一种基于计算机模拟获取可调控的TRPV5变异体的方法与流程
本发明属于分子动力学计算机模拟技术领域。更具体地,涉及一种基于计算机模拟获取可调控的trpv5变异体的方法。
背景技术:
瞬态感受器电位阳离子通道蛋白(transientreceptorpotentialcationchannels,trpchannels)是一类广泛存在于生物体的阳离子通道蛋白,其中trpv5是瞬时性受体电位(trp)通道超家族成员中trpv亚家族通道中一员,是该家族蛋白中子类为v,成员5的阳离子通道蛋白。该通道蛋白由729个氨基酸构成,分子量约为83kda。相应的基因由在7q34染色体排列的15个外显子组成,主要分布于肾脏、破骨细胞、胎盘等。其中,肾小管中分布较高,主要表达于远曲小管和收集小管中的肾上皮细胞的顶端细胞膜中,另在破骨细胞中也有表达,其表达量虽不如在肾脏的高表达,但也提示trpv5是介导破骨细胞骨吸收的重要通道蛋白。研究证实其与肾脏中ca2+主动重吸收相关,trpv5是trp家族中一个独特的钙选择性离子通道。首先,当该通道打开时,trpv5允许尿液中的钙离子沿浓度梯度进入细胞中,钙离子通过trpv5进入细胞后,结合到钙结合蛋白d28k上弥散到基底外侧膜。对小鼠的研究中敲除相关trpv5基因的小鼠都出现了不同程度的体内离子浓度失调和尿液含钙过高的情况。而对不同的人群的研究也表明,非裔美国人的低肾结石风险也与trpv5的表达紧密相关。因此肾脏表皮细胞中的trpv5钙离子主动再吸收过程对人体起重要作用。对trpv5的机理探讨、结构修饰与调控等研究将有利于促进现代医学对肾脏疾病的认知,为治愈钙离子失衡所导致的疾病提供新的研究角度。
目前,多使用益康唑(econazol)、携钙素(calmodulin,cam)与trpv5出口处残基结合,通过溶胶电泳等实验手段对trpv5的钙离子传导进行调节,研究trpv5的结构、电生理特性及调控因素。但是实验研究具有一定的局限性,影响导向较为单一,且逆反应可操作性较差,益康唑、携钙素等物质一旦与trpv5出口结合就不容易脱离,且往往需要多个化合物与残基进行结合才可达到理想的调控效果,成本较高。对trpv5通道蛋白的变异体研究由于实验繁杂,培养时间长,不易于操作,成为了trpv5的研究瓶颈。此外,由于trpv5传导钙离子的过程非常短暂,实验上难以捕捉离子传导过程,无法连续性观测trpv5的动态调控过程,使得对trpv5的传导性能和蛋白质结构设计研究受限。
利用计算机模拟构建蛋白质结构模型的方法可以节省实验成本,效率高。但通过计算机模拟蛋白质工程和酶催化残基磷酸化修饰技术来构建trpv5通道蛋白变异体结构的研究未见报道。理论上,可以基于反应分子动力学方法通过计算机模拟蛋白质与离子反应过程从而构建离子摄入过程的复杂蛋白变异体最终结构,但由于变异体数量较多,仅使用变异体的最终结构进行比较,难以全面评估不同变异体的性能,同时存在离子传导连续状态难以观测、反应亲和力不可控、模拟时间长、计算量大、速度慢等技术难题,不利于trpv5在生物化学、蛋白质工程学等学科进一步研究。
技术实现要素:
本发明的首要目的是克服上述现有技术的缺陷和不足,提供一种基于计算机模拟获取可人工调控的trpv5通道蛋白变异体的方法,通过分步模拟以及借助vmd、cpptraj模块等多种可视化工具对模拟体系进行连续性观测,从而可以更直观地观测到trpv5通道蛋白的离子摄入过程。
本发明上述目的通过以下技术方案实现:
一种基于计算机模拟获取可人工调控的trpv5通道蛋白变异体的方法,包括以下步骤:
s1.分析trpv5蛋白结构关键位点,对关键位点进行突变修饰,获得失去摄取钙离子能力的trpv5通道蛋白突变体a1,使通道关闭;
s2.对s1所述突变体a1的已突变位点进行磷酸化修饰,获得若干可摄入钙离子的trpv5通道蛋白突变体a2,使通道打开;
s3.将待摄入的钙离子分别置于s1所述突变体a1、s2所述突变体a2的通道入口附近,并使用charmm-gui软件将s1所述突变体a1、s2所述突变体a2分别构建成细胞膜结构模型;所述细胞膜结构模型包括变异体a1或变异体a2、以及待摄入钙离子、溶质离子、磷脂双分子层和溶剂;
s4.使用分子动力学模拟软件中的动力学模拟模块,结合最陡下降法和共轭梯度法对s3所述细胞膜结构模型分别进行能量最小化优化,获得能量优化后体系;
s5.对s4所述能量优化后体系中的蛋白质重原子和钙离子进行位置限制,随后使用分子动力学模拟软件中的动力学模拟模块对体系进行预平衡模拟,获得预平衡体系;
s6.在分子动力学模拟软件中的动力学模拟模块,对s5所得预平衡体系进行分子动力学模拟,获得钙离子运动轨迹;
s7.结合vmd程序和软件测距工具,对不同变异体的通道入口处进行观察钙离子是否进入其离子通道,并根据s6所得的钙离子运动轨迹分析其钙离子运动情况,获得不同变异路线中的初始最优变异体;
s8.得到初始最优变异体后,重新进行动力学模拟(包括能量最小化优化、预平衡模拟和分子动力学模拟,并且各参数与之前的模拟一致)从而进一步测试钙离子摄入能力,动力学模拟结束后获得含有若干晶体结构输出文件和体系各原子的运动轨迹文件,待进一步验证钙离子在更远的距离能否被通道蛋白突变体摄入;
s9.先使用ambpdb模块将s8所述若干晶体结构输出文件转化成适用于观察的pdb文件,再使用vmd可视化软件观测晶体结构输出文件中钙离子的最终位置,并使用分子动力学模拟软件中的分析模块对s8所述运动轨迹文件进行分析,根据分析结果选择确定最终最优的trpv5通道蛋白变异体。
目前实验研究均集中于trpv5离子通道出口处残基,由于变异体研究步骤繁多且耗时长,少有对trpv5通道蛋白入口处进行残疾修饰的研究;另外,现有的实验观测和计算机模拟都着眼于捕捉体系的中间态结构,而蛋白质通道内离子摄入是一个连续性过程,存在离子传导连续状态难以观测的不足。本发明方法首先采用charmm-gui对通道蛋白质进行变异处理,随后使用amber16中的分子动力学模拟模块pmemd.cuda对不同状态下的trpv5通道蛋白变异体阳离子进行模拟,反应度可控;最后结合vmd分子可视化软件和amber16中的cpptraj数据提取模块,分析阳离子的运动路径,通过在原子水平上对整个模拟体系的运动轨迹进行解析,克服了中间状态难以连续观测的难题,本发明能够更加直观地观测到阳离子在极短的时间尺度内的运动变化。整个模拟过程时间短、计算过程效率高,耗时短、效率高,操作简单,可以弥补实验研究的不足。
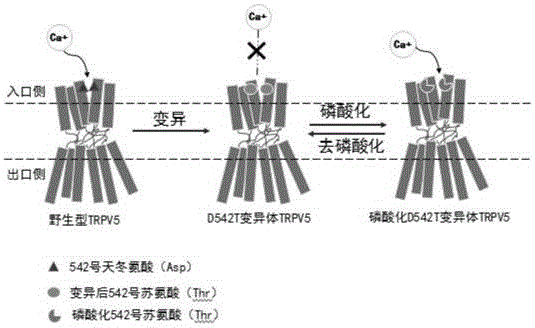
在本发明较佳的实施例中,s1所述关键位点为542号天冬氨酸(asp542)。
更进一步地,在本发明较佳的实施例中,所述对关键位点进行突变修饰为:将trpv5通道蛋白的542号天冬氨酸分别变异为丝氨酸突变体(d542s)、苏氨酸突变体(d542t)和酪氨酸突变体(d542y)。
在本发明较佳的实施例中,s2所述磷酸化修饰为:通过charmm-gui软件对所述变异体a1的542号残基进行磷酸化。
更进一步地,在本发明较佳的实施例中,经过s2所述磷酸化修饰得到磷酸化丝氨酸突变体(d542s-phos)、磷酸化苏氨酸突变体(d542t-phos)和磷酸化酪氨酸突变体(d542y-phos)。
在本发明较佳的实施例中,s2所述细胞膜结构模型呈电中性;所述溶质离子包括氯离子、钠离子。本发明采用最为常见的棕榈酰油酰磷脂酰胆碱(popc,1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)搭建磷脂双分子层。本发明选用钠离子和氯离子(na+/cl-)对体系进行调节,使体系维持电中性。
在本发明较佳的实施例中,s3采用vmd软件确定突变体通道入口的坐标信息,通过修改pdb格式的突变体晶体结构文件来设置钙离子的位置。
本发明所述动力学模拟优化包括能量最小化优化、预平衡模拟和动力学模拟。具体为:使用分子动力学模拟软件中的动力学模拟模块对体系进行限制性能量最小化的优化;对体系进行预平衡模拟,逐步放开对蛋白质和磷脂的限制;采用npt系综,在分子动力学模拟软件中的动力学模拟模块对不同变异体结构进行模拟,获得初始最优变异体结构。
在本发明较佳的实施例中,s4所述能量最小化优化包括对磷脂分子、蛋白质的氢原子以及溶液分子的位置和结构进行优化;采用charmm36m力场,在amber16动力学模拟工具中的pmemd模块中运行,先后采用最陡下降法和共轭梯度法对细胞膜结构模型一共进行5000~20000步的优化,优选进行5000~15000步的优化,以获得最佳的能量优化后体系。
为使得模拟过程尽可能接近真实情况,根据不同结构的运动需求,在正式分子动力学模拟开始前的结构优化模拟和预平衡模拟中,先对蛋白质重原子、磷脂分子和钙离子进行下述的位置限制,使体系能够充分适应模拟环境条件。经多次实验发现,采用本发明下述的位置限制权重能够比较准确地模拟蛋白质重原子、磷脂分子和钙离子的运动情况,从而正确预测钙离子与蛋白质等生物分子相互作用的过程。
更进一步地,在本发明较佳的实施例中,s4所述能量最小化优化过程采用
在本发明较佳的实施例中,s5所述预平衡模拟采用amber16的pmemd.cuda模块进行375~450ps的预平衡模拟;预平衡模拟分为六步进行,于303.15~330.5k的环境温度下逐步放开对蛋白质重原子和磷脂分子的位置限制。
在本发明较佳的实施例中,s5所述预平衡模拟的蛋白质重原子位置限制权重为
在本发明较佳的实施例中,s6所述分子动力学模拟压力控制采用montecarlo压强控制法,温度控制采用langevin法,环境设置为303.15~307.5k、一个大气压的npt系综,非键相互作用的截断值设置为10埃。
在本发明较佳的实施例中,s8所述轨迹文件保存为netcdf格式文件。
在本发明较佳的实施例中,s9采用amber16中的cpptraj模块对轨迹文件进行解析,并使用distance指令获取钙离子与通道入口中心处的距离数据。
与现有技术相比,本发明具有以下有益效果:
(1)本发明计算机模拟获取可人工调控trpv5通道蛋白变异体的方法,拥有效率高、操作简单的优势,克服了传统实验流程复杂以及实验时间长的局限。
(2)本发明利用charmm-gui对trpv5通道蛋白质进行不同变异,便于研究者能够在短时间内对多种变异可能性进行充分研究,操作灵活性高。
(3)本发明使用了amber16中的pmemd.cuda模块,借助图形处理器(gpu)进行分子动力学模拟,与常规使用中央处理器(cpu)进行分子动力学模拟相比,本发明有更高的工作效率,因此解决了常规分子动力学计算模拟在毫秒级时间尺度内模拟时间过长的问题,有助于在生物化学、蛋白质工程学的广泛应用。
(4)本发明可根据模拟体系大小调节过程中能量最小化优化、预平衡模拟、动力学模拟的时间长短,因此本发明可同时适用于分子数目较多的大体系和分子数目较小的小体系,适用性广,具有广泛的应用空间。
(5)本发明结合vmd分子可视化软件和amber16中的cpptraj数据提取模块,在原子水平上对整个体系的运动轨迹进行解析,相较于传统实验的观测手段而言,本发明克服了中间状态难以连续观测的难题,能够更加直观地观测到阳离子在极短的时间尺度内离子运动的整个过程。
附图说明
图1为本发明的流程示意图。
图2为trpv5蛋白质结构的设计示意图。
图3为trpv5通道蛋白变异体的示意图。
图4为磷酸化苏氨酸突变体(d542t-phos)经过一段时间计算模拟后,钙离子进入离子通道入口当中的示意图。
图5为苏氨酸突变体(d542t)经过一段时间计算模拟后,钙离子并没有进入离子通道入口处,流入周边空穴当中的示意图。
具体实施方式
以下结合具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。
实施例1
本发明提供了一种基于计算机模拟获取trpv5通道蛋白变异体的方法,包括以下步骤:
s1.分析trpv5蛋白结构关键位点,对关键位点进行突变修饰,获得失去摄取钙离子能力的trpv5通道蛋白突变体a1,使通道关闭;
s2.对s1所述突变体a1的已突变位点进行磷酸化修饰,获得若干可摄入钙离子的trpv5通道蛋白突变体a2,使通道打开;
s3.将待摄入的钙离子分别置于s1所述突变体a1、s2所述突变体a2的通道入口附近,并使用charmm-gui软件将s1所述突变体a1、s2所述突变体a2分别构建成细胞膜结构模型;所述细胞膜结构模型包括变异体a1或变异体a2、以及待摄入钙离子、溶质离子、磷脂双分子层和溶剂;
s4.使用分子动力学模拟软件中的动力学模拟模块,结合最陡下降法和共轭梯度法对s3所述细胞膜结构模型分别进行能量最小化优化,获得能量优化后体系;
s5.对s4所述能量优化后体系中的蛋白质重原子和钙离子进行位置限制,随后使用分子动力学模拟软件中的动力学模拟模块对体系进行预平衡模拟,获得预平衡体系;
s6.在分子动力学模拟软件中的动力学模拟模块,对s5所得预平衡体系进行分子动力学模拟,获得钙离子运动轨迹;
s7.结合vmd程序和软件测距工具,对不同变异体的通道入口处进行观察钙离子是否进入其离子通道,并根据s6所得的钙离子运动轨迹分析其钙离子运动情况,获得不同变异路线中的初始最优变异体;
s8.得到初始最优变异体后,重新进行动力学模拟从而进一步测试钙离子摄入能力,动力学模拟结束后获得含有若干晶体结构输出文件和体系各原子的运动轨迹文件,待进一步验证钙离子在更远的距离能否被通道蛋白突变体摄入;
s9.先使用ambpdb模块将s8所述若干晶体结构输出文件转化成适用于观察的pdb文件,再使用vmd可视化软件观测晶体结构输出文件中钙离子的最终位置,并使用分子动力学模拟软件中的分析模块对s8所述运动轨迹文件进行分析,根据分析结果选择确定最终最优的trpv5通道蛋白变异体。
本发明所用工具软件为分子动力学模拟软件,本发明对所述分子动力学模拟软件的种类没有限定,只要能够实现分子动力学模拟即可,在本发明实施过程中采用的是amber16计算软件。
本发明流程如图1所示,首先采用charmm-gui对trpv5蛋白质文件进行残基变异或磷酸化修改,在使用vmd可视化软件确定入口坐标后通过修改蛋白质变异体pdb文件的方式插入钙离子,最后再使用charmm-gui为蛋白质变异体构建细胞膜结构模型;而后进行一系列分子动力学模拟,包括能量最小化、平衡态模拟、动力学模拟;最后对各个最终结构进行分析,得出该蛋白质工程方案的可行性,为trpv5的人工调控方案提供新思路。
本发明中,trpv5通道蛋白的晶体结构数据均来源于pdb数据库(全称proteindatabank),从该数据库中下载所需要的蛋白质通道晶体结构。
本发明在charmm-gui(http://www.charmm-gui.org/)中对trpv5入口残基进行残基变异和磷酸化。根据之前研究者报道,trpv5通道蛋白是一种四聚体蛋白,其入口由四个天冬氨酸(简称asp542或称d542)构成,因此可对该部位进行蛋白质工程改造,赋予其符合实验预期的人工调控功能。
本发明通过残基变异的方式使trpv5暂时失去从环境摄取钙离子的能力,起到关闭通道入口的作用。因此,如图2和图3所示,将trpv5通道蛋白的542号天冬氨酸分别变异丝氨酸(ser,s),苏氨酸(thr,t)和酪氨酸(tyr,y),同时因为只有以上三种氨基酸能够进行下一步磷酸化,才可能满足人工调控目的。将入口处天冬氨酸(asp542)进行突变过后,共获得三个trpv5突变体结构,即丝氨酸突变体(d542s)、苏氨酸突变体(d542t)和酪氨酸突变体(d542y)。
本发明为实现可人工调控的通道打开,如图2和图3所示,在步骤5的基础上分别对上述蛋白质变异体通道入口残基进行磷酸化,重新赋予该通道蛋白trpv5的吸引钙离子进入的作用,通过charmm-gui对已变异的trpv5蛋白质磷酸化,因此获得磷酸化丝氨酸突变体(d542s-phos)、磷酸化苏氨酸突变体(d542t-phos)、磷酸化酪氨酸突变体(d542y-phos)。
本发明为探究钙离子能否进入离子通道,借助vmd等分子可视化软件确定离子通道入口的坐标,并通过手动修改蛋白质晶体结构(pdb)文件的方式,将钙离子放置于垂直距离入口约3埃的位置。本发明将对所获得的六个蛋白质变异体晶体结构分别插入待摄入的钙离子,为后续构建细胞膜体系做好准备。
本发明在charmm-gui中对所获得的含钙离子的蛋白质变异体晶体结构构建完整细胞膜体系。该体系内主要包括有蛋白质、钙离子、磷脂双分子层、氯离子、钠离子和溶剂。所述溶剂为水。本发明采用最为常见的棕榈酰油酰磷脂酰胆碱(popc,1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)搭建磷脂双分子层,磷脂双分子层的x轴和y轴方向尺寸均为150埃。本发明选用钠离子和氯离子(na+/cl-)对体系进行调节,使体系维持电中性。由于构建模拟体系过程中不可避免地产生了许多非自然接触,后续将通过动力学模拟手段将体系势能降低。
本发明为降低体系势能过高以及消除原子位置重叠等异常情况,在动力学模拟开始前,先对体系进行限制性能量最小化优化。该优化针对磷脂分子,蛋白氢原子和溶剂分子进行,对其原子所在位置和结构进行优化,消除体系中原子重叠、结构扭曲等不合理现象,进而使体系势能降低。在本发明中对体系采用charmm36m力场。该能量最小化优化采用amber16动力学模拟工具中的pmemd模块运行,依序采用最陡下降法和共轭梯度法对体系进行能量最小化优化。先采用2500步的最陡下降法对体系进行优化,消除体系中严重不合理的原子接触,而后再采用2500步的共轭梯度法对体系进行进一步优化,使体系势能进一步降低。优化过程采用
本发明在结构能量最小化优化过后,再对结构进行预平衡模拟,使体系在预设的实验环境温度和压强下进一步降低体系势能。本发明使用amber16的pmemd.cuda模块进行预平衡模拟,体系将在此步骤进行六步的预平衡模拟,预平衡模拟过程中逐步放开对蛋白质重原子和磷脂分子的位置限制,其中所述蛋白质重原子的位置限制权重依次为
本发明将对已平衡好的体系进行1ns的动力学模拟,以此获得尽可能接近真实环境下的离子运动情况和结构变化。为充分探究动力学模拟过程钙离子的运动路径,将动力学模拟分成10部分依次进行,每一部分进行0.1ns的动力学模拟,并输出一个结果文件以便后期使用vmd观察。本发明使用amber16的pmemd.cuda模块对体系进行动力学模拟,压力控制采用montecarlo压强控制法,温度控制采用langevin法,环境设置为303.15k,一个大气压的npt系综,非键相互作用的截断值设置为10埃。
本发明将在模拟计算完成后,使用amber16的ambpdb模块,将模拟计算各个步骤最终生成的结果文件转换成pdb文件用于后续观察。
本发明采用vmd软件对试验后的钙离子位置和通道入口结构变化进行分析,借助其软件测距工具测量通道入口宽度、以及钙离子与通道入口处、通道内部关键残基的距离,分析结构是否满足需求。实验发现,以上几个蛋白质变异体结构存在一定的缺陷,其中丝氨酸突变体(d542s)由于丝氨酸位阻小,以至于无法有效的阻挡离子的进入,无法满足关闭通道入口的功能。而磷酸化酪氨酸突变体(d542y-phos)由于位阻过大,将通道入口完全堵塞,因此钙离子止步于通道入口附近难以进入。
而本发明最终得到苏氨酸突变体(d542t)及其磷酸化酪氨酸突变体(d542y-phos)满足人工调控通道开关的需求,见图4、图5。然后,将钙离子逐步置于距离苏氨酸突变体(d542t)及其磷酸化酪氨酸突变体(d542y-phos)通道入口更远的位置(如:6埃、9埃、12埃、20埃等),在此基础上采用同样的参数重复以上能量最小化优化、预平衡模拟、动力学模拟三个步骤。本发明在动力学模拟过程中,设置每5000步记录一次晶体结构坐标信息,信息记录于netcdf格式的轨迹文件当中。
本发明在获得netcdf格式的轨迹文件后,将使用分子动力学模拟软件中的分析模块对轨迹文件进行分析,以及使用vmd对离子运动状况的直接观察。采用amber16当中的cpptraj模块对轨迹文件进行解析,使用cpptraj模块当中的distance指令获取钙离子与通道入口中心处的距离变化数据,可通过具体的数值变化分析离子运动趋势和摄入速度。此外,再使用ambpdb模块将每一中间步骤的结果文件转化成pdb格式,并使用vmd软件对其进行观察,从而更直观地分析动力学模拟过程中每一个步骤的蛋白质结构变化和离子移动情况,验证钙离子是否能够被苏氨酸突变体(d542t)及其磷酸化酪氨酸突变体(d542y-phos)正常摄入。通过验证实验发现,苏氨酸突变体(d542t)及其磷酸化酪氨酸突变体(d542y-phos)能够满足实验预期,通过磷酸化和去磷酸化控制trpv5通道蛋白变异体的钙离子摄入能力,为最优的trpv5通道蛋白变异体。
申请人声明,以上具体实施方式为便于理解本发明而说明的较佳实施例,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!