,此后压制;以及挤出滚圆成分,此后将得到的小球填入胶囊。在优选的实施方案中,本发明提供用于制备如上所定义的调释药物组合物的方法,它包括下列步骤:a)混合卡利拉嗪与适合的赋形剂,和b)将它们直接压制成片剂。在另一个优选的实施方案中,本发明提供用于制备如上所定义的调释药物组合物的方法,它包括下列步骤:a)在流化床设备中混合卡利拉嗪与适合的赋形剂;b)用溶于适合溶剂中的适合赋形剂喷雾该混合物;c)干燥颗粒;d)用适合的赋形剂覆盖颗粒;e)混合颗粒与适合的赋形剂;以及f)将得到的混合物压制成片剂。在另一个优选的实施方案中,本发明提供用于制备如上所定义的调释药物组合物的方法,它包括下列步骤:a)混合卡利拉嗪与适合的赋形剂;b)湿润得到的混合物;c)通过挤出形成圆柱状聚集物;d)打碎和通过滚圆将挤出物滚圆成圆形小球;e)干燥得到的小球;以及f)将小球填入适合的胶囊。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括对有此需要的患者以低于每日一次的频率施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括以2-14体内期限内施用1次如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每2天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每3天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每4天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每7天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每10天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明涉及治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中该方法包括每14天1次施用如上所定义的药物组合物。在另一个优选的实施方案中,本发明提供一种治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中将所述的药物组合物分成2-15个月剂量。在另一个优选的实施方案中,本发明提供一种治疗患有需要调节多巴胺受体的病理学状况的患者的方法,其中将所述的药物组合物分成2、3、4、5、6、7、8、9、10、11、12、13、14或15个月剂量。在优选的实施方案中,本发明涉及提供一种治疗患有需要调节多巴胺受体的病理学状况的患者的方法,所述的病理学状况例如精神病(例如精神分裂症,分裂情感障碍等),药物滥用(例如,酒精,可卡因,尼古丁,阿片类药物等),伴有认知缺损的精神分裂症(包括阳性症状,例如妄想和幻觉,以及阴性症状,例如缺乏动力和回避社交,以及认知症状,例如具有注意力和记忆的问题),轻度至中度认知缺陷,痴呆,与痴呆相关的精神病状态,进食障碍(例如贪食症等),注意缺陷障碍,儿童多动症,精神病性抑郁症,躁狂症,偏执和妄想障碍,运动障碍(例如帕金森病,精神安定药诱发的帕金森综合征,迟发性运动障碍),焦虑症,性功能障碍,睡眠障碍,呕吐,攻击行为和孤独症。在一个更优选的实施方案中,本发明提供一种治疗患有精神分裂症和/或躁狂症的患者的方法。除非本说明书中另有说明,否则术语“药学上可接受的盐”是指通过使作为碱起作用的主要化合物与无机酸或有机酸反应形成盐而获得的盐,例如,盐酸、硫酸、磷酸、甲磺酸、樟脑磺酸、草酸、马来酸、琥珀酸、柠檬酸、甲酸、氢溴酸、苯甲酸、酒石酸、富马酸、水杨酸、扁桃酸和碳酸的盐。药学上可接受的盐还包括主要化合物作为酸起作用并且与适当的碱反应形成例如钠、钾、钙、镁、铵和胆碱盐的那些。本领域技术人员将进一步认识到,可以通过许多已知方法中的任一方法使化合物与适当的无机酸或有机酸反应来制备酸加成盐。或者,可以通过多种已知方法通过使本发明的化合物与适当的碱反应来制备碱金属盐和碱土金属盐。此外,可以通过与无机酸或有机酸反应获得几种酸盐,即乙酸盐、己二酸盐、海藻酸盐、柠檬酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、硫酸氢盐、丁酸盐、樟脑酸盐、二葡萄糖酸盐、环戊烷丙酸盐、十二烷基硫酸盐、乙磺酸盐、葡庚糖酸盐、甘油磷酸盐、半硫酸盐、庚酸盐、己酸盐、富马酸盐、氢溴酸盐、氢碘酸盐、2-羟基-乙磺酸盐、乳酸盐、马来酸盐、甲磺酸盐、烟酸盐、2-萘磺酸盐、草酸盐、棕榈酸盐、果质酸盐、过硫酸盐、3-苯基丙酸盐、苦味酸盐、新戊酸盐、丙酸盐、琥珀酸盐、酒石酸盐、硫氰酸盐、甲苯磺酸盐、甲磺酸盐和十一酸盐。例如,药学上可接受的盐可以是盐酸盐、氢溴酸盐或甲磺酸盐。关于术语“可经口递送”,我们包括适合于口服的含义,包括经口和口内(例如舌下或颊粘膜)施用。优选,将本发明的组合物设计用于经口施用于患者,即通过吞咽(例如吃或饮用)。术语“少于每日一次”是指以低于每日1次(od)的频率施用的适合于调释剂量方案的组合物。关于低于od的频率的剂量方案,我们包括每2天和/或每3天和/或每4天和/或每5天和/或每6天和/或每7天和/或每8天和/或每9天和/或每10天和/或每11天和/或每12天和/或每13天和/或每14天1次,例如在2-14天期限内的任何时间1次剂量。换句话说,关于低于od的频率,我们包括将组合物分为2-15个月剂量,包括2、3、4、5、6、7、8、9、10、11、12、13、14或15个月剂量。如本说明书中所用,“生物利用度”是口服生物利用度,其是达到全身循环的不变药物的施用口服剂量的一部分。如本说明书中所用,化合物的“治疗有效量”意指在包括施用所述化合物的治疗干预中足以治疗、缓解或部分阻止给定疾病及其并发症的临床表现的量。治疗有效量不是固定不变的,特别取决于疾病及其严重程度以及待治疗患者的年龄、体重、身体状况和反应性。如本说明书中所用,“治疗”是指为对抗病症,例如疾病或障碍的目的而对患者的管理和护理,该术语旨在包括对患者所患的给定病症的全谱处理。例如,施用活性化合物以缓解症状或并发症,延缓疾病、障碍或病状的进展,缓解或减轻症状和并发症,和/或治愈或消除疾病、障碍或病状以及预防病症,其中预防还应理解为以对抗疾病、病症或障碍为目的的对患者进行管理和护理,包括施用活性化合物以预防症状或并发症的发作。在药物动力学领域,“曲线下面积(auc)”是血浆中药物浓度对时间作图的曲线下面积(数学上称为定积分)。典型地,在施用药物的时间开始且在血浆中浓度可忽略不计时结束,计算该面积。实际上,在某些离散的时间点处测量药物浓度,并使用梯形法则来估计auc。如本说明书中所用,“cmax”是在给药后以及在施用第二个剂量前,药物在身体的特定隔室或测试区域中达到的最大(或峰值)血清浓度。短期药物副作用最有可能在cmax或其附近发生。如本说明书中所用,药理学和医学中使用的“功效”一词是指在研究环境中可从制药药品获得的最大响应,以及对临床环境中足够治疗效果或有益改变的能力。如本说明书中所用,措词“稳态”是指药物输入的速率等于药物清除速率时的情况。如本说明书中所用,卡利拉嗪的“即释(ir)剂型”一词包括如下含义:例如在施用30分钟内,该剂型立即释放出其中所含的基本上全部卡利拉嗪及其药学上可接受的盐。该定义旨在包括在本说明书的简介页面中描述的卡利拉嗪的组合物,其目前用于治疗和/或预防需要调节多巴胺受体的病理学状况。如本说明书中所用,“调释片”是包含特殊赋形剂或通过特殊方法制备或它们二者的包衣片剂或未包衣片剂,被设计改变活性物质释放的速率、位置或时间。这包括延迟释放剂量,延长释放[er、xr、xl]剂量和靶向释放剂量。延长释放剂量由持续释放(sr)剂量组成,其在持续时间段内维持药物释放,但不是以恒定的速率;以及受控释放(cr)剂量,其在持续时间段内以几乎恒定的速率维持药物释放。与目前使用的在治疗剂量范围中应用的每日一次ir制剂相比,这种调释还可以在本发明组合物中伴有更高单剂量的卡利拉嗪。本发明的制剂被设计用于口服,包括但不限于片剂,胶囊,颗粒,粉剂,微球,微丸,小丸。为了获得改进的释放特性,可以用多种不同方式将配制治疗有效量的卡利拉嗪,包括但不限于溶出受控型制剂,扩散受控型制剂,基于渗透的制剂,基于离子交换的制剂和漂浮药物递送系统。本发明的组合物可以是溶出受控型制剂,包括但不限于包封的溶出系统和基质溶出系统。在包封的溶出系统(储库系统)中,可以通过改变围绕药物芯的聚合物膜的厚度和溶出速率来改变药物的释放。在基质溶出系统中,卡利拉嗪均匀地分布在整个聚合物基质中。在这些系统中,卡利拉嗪通过扩散机理释放,也可以基于所应用聚合物的性质释放。本发明的组合物可以是扩散受控型制剂,包括但不限于储库系统和整体装置。在储库系统中,卡利拉嗪被聚合物膜包围,在整体装置中,卡利拉嗪分布在聚合物基质中。储库系统可以是无孔膜储库或微孔膜储库;整体装置(溶液或分散体)可以是无孔基质或微孔基质系统。本发明的组合物可以是基于渗透的制剂,其中释放速率取决于释放介质的渗透压。本发明的组合物可以是基于离子交换的制剂,其中释放调节材料是离子交换树脂,其为包含离子基团的水不溶性聚合物材料,例如聚(苯乙烯磺酸)。还可以通过在具有小于胃液的堆积密度的堆积密度的漂浮药物递送系统中将卡利拉嗪递送至胃来改变药物释放速率,该系统长时间保持浮在胃中并且增加胃滞留时间(grt)。典型地,因为此类系统漂浮在胃液上时,卡利拉嗪以期望的速率缓慢释放,并且在释放药物后,残留系统从胃中排空,导致更好控制血浆药物浓度的波动。漂浮药物递送系统包括非泡腾系统和产气系统。非泡腾型漂浮系统包括双层压制胶囊,多层柔性片状药物装置,丙烯酸树脂空心微球,聚苯乙烯可漂浮壳,具有漂浮隔室和微孔隔室的单单元/多单元装置以及浮力控释粉末制剂,或浸入水中时膨胀至其脱水形式数百倍的水凝胶。由这些凝胶制成的口服药物递送制剂在胃中迅速膨胀,导致药物从胃到肠的移动更加缓慢,并被身体更有效地吸收。可以通过组合较高分子量脂肪醇或脂肪酸甘油酯和阻滞释放的聚合物和/或可溶胀的聚合物例如黄原胶和聚氧化乙烯的优化固体分散体,制备非泡腾型漂浮片。气体发生系统典型地使用泡腾成分:碳酸盐源,任选地酸源。与胃液接触时,这些成分形成co2,其被俘获在典型地与这些材料一起使用的聚合物基质中。这导致剂型的总密度降低,因此导致漂浮。漂浮剂型的酸源包括但不限于柠檬酸,酒石酸,苹果酸,富马酸,己二酸,琥珀酸;所述酸的酸酐;酸盐,包括但不限于磷酸二氢钠,焦磷酸二氢二钠和酸式亚硫酸钠以及酸、酸酐和酸盐的混合物。碳酸盐源包括但不限于碳酸氢钠,碳酸钠,碳酸氢钾,碳酸钾,倍半碳酸钠,甘氨酸碳酸钠及其混合物。也可以通过配制生物黏附多颗粒系统来实现调节的药物释放模式,该系统能够将药物保留在小肠道中以便防止小颗粒过早消除。适合的释放调节剂可以选自亲水性聚合物和/或疏水性聚合物和/或材料(脂质基质和不溶性聚合物基质)。亲水性聚合物的实例包括但不限于聚氧化乙烯(peo)、环氧乙烷-环氧丙烷共聚物、聚乙烯-聚丙二醇(例如泊洛沙姆)、卡波姆、聚卡波非、壳聚糖、聚乙烯吡咯烷酮(pvp)、聚乙烯醇(pva)、羟烷基纤维素例如羟丙基纤维素(hpc)、羟乙基纤维素(hec)、羟甲基纤维素和羟丙基甲基纤维素(hpmc)、羧甲基纤维素、羧甲基纤维素钠、甲基纤维素、羟乙基甲基纤维素、羟基丙基甲基纤维素、聚丙烯酸酯例如卡波姆、聚丙烯酰胺、聚甲基丙烯酰胺、聚磷腈、聚噁唑烷、聚羟烷基羧酸、海藻酸及其衍生物例如角叉菜胶海藻酸盐、海藻酸铵和海藻酸钠、淀粉和淀粉衍生物、多糖、羧基聚亚甲基、聚乙二醇、天然树胶例如瓜尔胶、阿拉伯树胶、黄蓍胶、刺梧桐树胶和黄原胶、聚维酮、明胶等。疏水性聚合物的实例包括但不限于基于烯酸的聚合物、基于甲基丙烯酸的聚合物和基于丙烯酸-甲基丙烯酸的共聚物。如本说明书中所用,短语“基于丙烯酸的聚合物”是指包括一个或多个包括和/或衍生自丙烯酸的重复单元的任何聚合物。如本说明书中所用,短语“基于甲基丙烯酸的聚合物”是指包括一个或多个包括和/或衍生自甲基丙烯酸的重复单元的任何聚合物。丙烯酸和甲基丙烯酸的衍生物包括但不限于烷基酯衍生物、烷基醚酯衍生物、酰胺衍生物、烷基胺衍生物、酸酐衍生物、氰基烷基衍生物和氨基酸衍生物。基于丙烯酸的聚合物、基于甲基丙烯酸的聚合物和基于丙烯酸-甲基丙烯酸的共聚物的实例包括但不限于:l100、l100-55、l30d-55、s100、4135f、rs、丙烯酸和甲基丙烯酸共聚物、甲基丙烯酸甲酯聚合物、甲基丙烯酸甲酯共聚物、聚甲基丙烯酸乙氧基乙酯、聚甲基丙烯酸氰基乙酯、甲基丙烯酸氨基烷基酯共聚物、聚丙烯酸、聚甲基丙烯酸、甲基丙烯酸烷基胺共聚物、聚甲基丙烯酸甲酯、聚甲基丙烯酸酐、聚甲基丙烯酸烷基酯、聚丙烯酰胺和聚甲基丙烯酸酐和甲基丙烯酸缩水甘油酯共聚物。与水接触形成在通过胃肠道的过程中保持完整的水化凝胶的亲水胶体为亲水性制剂的适合基质形成剂。亲水性胶体的实例包括纤维素衍生物、羟丙基甲基纤维素、羧甲基纤维素钠、海藻酸盐、黄原胶、聚丙烯酸聚合物。这些试剂的比例通常为组合物的20-80%,实际用量取决于药物和期望的释放时间。生物黏附剂和粘膜粘附剂是含有药物的聚合物材料,与水分或粘液化合物组合后具有粘附生物膜的能力。这些药物递送系统的主要优点在于它们延长在药物吸收部位的停留时间的潜力,因此它们可以降低调释药物制剂中的给药频率。这些剂型还可以增强其药物内容物与潜在粘膜屏障的接触,并改善药物通过粘膜的上皮运输,尤其是在药物吸收不良的情况下(ludwig,2005;lehr,2000)。合成聚合物,例如丙烯酸衍生物,卡波姆和聚卡波非;天然聚合物,例如角叉菜胶,果胶,阿拉伯胶和海藻酸盐;以及半合成聚合物,如壳聚糖和纤维素衍生物,可用于生物黏附制剂(deshpande等人,2009;grabovac等人,2005)。优选,在生物粘合剂中使用纤维素衍生物,尤其是纤维素醚。更优选使用非离子纤维素醚,例如乙基纤维素(ec),羟乙基纤维素,羟丙基纤维素(hpc),甲基纤维素(mc),羧甲基纤维素(cmc)或羟丙基甲基纤维素(hpmc)和阴离子醚衍生物,如羧甲基纤维素钠(nacmc)。本发明的组合物可包含增溶剂(例如聚乙二醇,多元醇,表面活性剂)和ph调节剂(例如柠檬酸,酒石酸)以促进活性成分的溶解。所述的组合物还可包含一个或多个包衣层:a)涂覆在芯上的包衣层,该包衣层是由至少一种包衣聚合物形成的内部密封包衣;b)第二包衣层,其设置在内部密封包衣上,由药物和至少一种包衣聚合物形成;以及任选地c)外部保护包衣层,其设置在第二包衣层上,由至少一种包衣聚合物形成。所述的包衣制剂可以包含至少一种包衣层材料和包衣溶剂,该溶剂优选为水,其用于加工和通过干燥除去。包衣层材料可以为二硬脂酸甘油酯;包衣层聚合物,例如羟丙基甲基纤维素,聚乙烯醇(pva),乙基纤维素,甲基丙烯酸聚合物或羟丙基纤维素。包衣层还可以任选地包括增塑剂,例如三醋精,邻苯二甲酸二乙酯,癸二酸三丁酯或聚乙二醇(peg),优选peg;以及抗粘着剂或助流剂,例如滑石粉,蒸气沉积二氧化硅或硬脂酸镁,遮光剂,例如二氧化钛。包衣层还可以包括基于氧化铁的着色剂。除上述成分之外,本发明的组合物还可包含适合量的其他药学上可接受的赋形剂,例如稀释剂,润滑剂,粘合剂,制粒助剂,成膜剂,着色剂和助流剂。可以以常规方式单独或以任意组合的方式使用这些赋形剂。示例性的润滑剂包括,但不限于硬脂酸钙、山萮酸甘油酯、硬脂酸镁、矿物油、聚乙二醇、硬脂富马酸钠、硬脂酸、滑石粉、植物油、硬脂酸锌及其组合。示例性的稀释剂包括,但不限于微晶纤维素,乳糖和淀粉。示例性的粘合剂包括,但不限于羟丙基纤维素,羟丙基甲基纤维素,乙基纤维素,甲基纤维素,羟乙基纤维素,糖,聚乙烯吡咯烷酮,聚乙烯醇,阿拉伯树胶粉,明胶,普鲁兰及其组合。示例性的助流剂包括,但不限于二氧化硅、滑石粉和淀粉。本发明的组合物可以用于治疗和/或预防需要调节多巴胺受体的病理学状况,例如精神病(例如精神分裂症,分裂情感障碍等),药物滥用(例如酒精,可卡因,尼古丁,阿片类药物等),伴有认知缺损的精神分裂症(包括阳性症状,例如妄想和幻觉,以及阴性症状,例如缺乏动力和回避社交,以及认知症状,例如具有注意和记忆的问题),轻度至中度认知缺陷,痴呆,与痴呆相关的精神病状态,进食障碍(例如贪食症等),注意缺陷障碍,儿童多动症,精神病性抑郁症,躁狂症,偏执和妄想障碍,运动障碍(例如帕金森病,精神安定药诱发的帕金森综合征,迟发性运动障碍),焦虑症,性功能障碍,睡眠障碍,呕吐,攻击行为,孤独症。因此,根据现有技术,在包含以ph依赖性溶解性为特征的活性成分的药物制剂的开发过程中;微环境ph调节或溶解度增强对于实现药物的完全溶解至关重要。考虑到卡利拉嗪的特性,本领域技术人员可以预期,对于调释制剂,需要一种具有额外添加剂(如ph调节剂)的复杂递送系统,以获得少于每日剂量方案同时保持与即释制剂的相同暴露的方案。因此,我们的目标在于提供一种卡利拉嗪制剂,该制剂提供非即释(调释)特性,作为口服储库制剂,具有有效且耐受性好,频率较低的非每日给药方案的潜力。如下文将更详细地描述,可以相对于它们的体外或体内释放特性或相关值例如cmax和auc来定义组合物的调释特性。在临床前研究中测试了几种包含ph调节剂和/或药学上可接受的酸和/或药学上可接受的生物黏附聚合物和/或药学上可接受的ph依赖性聚合物和/或旨在长期吸收的保留在胃肠系统中的任意成分的制剂。在研发的药物动力学阶段,在血浆样品中测试了多种制剂,这些血浆样品取自接受不同制剂的七只狗,并分析了卡利拉嗪浓度以比较口服制剂后暴露的速率(cmax)和程度(auc),以及tmax。在i期临床研究和所有调节释放中测试了两种不同的调释组合物和作为参比样品的即释胶囊。实施例下面参照实施例更具体地解释本发明。然而,本发明不限于这些实施例。一组开发的制剂(f1和f2)能够将药物保留在胃的酸性介质中延长的时间期限。可以通过漂浮递送装置来确保这种所谓的胃滞留,该漂浮递送装置保持漂浮在胃内容物上并且因此防止其穿过幽门。为了实现这种漂浮行为,以不同的分子量形式和数量测试了几种亲水性可溶胀聚合物和气体形成剂。这类聚合物还通过缓慢侵蚀且因此阻碍活性成分通过溶胀的凝胶层扩散而导致调释。从理论上讲,由于防止剂型过早从胃肠道中消除(在大部分活性成分可以被释放之前),开发的片剂的胃滞留特征也是有利的。通过使用海藻酸作为粘合剂和柠檬酸水溶液作为制粒液将活性成分制粒以确保形成气体促进早期漂浮,制备胃滞留型调释卡利拉嗪盐酸盐片剂。在最后步骤中将筛分的颗粒与控制释放的基质形成剂和其他赋形剂共混。此外,我们开发了生物黏附多颗粒系统(f3),该系统能够将药物物质保留在上胃肠道中以便防止小颗粒过早消除。所述小球包含弱酸和聚丙烯酸聚合物。此外,开发了即释制剂(f4)和基质制剂(f5、f6和f7)作为参比组合物。通过将卡利拉嗪与适合的赋形剂混合并将该混合物填入胶囊中,制备即释组合物。在基质制剂中,将卡利拉嗪包埋在赋形剂中,该赋形剂形成了一个非崩解性芯,称为基质。(溶解的)卡利拉嗪的扩散通过该芯发生。开发并测试了几种不同的基质制剂,即含有生物黏附聚合物的基质片剂,未包衣的和肠溶剂包衣的基质片剂,其包含非ph依赖性聚合物。实施例1:漂浮片(f1)通过流化制粒制备f1漂浮片,其中将卡利拉嗪与微晶纤维素和海藻酸在流化床设备中混合;然后用柠檬酸水溶液喷雾混合物。通过加热颗粒用二硬脂酸甘油酯覆盖干燥的颗粒。在最终的步骤中,将颗粒与外相(羟丙甲纤维素,碳酸氢钠,胶态无水二氧化硅,硬脂酸镁)混合,并使用旋转压片机设备将其压制成片剂。该组合物包含气体形成剂和释放调节剂以增加整个8小时溶出时间过程在胃中的停留时间。表4:f1漂浮片的定性和定量组成f1漂浮片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约15-35%,在4小时内释放不超过总卡利拉嗪的约50-70%,且在8小时内释放不少于总卡利拉嗪的约80%。溶出方法:设备nr.1(篮);介质-900ml0.001nhcl–运行时间8小时;温度:37±0.5℃;转速:50rpm。表5:f1漂浮片的溶出试验结果实施例2:漂浮片(f2)通过流化制粒制备f2漂浮片,其中将卡利拉嗪与微晶纤维素和海藻酸在流化床设备中混合;然后用柠檬酸水溶液喷雾混合物。通过加热颗粒用二硬脂酸甘油酯覆盖干燥的颗粒。在最终的步骤中,将颗粒与外相(一水合乳糖,羟丙甲纤维素,碳酸氢钠,胶态无水二氧化硅,硬脂酸镁)混合,并使用旋转压片机设备压制成片剂。该组合物包含气体形成剂和释放调节剂以增加整个8小时溶出时间过程在胃中的停留时间。表6:f2漂浮片的定性和定量组成f2漂浮片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约20-40%,在6小时内释放不超过总卡利拉嗪的约45-65%,且在12小时内释放不少于总卡利拉嗪的约75%。溶出方法:设备nr.1(篮);介质-900ml0.001nhcl–运行时间12小时;温度:37±0.5℃;转速:50rpm表7:f2漂浮片的溶出测试结果实施例3:包含生物黏附小球的胶囊(f3)通过下列步骤制备f3胶囊组合物:在高剪切混合器中将卡利拉嗪与微晶纤维素和聚丙烯酸聚合物混合;在用液体造粒后,将粒化的混合物挤出形成适合的圆柱形聚集物,然后将其滚圆成圆形小球。包囊前,将小球在流化床设备中干燥;将小丸筛分至目标粒度,并用滑石粉和硬脂酸钙润滑。将得到的小球填入硬明胶胶囊中。表8:用于胶囊的f3生物黏附小球的定性和定量组成f3胶囊表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在1小时内释放不超过总卡利拉嗪的约55-65%,在3小时内释放不超过总卡利拉嗪的约74-86%,且在6小时内释放不少于总卡利拉嗪的约85%。溶出方法:设备nr.1(篮);介质-900ml0.001nhcl–运行时间6小时;温度:37±0.5℃;转速:50rpm表9:f3胶囊的溶出试验结果实施例4:即释胶囊(f4)通过混合成分,然后将获得的混合物填充到硬明胶胶囊壳中制备该参比样品。表10:f4参比实施例的定性和定量组成该参比胶囊表现出体外释放特征,其中在放置于标准溶出度测试装置中后30分钟内平均释放超过总卡利拉嗪的约85%。溶出方法:设备nr.2(桨);介质-900ml0.001nhcl–运行时间30分钟;温度:37±0.5℃;转速:50rpm表11:f4参比胶囊的溶出测试结果实施例5:包含生物黏附聚合物的基质片(f5)通过下列步骤制备f5基质片:混合所述成分并且将它们直接混合压制成片剂,未使用制粒或辊压。通过筛器(开口尺寸:1.0mm)将卡利拉嗪盐酸盐、磷酸氢二钙和胶态无水二氧化硅一起过筛,然后将粉末与聚丙烯酸聚合物(卡波普974p)在双锥混合器中共混,并用硬脂酸镁润滑。使用旋转压片机设备将经润滑的粉末压制成片剂。表12:含生物黏附聚合物的f5基质片的定性和定量组成f5基质片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约35-45%,在4小时内释放不超过总卡利拉嗪的约60%,且在8小时内释放不少于总卡利拉嗪的约75%。溶出方法:设备nr.1(篮);介质-500ml0.001nhcl–运行时间8小时;温度:37±0.5℃;转速:50rpm。表13:f5基质片的溶出测试结果实施例6:包含非ph依赖性聚合物的基质片(f6)f6基质片通过混合所述成分并且将混合物压制成片剂制备,未使用制粒或辊压。将卡利拉嗪盐酸盐、微晶纤维素、胶态无水二氧化硅和一水合乳糖通过筛过筛(开口尺寸:1.0mm),然后将粉末与羟丙甲纤维素在双锥混合器中共混,并用硬脂酸镁润滑。使用旋转压片机设备将经润滑的粉末压制成片剂。表14:包含非ph依赖性聚合物的f6基质片的定性和定量组成f6基质片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约55-70%,在4小时内释放不超过总卡利拉嗪的约90%,且在8小时内释放不少于总卡利拉嗪的约95%。溶出方法:设备nr.1(篮);介质-500ml0.001nhcl–运行时间8小时;温度:37±0.5℃;转速:50rpm。表15:f6基质片的溶出测试结果实施例7:包含非ph依赖性聚合物的肠溶剂包衣的基质片(f7)未使用制粒或辊压,通过混合所述成分并且将混合物压制成片剂,制备f7基质片。将卡利拉嗪盐酸盐、微晶纤维素、胶态无水二氧化硅和一水合乳糖通过筛器(开口尺寸:1.0mm)一起过筛,然后将粉末与羟丙甲纤维素在双锥混合器中共混,并用硬脂酸镁润滑。使用旋转压片机设备将经润滑的粉末压制成片剂。用常规包衣方法用sureleasecleare-7-19040给片剂包衣。表16:包含非ph依赖性聚合物的f7肠溶剂包衣的基质片的定性和定量组成f7基质片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约45-55%,在4小时内释放不超过总卡利拉嗪的约70%,且在8小时内释放不少于总卡利拉嗪的约90%。溶出方法:设备nr.1(篮);介质-500ml0.001nhcl–运行时间8小时;温度:37±0.5℃;转速:50rpm表17:f7基质片的溶出测试结果体内研究的目的在于在对雄性比格(beagle)犬口服片剂施用后,提供含有卡利拉嗪的口服剂量制剂的比较药物动力学数据。此外,狗pk研究的目的在于从测试的原型中鉴别在人体生物利用度研究中进一步被评估的候选制剂。每组动物接受目标剂量水平为18毫克卡利拉嗪的单个口服片剂形式的卡利拉嗪制剂。完成了给药,没有任何偶发事件。下表18显示了所述制剂的药物动力学参数:表18:单剂量口服后卡利拉嗪的平均(cv%)pk参数(tmax和frel的中位值和最小值-最大值)(na:不适用)令人惊讶地,在不同制剂的frel(相对生物利用度,给定制剂的auc0-t与参比制剂的auc0-t之比)值之间没有检测到统计学显著差异,并且与f4参比ir胶囊相比每种所述的制剂均降低了cmax值。此外,关于与暴露有关的pk参数,也不能检测到差异。f1-f7制剂的临床前研究显示了比格犬中有利的pk结果。但是,为了满足人体临床试验的要求,必需开发用于i期研究的相似制剂。因此,开发并且在体外测试了不同类型的制剂(pra-e),特别是漂浮片、基质片和生物黏附小球,以找到用于i期研究的最适合组合物。实施例8:漂浮片(pra):通过流化制粒制备pra漂浮片,其中将卡利拉嗪与微晶纤维素和海藻酸在流化床设备中混合;然后用柠檬酸水溶液喷雾混合物。通过加热颗粒,用二硬脂酸甘油酯覆盖干燥的颗粒。在最终的步骤中,将颗粒与外相(羟丙甲纤维素、碳酸氢钠、胶态无水二氧化硅和硬脂酸镁)混合,并使用旋转压片机设备将其压制成片剂。在1.5至24毫克卡利拉嗪含量的范围内,不同pra制剂在定性上相同并且在定量方面它们比较地相似。所有不同pra制剂均具有相同的标称质量和定性组成。通过改变卡利拉嗪和微晶纤维素的量获得不同剂量强度。表19:1.5毫克至9.0毫克剂量范围内pra制剂的定性和定量组成表20:12毫克至24毫克剂量范围内pra制剂的定性和定量组成pra漂浮片表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约20-40%,在4小时内释放不超过总卡利拉嗪的约48-75%,且在8小时内释放不超过总卡利拉嗪的约80%。溶出方法:设备nr.2(桨);介质-900ml0.001nhcl溶液–运行时间12小时;温度:37±0.5℃;转速:50rpm。表21:不同pra制剂的溶出测试结果还在含有不同量乙醇的介质中检查了“酒精诱发的剂量倾泻”,发现药物的溶出没有改变。因此,该组合物为在治疗期间消费水-醇液体的患者提供安全应用。表22:pra制剂的剂量倾泻溶出测试结果溶出方法:设备nr.1(篮);介质1-500ml0.1nhcl溶液–运行时间2小时;介质2-500ml乙醇/hcl0.1n(5%)–运行时间2小时;介质3-500ml乙醇/hcl0.1n(20%)–运行时间2小时;介质4-500ml乙醇/hcl0.1n(40%)–运行时间2小时;温度:37±0.5℃;转速:50rpm。实施例9:包含非ph依赖性聚合物的基质片(prb)通过混合所述成分并且将该混合物压制成片剂制备prb基质片,未使用制粒或辊压。将卡利拉嗪盐酸盐、微晶纤维素、胶态无水二氧化硅和一水合乳糖通过筛器(开口尺寸:1.0mm)一起过筛,然后将粉末与羟丙甲纤维素在双锥混合器中共混,并用硬脂酸镁润滑。使用旋转压片设备将经润滑的粉末压制成片剂。在1.5至24毫克卡利拉嗪含量的范围内,不同的prb制剂在定性上相同并且在定量方面它们比较地相似。所有不同的prb制剂均具有相同的标称质量和定性组成。通过改变卡利拉嗪和一水合乳糖的量获得不同剂量强度。表23:1.5毫克至9.0毫克剂量范围内prb制剂的定性和定量组成表24:12毫克至24毫克剂量范围内prb制剂的定性和定量组成prb组合物表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在1小时内释放不超过总卡利拉嗪的约15-35%,在3小时内释放不超过总卡利拉嗪的约40-60%,且在12小时内释放不超过总卡利拉嗪的约75%。溶出方法:设备nr.1(篮);介质1-500ml0.1nhcl溶液–运行时间2小时;介质2-500ml乙酸盐缓冲液,ph=5.0–运行时间10小时;温度:37±0.5℃;转速:50rpm。表25:不同prb制剂的溶出测试结果在含有不同量乙醇的介质中,还检查了“酒精诱发的剂量倾泻”,发现药物的溶出度没有改变。因此,该组合物为在治疗期间消耗水-醇液体的患者提供安全应用。表26:不同prb制剂的剂量倾泻溶出测试结果溶出方法:设备nr.1(篮);介质1-500ml0.1nhcl溶液–运行时间2小时;介质2-500ml乙醇/hcl0.1n(5%)–运行时间2小时;介质3-500ml乙醇/hcl0.1n(20%)–运行时间2小时;介质4-500ml乙醇/hcl0.1n(40%)–运行时间2小时;温度:37±0.5℃;转速:50rpm。实施例10:包含非ph依赖性聚合物的基质片(prc)通过混合所述成分并将该混合物压制成片剂,制备prc基质片,未使用制粒或辊压。将卡利拉嗪盐酸盐、微晶纤维素和/或一水合乳糖和/或磷酸氢钙和胶态无水二氧化硅通过筛器(开口尺寸:1.0mm)一起过筛,并将粉末与羟丙甲纤维素或乙基纤维素在双锥混合器中混合,用硬脂酸镁润滑。使用旋转压片机设备将经润滑的粉末压制成片剂。任选地用任意常规方法将获得的片剂用欧巴代(opadry)和acryleze包衣。表27:prc制剂的定性和定量组成prc表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在1小时内释放不超过总卡利拉嗪的约15-35%,在3小时内释放不超过总卡利拉嗪的约40-70%,且在7小时内释放不超过总卡利拉嗪的约75%。溶出方法(11301,11302):设备nr.1(篮);介质1-500ml0.1nhcl溶液–运行时间2小时;介质2-500ml乙酸盐缓冲液,ph=5.0–运行时间4小时;温度:37±0.5℃;转速:50rpm。溶出方法(11406):设备nr.1(篮);介质1-500ml0.1nhcl溶液–运行时间2小时;介质2-500ml乙酸盐缓冲液,ph=5.5–运行时间12小时;温度:37±0.5℃;转速:50rpm。表28:prc制剂的溶出测试结果实施例11:包含生物黏附小球的胶囊(prd)在高剪切混合器中通过将卡利拉嗪与微晶纤维素和聚丙烯酸聚合物混合,制备prd胶囊;在用液体制粒之后,将成粒的混合物挤出形成适合的圆柱形聚集物,然后将其滚圆成圆形小球。在包囊之前,将小丸在流化床设备中干燥,然后将小丸筛分至目标粒度并用滑石粉和硬脂酸钙润滑。将获得的小球填入硬明胶胶囊中。表29:prd制剂的定性和定量组成prd制剂表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在1小时内释放不超过总卡利拉嗪的约15-45%,在3小时内释放不超过总卡利拉嗪的约48-80%,且在8小时内释放不少于总卡利拉嗪的约80%。溶出方法:设备nr.1(篮);介质-900ml乙酸盐缓冲液,其中ph=5.0–运行时间8小时;温度:37±0.5℃;转速:50rpm。表30:不同prd制剂的溶出测试结果实施例12:包含生物黏附小球的胶囊(pre)按照与prd胶囊(实施例11)类似的方式制备pre制剂。组合物之间的区别在于pre制剂不含任何电解质,例如cacl2,以便获得小球的更好弹性。表31:不同pre组合物的定性和定量组成pre制剂表现出体外释放特性,其中在放置于标准溶出度测试装置中后,平均在2小时内释放不超过总卡利拉嗪的约15-45%,在10小时内释放不超过总卡利拉嗪的约48-80%,且在16小时内释放不少于总卡利拉嗪的约80%。溶出方法:设备nr.1(篮);介质-900ml乙酸盐缓冲液,其中ph=5.0–运行时间8小时;温度:37±0.5℃;转速:50rpm。表32:不同pre组合物的定性和定量组成液体与固体材料之比以及尺寸、粒度分布和挤出机孔表面的光滑度一起显著决定了挤出物的质量。最终干燥确保粒丸硬度。已知该方法通过干扰相邻聚合物分子上的羧酸酯基团之间的相互作用,由此降低生物黏附能,降低聚丙烯酸的粘度,但是它显著降低了挤出物的弹性,这在滚圆步骤中是关键。为了获得最高的收率和球形度,需要优化水量、挤出速度、滚圆速度和时间。在存在电解质(例如氯化钙)的情况下,加工过程更容易,但是电解质对生物黏附和药物释放具有负面影响。此外,电解质的使用不能完全解决问题,因为小丸的形状不是球形,并且在加工过程中粘附引起团聚。此外,尝试了非离子型表面活性剂,以确保在长释放时间内卡利拉嗪在上肠道中完全溶出。令人惊讶地发现,使用改善溶解度的液体完全解决了粘附问题,并且可以获得球形小丸。本发明的组合物包含溶解度增强剂溶剂,其选自由以下各项组成的组:辛基己酰基聚乙二醇甘油酯、1,2,3-丙三醇、乳酸、月桂酰基聚氧乙烯甘油酯、聚氧乙烯甘油酯、聚氧化乙二醇、2-羟基丙醇。然而,制备生物黏附小球的方法复杂且经济上没有优势,因此它不是本发明的最优选实施方案。实施例13:单剂量i期研究基于临床前pk结果和经济考量,选择了两种不同类型的制剂(pra和prb)进行临床研究以将它们进行比较,因为它们具有最适合的auc(暴露)结果与低cmax值。单剂量i期研究旨在评估健康男性中上述两种调释制剂与即释制剂相比的药物动力学特性。下表33中给出了在健康男性志愿者中单剂量施用ir、pra和prb制剂后主要卡利拉嗪pk参数的描述性统计值的概述(cmax:最大观测血浆浓度;tmax:观察cmax的时间;auc0-t:从时间零到最后可量化浓度的血浆浓度-时间曲线下面积;t最终:最终可量化浓度的时间;auc0-∞:从时间零到无穷大(外推)的血浆浓度-时间曲线下面积;auc%外推:外推面积与auc0-∞的百分比;mrt0-∞:从时间零到无穷大(外推)的平均停留时间;t1/2:表观终末半衰期;cl/f:表观口服清除率;vz/f表观分布容积):表33:在对健康男性志愿者单次口服施用1.5毫克剂量的ir、pra或prb制剂后总卡利拉嗪的药物动力学参数的描述性统计值总体而言,两种延长释放制剂表现出总卡利拉嗪暴露(auc0-∞)与ir制剂的相当,同时延迟和降低了最大血浆浓度(cmax)。令人惊讶地发现两种延长释放制剂:位于酸性环境中的漂浮片和简单的基质片彼此非常相似。ir制剂的总卡利拉嗪的tmax值的中位值为3小时,而延长释放制剂(pra和prb)的中位tmax值延迟至36小时。ir制剂的总卡利拉嗪的平均(±sd)cmax为2.834(±0.902)nmol/l,延长释放制剂pra和prb的平均(±sd)cmax分别降低至1.027(±0.428)和0.950(±0.272)nmol/l。ir制剂的总卡利拉嗪的平均(±sd)auc0-∞值为329(±84)h*nmol/l,pr制剂pra和prb的平均(±sd)auc0-∞值分别为286(±70)和284(±86)h*nmol/l。附图1中显示了对健康男性志愿者单次口服1.5毫克剂量的ir、pra和prb制剂后总卡利拉嗪的平均血浆浓度–时间曲线。结果显示,在健康志愿者中,以pra片剂和prb片剂形式口服的单剂量卡利拉嗪1.5毫克得到了相似的pk。在禁食条件下两种pr片剂导致的卡利拉嗪系统暴露(auc0-∞或auc0-t)与ir胶囊的相当,而相较于ir胶囊,每种分析物的cmax更低,tmax更迟。在禁食和进食条件下两种pr制剂均显示出相当的系统暴露(auc0-∞)。总的说来,两种调释制剂具有相似的食物效应。根据现有技术,本领域技术人员将期望聚合物化合物和酸化剂和/或确保定位胃的试剂(例如碳酸盐源和/或生物黏附化合物)是开发包含卡利拉嗪的药物制剂的必要成分。与此相反,已经出乎意料地发现,不使用任何特殊添加剂的最简单基质片制剂仍然能够为卡利拉嗪盐酸盐组合物提供具有不依赖于ph的生物利用度的适合延长释放系统。不含任何ph调节剂和/或气体形成剂和/或生物黏附材料的基质片形式表现出与包含多种特殊添加剂的更复杂的复合系统相同的特性;即auc值没有降低且cmax值没有增加。考虑到两种不同缓释制剂的最重要pk参数相同,这些结果是令人惊讶的。得出的结论是,不含任何添加剂的简单prb组合物和简单制造工艺显示出所期望的调释卡利拉嗪制剂的特性,且实际上prb组合物挑战了更复杂的pra组合物。由于prb片剂实现了开发目标并很好地完成任务,因此对于包括卡利拉嗪盐酸盐或其药学上可接受的盐的新剂量方案的完美操作,不需要使用更复杂递送系统例如漂浮片或生物黏附小球。因此,这种开发带来了真正的惊喜,因为证明了不需要开发复杂的胃滞留漂浮系统和在不同的递送系统中使用生物黏附性聚合物、ph依赖性赋形剂和ph调节剂,因为令人惊讶的是prb片符合所有预期。同时,在要求的稳定性测试中,应用更复杂的递送系统通常容易受到影响并且对技术的要求更高(由于使用了特殊添加剂、困难制造和设备系统),而它们不能提供任何显著益处。从临床观点来看,口服制剂的较低频率(少于每日)施用的是有利的,特别是长期针对包括精神分裂症在内的中枢神经系统的疾病等。为了达到这个目标,需要卡利拉嗪的调释制剂,其具有ir胶囊的几乎相同的系统暴露且cmax不高于ir胶囊的cmax。几种调释系统的分析测试、临床前研究和临床研究的所有结果得出的结论是,使用不含任何特殊试剂的简单基质组合物良好地实现了具有低于每天给药具有适合系统暴露的调释卡利拉嗪制剂的所期望的特征。我们发现这些结果是最出乎意料和令人惊讶的结果。实施例14:药物动力学建模卡利拉嗪的即释(ir)制剂典型地以低剂量(例如1.5–6毫克/天)施用,并以随时间递增的频率和剂量进展性地施用,以达到治疗有效的稳态血清浓度。根据fda批准的标签,卡利拉嗪的即释(ir)制剂首先以每天1.5毫克的剂量给予受试者。使用包含更高剂量的卡利拉嗪的调释制剂,可以在不使用剂量递增方案的情况下,基本上更快地达到治疗有效的稳态浓度,但在开发的此阶段这还是不可接受的。因此,即使所施用的剂量大于即释制剂,与即释制剂相比,调释制剂的cmax降低。为了确定最适合的制剂,使用模拟程序计算了本发明药物组合物的药物动力学血液分布。在该模型中,基于对健康志愿者施用1.5毫克/天单剂量,预测了施用更高剂量调释卡利拉嗪制剂的pk参数。使用实施例13中所述的制剂和溶出特性,以及单次施用卡利拉嗪产生的血清浓度,使用药物动力学软件gastroplustm计算了auc和cmax值,以便预测与相应的ir剂量相比,生理学和生物化学过程对使用不同剂量和方案的调释制剂的口服药物生物利用度的影响。gastroplustm软件用于模拟比临床研究中施用的更高剂量的卡利拉嗪的血浆浓度。gastroplustm是高级软件程序,其可模拟人和临床前物种中经由静脉、口、眼和肺途径给药的药物的吸收、药物动力学和药效学。基本模型是高级隔室吸收和运输(acat)模型。自1997年以来,simulationsplus已将acat模型发展到高度精细化,为业界提供了最准确、灵活和强大的模拟程序。对于模型建立,测定了物理化学(pka,溶解度-ph数据,包括生物相关的溶解度,logp,caco2细胞的通透性,分布的粒径)和生物药剂学参数(时间-血浆浓度曲线,血液/血浆浓度比,血浆未结合分数(%))。为测定包括清除率在内的药物动力学参数,通过在时间血浆浓度曲线上的拟合二室模型(使用gastroplus软件中的pkplus模块),得到了分布容积、k12和k21速率常数,以确定时间-体内释放(%)特性。使用了gastroplus软件中的ivivc模块。表34显示了在31天间隔中测定的gastroplustm模拟的结果。表34:gastroplustm模拟(31天间隔)。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1