一种基于深度学习的植物蛋白质互作网络构建方法与流程
本发明涉及生物技术领域的深度学习技术,尤其是涉及一种基于深度学习的植物蛋白质互作网络构建方法。
背景技术:
蛋白质相互作用在细胞的生物过程中必不可少,大部分基因在蛋白质水平通过与其它蛋白质的相互作用来行使其生物学功能。后基因组时代的到来为在全基因组范围预测蛋白质相互作用提供了丰富的数据信息,且随着高通量实验技术、生物信息学的发展,大大提高了复杂生物网络的研究进展。
构建分类模型需要利用统计学、机器学习等方法从大量数据中提取有价值的信息,该过程包括了对数据的预处理、分类和异常检测等方面。随着生物数据的爆炸式增长,许多机器学习方法经过改进后可以适用于生物数据的分析,帮助我们从多个组学数据中提取有效信息。其中,深度学习是机器学习研究中的一个新领域,其动机在于建立、模拟人脑进行分析学习的神经网络,模仿人脑的机制来解释数据,比如图像,声音和文本等。目前深度学习已成功应用于计算机视觉、语音识别、记忆网络、自然语言处理等其他领域。然而针对植物蛋白质互作网络构建的研究,大多使用如决策树、朴素贝叶斯、支持向量机及随机森林等传统机器学习手段进行建模,利用深度学习方法搭建蛋白质互作分类模型的研究少之又少,大大限制了提高预测精度的可能性。
此外,深度学习模型参数量大,所需训练数据多,导致模型复杂,计算量极大,且易造成训练过拟合现象。因此,如何快速选择最优参数组合,在减少训练任务前提下大大提高模型预测精度成为将深度学习广泛应用于复杂生物网络构建的主要研究问题。
技术实现要素:
本发明的目的就是为了克服上述现有技术存在的缺陷而提供一种基于深度学习的植物蛋白质互作网络构建方法。
本发明的目的可以通过以下技术方案来实现:
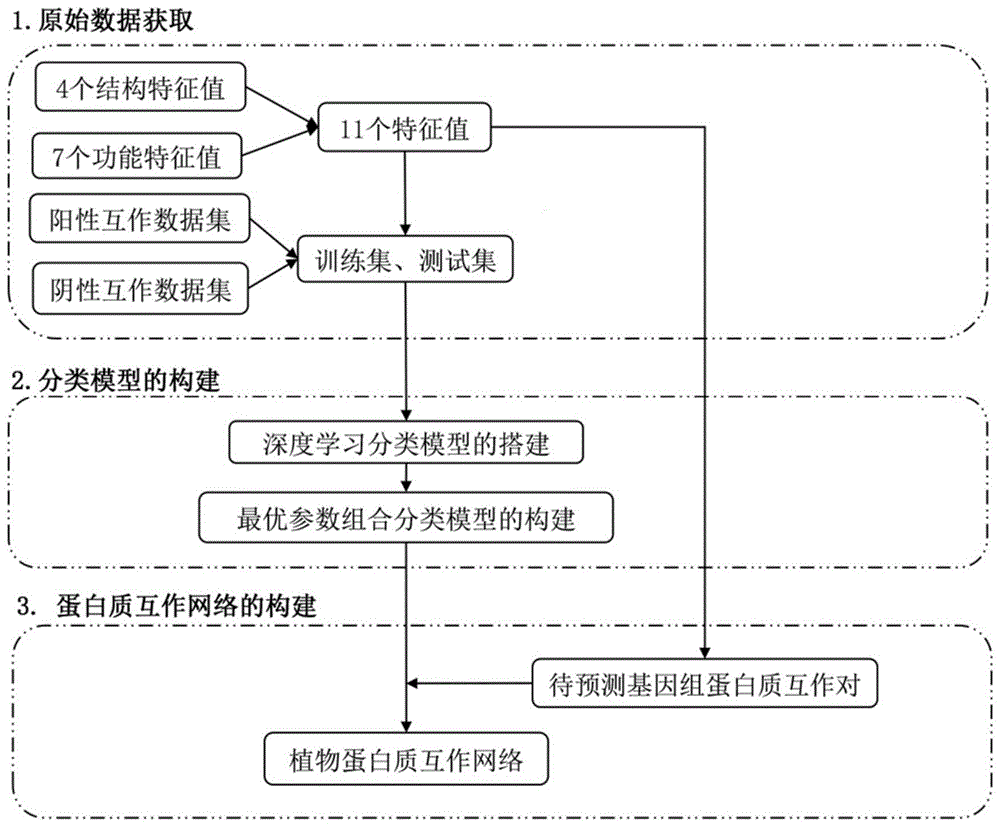
一种基于深度学习的植物蛋白质互作网络构建方法,包括以下步骤:
1)获取蛋白质互作对11个特征数据;
2)筛选获得训练集及测试集;
3)构建深度学习分类模型;
4)对深度学习分类模型的参数进行批量优化,获得最佳优化参数组合的分类模型;
5)根据最佳优化参数组合分类模型对全基因组所有可能两两互作蛋白对进行互作关系预测;
6)根据互作关系预测结果构建蛋白质互作网络。
所述的步骤1)中,特征数据包括4个结构特征信息和7个功能特征信息,所述的结构特征信息包括蛋白质同源模型与复合体模板之间的rmsd值、tmscore值、互作界面保守残基数目以及保守残基比例,所述的功能特征信息包括基因共表达、基因功能相似性、基因系统发生谱、蛋白质相互作用跨物种保守性以及基因融合信息,所述的基因功能相似性包括细胞组分、分子功能和生物过程。
所述的步骤2)具体为:
从多个蛋白质互作数据库中获取蛋白质阳性互作数据,将筛选出的严谨阳性数据集与不互作蛋白的阴性数据集以不同比例进行合并构成训练集,剩余阳性数据集与阴性数据集以不同比例合并构成测试集,其中,严谨阳性数据集的筛选标准为:
蛋白质互作数据由低通量实验支持或至少经过两次不同的独立高通量实验证据支持。
所述的步骤2)中,蛋白质互作数据库包括biogrid、intact、dip、mint及bind数据库。
所述的步骤3)中,深度学习分类模型为采用keras搭建的深度学习模型,其模型类型为序贯模型。
所述的步骤4)中,对深度学习分类模型的参数进行批量优化具体为:
在scikit-learn模型中采用网格搜索进行优化,需要调节的参数包括批尺寸、训练周期、优化算法、学习速率、动量因子、网络权值初始化、神经元激活函数、dropout正则化及隐藏层中神经元数量。
所述的步骤5)中,根据最佳优化参数组合的分类模型对全基因组所有可能两两互作蛋白对进行互作关系预测的筛选阈值不小于0.5,该阈值根据频率累计mpqs分数获得。
所述的步骤6)中,采用cytoscape软件构建蛋白质相互作用网络。
与现有技术相比,本发明具有以下优点:
在现有技术的基础上,提出了利用深度学习手段预测蛋白质互作可能的方法,并采用批量优化参数的手段降低调参工作量,在提高建模效率的前提下增加了蛋白质互作关系的预测准确性,构建所得蛋白质互作网络对未知蛋白质功能的试验验证提供了可靠预测手段。
附图说明
图1为本发明的方法流程示意图。
图2为深度学习模型搭建的流程示意图。
图3为深度学习模型搭建中批量优化参数的流程示意图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明。
实施例
如图1所示,以拟南芥全基因组水平的蛋白质互作网络构建为例,本发明提供一种基于深度学习蛋白质互作网络构建方法,具体包括以下步骤:
(1)同源建模、空间结构比对及结构特征计算
从modbase数据库中收集拟南芥蛋白质的同源结构模板并按照以下标准进行筛选:mpqs(modpipe质量得分)>=0.5;或ga341>=0.5;或e-value<0.0001;或z-dope<0。此外,从pdb和pisa两个数据库中收集同源或异源蛋白质复合体空间结构数据,利用pibase软件包计算得到蛋白质复合体各链间互作界面的结构及对应互作残基。随后采用modeller软件对拟南芥基因组所有编码基因进行蛋白质同源建模,并用tm-align软件对蛋白质同源模型与复合体模板进行结构叠加。将tm值控制在0.4以下,最终可以获得超过8800万个同源模板-链对比。最终,对蛋白质结构同源模型和模板复合物进行结构比对,计算蛋白质同源模型与复合体模板之间的rmsd值、tmscore值、互作界面保守残基数目以及保守残基比例作为结构特征。
(2)功能特征信息计算
功能特征信息为基因共表达、基因功能相似性、基因系统发生谱、蛋白质相互作用跨物种保守性以及基因融合等信息,
基因共表达特征值的具体计算方法为:从ncbi的sra数据库中收集拟南芥的所有实验设计结果,利用fastq质控编码检测程序筛选质控文件字符编码形式为33位的数据,并建立拟南芥参考基因组的bowtie2索引文件并下载拟南芥基因坐标文件,随后针对单双末端采用不同的tophat2与htseq-count指令,将rna-seq数据映射到拟南芥参考基因组上并计算基因表达量。利用坐标文件以及脚本文件算出每个位点基因片段长度,和所有基因组各个位点的rpkm值。最终通过比较计算两两基因表达丰度,计算其皮尔逊相关系数,以此代表表达谱之间的相似程度。
基因同源映射特征值的具体计算方法为:从biogrid、intact、dip、mint及bind五个数据库中收集大肠杆菌、酵母、线虫、果蝇、小鼠以及人6个物种的蛋白质互作数据。下载拟南芥及以上六种模式生物的基因组编码蛋白序列,并根据inparanoid方法计算获得的与上述物种直系同源的拟南芥基因,最终计算代表拟南芥基因之间互作可能性大小的interolog分值。
基因系统发生谱特征值的具体计算方法为:从kegg数据库中下载基因组完整且注释全的物种的蛋白质序列,对已完成测序的拟南芥基因组与所有编码蛋白质序列进行blast比对(e<10-10),如有匹配序列为1,反之为0,结果产生一个代表同源序列存在与否的n维向量的基因系统进化谱。
基因融合特征值的具体计算方法为:将拟南芥2.7万个序列与nr蛋白质数据库中所有物种的基因序列进行blast比对(e<10-10)。若两个互为非同源的拟南芥蛋白质与第三个其他物种蛋白质序列部分有至少70%的序列同源,则第三种蛋白质被称为rosettastone蛋白质。两个拟南芥蛋白质的互作可能p值由公式计算得出:
基因功能相似性特征值的具体计算方法为:通过go数据分别计算两两基因在三大本体(molecularfunction、biologicalprocess及cellularcomponent)中的最小共享生物途径中所含基因数。基因功能相似性特征值s的计算公式为:
(3)拟南芥蛋白质互作数据收集
从biogrid、intact、dip、bind以及mint五个公开数据库中收集整合拟南芥蛋白质相互作用数据集,获得阳性数据集。按照以下要求对严谨阳性数据集进行筛选:蛋白质互作数据需由低通量实验支持,或至少经过两次不同的独立高通量实验证据支持。不同的独立实验情况包括:由不同文献报道或同一文献中多种实验方法支持。经筛选后获得的严谨阳性数据集与不含互作蛋白对的随机数据构成的阴性数据集以不同比例进行合并构成训练集,剩余阳性数据集与阴性数据集以不同比例合并构成测试集;
(4)深度学习模型的搭建
使用keras搭建深度神经网络具体流程如图2所示。该深度学习模型采用了序贯模型,随后构建网络层的输入层、隐藏层和输出层,编译后给定输入数据集并对模型进行测试与验证,得到一个深度学习的初始分类模型及对该模型的评估结果。其中该模型的参数初始值设置为:批尺寸为20,训练周期为100,优化算法为sgd,学习速率为0.01,动量因子为0.0,网络权值初始化为uniform,神经元激活函数为sigmoid,dropout正则化为0.5,最大范数权值约束为0,隐藏层内神经元的个数为3个。随后采用scikit-learn模型中的网格搜索功能对该初始模型进行参数的批量优化,默认全部参数调优使用的是scikit-learn的三折交叉验证。首先设置批尺寸和训练周期的取值列表,各值单调递增,随后采用网格搜索方法,在其他参数不变的前提下,得到这两个参数的不同取值对模型效果的影响评估。取得效果最好的批尺寸和训练周期参数值后,在模型中固定该参数,随后继续采用网格搜索办法搜索训练优化算法的最佳选择。按照如图3所示顺序依次得到各个参数的最佳选择。最终该拟南芥蛋白质互作分类模型的最佳参数组合为:批尺寸为10,训练周期为100,优化算法为rmsprop,学习速率为0.001,动量因子为0.0,网络权值初始化为uniform,神经元激活函数为linear,dropout正则化为0.0,最大范数权值约束为3,隐藏层内神经元的个数为4个。最终获得最佳参数组合的深度学习分类模型并保存该模型。
(5)利用最优分类模型对拟南芥基因组2.7w个蛋白质基因的所有可能两两互作蛋白对进行关系预测,其筛选阈值>=0.5,将获得的预测结果利用cytoscape软件构建最终拟南芥基因组水平蛋白质相互作用网络。
- 还没有人留言评论。精彩留言会获得点赞!