一类自由基敏感的尿嘧啶类ProTide前药及其药物用途的制作方法
本发明属于制药领域,提供一类自由基敏感的尿嘧啶类protide前药及其药物用途。
背景技术:
尿嘧啶类抗肿瘤药物如5-氟尿嘧啶(fluorouracil)、5-氟脱氧尿苷(floxuridine)、曲氟脲苷(trifluridine)是临床常见的抗肿瘤药物。5-氟尿嘧啶在细胞内转化成几个活性代谢物:5-氟脱氧尿苷单磷酸盐(fdump),5-氟脱氧尿苷二磷酸盐(fdutp)以及5-氟尿苷三磷酸盐(futp)。fdump结合到胸腺嘧啶合成酶(ts)核苷酸结合位点,与5-亚甲基四氢叶酸(ch2thf)形成稳定的三聚体,进而阻断了正常的底物dump的结合,抑制了dtmp的合成。该类药物容易发生耐药,其protide前药能够增加药效、减少耐药发生,具有良好的抗肿瘤作用,其中:5-氟脱氧尿苷protide前药nuc-3373已经进入临床研究。但是protide前药不能减少药物对非肿瘤组织的毒副作用。
由于肿瘤快速增长,肿瘤组织或者肿瘤细胞中的自由基浓度较高,因此,如果能够将抗肿瘤药物结构修饰为没有或者活性较小的前药,在自由基作用下,释放出活性较强的活性成分,就能够实现在保持高的抗肿瘤活性的同时,减少药物对正常细胞的毒性。
技术实现要素:
解决的技术问题:本发明提供一类自由基敏感的尿嘧啶类protide前药及其药物用途。该类前药对肿瘤具有优异的抗癌作用和良好的安全性,可用于制备治疗肿瘤的药物。
技术方案:一类自由基敏感的尿嘧啶类protide前药,其化学结构符合通式(i):
作为优选方案,上述自由基敏感的尿嘧啶类protide前药,结构如下所示:
上述化合物或其药学上可接受的盐在制备治疗肿瘤治疗药物中的应用。
治疗肿瘤药物,有效成分为上述自由基敏感的尿嘧啶类protide前药或其药学上可接受的盐。
需要指出的是,我们的研究发现:化合物1-4在预先清除自由基条件下的细胞毒性明显小于正常条件下的细胞毒性,提示该类药物的抗肿瘤活性与自由基的存在密切相关。
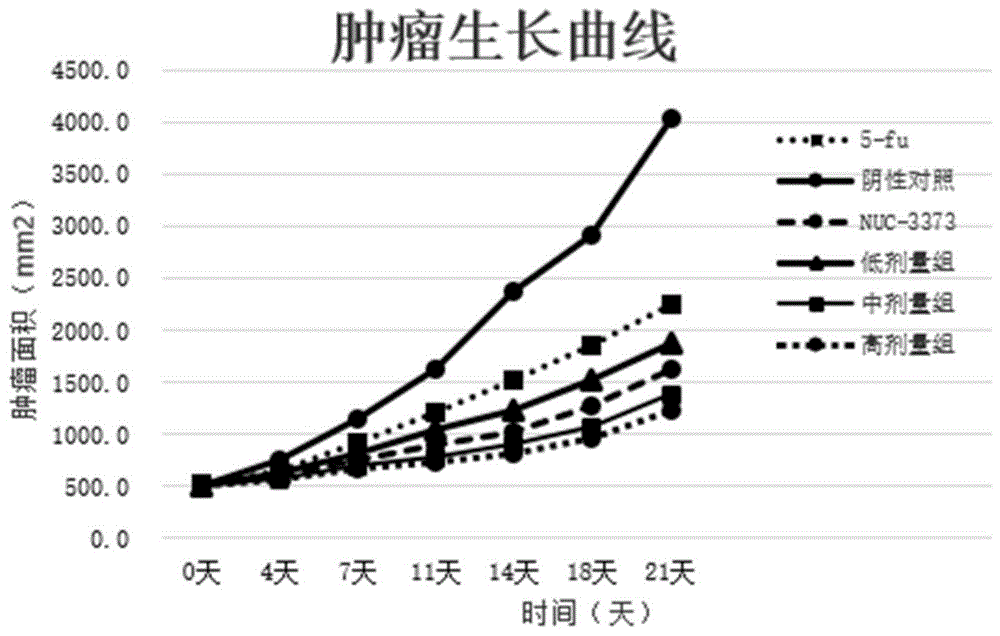
以化合物1为例,考察了目标化合物的抗肿瘤效果和安全性,化合物1显示了和5-氟尿嘧啶类似的抗肿瘤生长作用(见图1,表2),在4倍治疗剂量下,与对照组相比,动物体重没有明显差异,说明目标化合物具有良好的安全性(见表2)。
有益效果:本发明获得的自由基敏感的嘧啶类protide前药,在无自由基条件下具有较小的细胞毒性,在正常条件下,具有较大的细胞毒性。由于肿瘤细胞自由基浓度较高,本发明获得的自由基敏感的尿嘧啶类protide前药能够特异性的对肿瘤区域的肿瘤发挥抗肿瘤作用,减少对其他组织的毒副作用,对肿瘤具有优异的抗癌作用和良好的安全性。
附图说明
图1目标化合物1对祼鼠ht-29结肠癌移植瘤的生长抑制作用示意图(以5-氟尿嘧啶为对照)。
具体实施方式
下面的实施例可使本专业技术人员可全面地理解本发明,但不以任何方式限制本发明。
实施例1:目标化合物的合成:
合成路线(以化合物1为例):
1.1:丙氨酰噻唑酮盐酸盐(m-1)的合成
在配有磁子的100ml干燥茄形瓶中,依次加入n-叔丁氧羰基-l-丙氨酸(10g,52.8mmol),1,3-噻唑烷-2-酮(5.44g,52.8mmol),edci(10.1g,52.8mmol),hobt(7.13mg,52.8mmol),dipea(6.82g,52.8mmol)和二氯甲烷300ml,室温搅拌反应4h。反应结束后,加入水(300ml),用乙酸乙酯(300ml×3)萃取,收集有机相,后用饱和食盐水(300ml×3)洗涤,无水硫酸钠干燥,过滤,减压蒸除溶剂,得白色油状液体。粗产物经硅胶色谱柱纯化(洗脱剂:石油醚:乙酸乙酯=5:1),得白色固体化合物(7.1g)。加入氯化氢/乙酸乙酯溶液(50ml,2mmol/ml)。室温搅拌3h后,有大量白色固体析出,过滤,干燥,得到白色固体丙氨酰噻唑酮盐酸盐(m-1,4.5g)。1hnmr(400mhz,dmso-d6)δ(ppm)8.54(s,3h),4.64(d,1h),4.133.99(m,2h),3.473.39(m,2h),1.35(d,3h)。
1.2:4-硝基苯基苯基(1-氧代-1-(2-氧代噻唑烷-3-基)丙-2-基)氨基磷酸酯(m-2)的合成在-20℃下,向配有磁子和温度计的100ml二颈烧瓶中加入m-1(3.6g,17mmol)和二氯甲烷40ml。随后,向上述混悬液中滴加二氯化磷酸苯酯(3.6g,17mmol),搅拌15min后,将三乙胺(3.4g,34mmol)缓慢滴入上述反应液中(控制在15min内),并使溶液内温度保持在-10℃--3℃。反应3h后,加入对硝基苯酚(2.5g,17mmol),并将三乙胺(1.7g,17mmol)缓慢滴入反应液中(控制在15min内)。升至室温反应5h后,加入甲基叔丁基醚(40ml),有大量白色固体析出,过滤,收集有机相,减压蒸除溶剂,得黄色油状液体。粗产物经硅胶色谱柱纯化(洗脱剂:石油醚:乙酸乙酯=1:1),得白色固体化合物4-硝基苯基苯基(1-氧代-1-(2-氧代噻唑烷-3-基)丙-2-基)氨基磷酸酯(m-2)(1.5g,产率20%)。1hnmr(400mhz,dmso-d6)δ(ppm)8.29–8.23(m,2h),7.42(dd,2h),7.40–7.33(m,2h),7.22–7.13(m,3h),6.65(dd,1h),4.96(m,1h),4.00–3.85(m,2h),3.45–3.30(m,2h),1.12(dd,3h).1.3:目标化合物1的合成
在配有磁子和温度计的100ml三颈瓶中,加入5-氟脱氧尿苷(0.41g,3.28mmol),四氢呋喃15ml和n-甲基咪唑4ml,并用氮气保护,置于室温中,搅拌至完全溶解。逐滴滴加叔丁基氯化镁(2.46ml,2.46mmol),搅拌20min后,加入用四氢呋喃5ml溶解的m-2(1.48g,3.28mmol),随后将温度缓慢提升至50℃,搅拌5h后,加入甲醇5ml,使析出的白色固体完全溶解。通过快速柱层析(先用70%乙酸乙酯/石油醚将极性较小的物质冲洗下来,再用10%甲醇/二氯甲烷)得到白色固体1(0.65g,产率35%)。1hnmr(400mhz,dmso-d6)δ(ppm)8.22-8.21(t,1h),7.40–7.34(m,2h),7.23–7.14(m,3h),6.66(dd,1h),6.15-6.11(m,1h),5.25-5.10(m,2h),4.97(m,1h),4.25-4.22(m,1h),4.01–3.87(m,2h),3.80-3.77(m,1h),3.65-3.55(m,2h),3.47–3.31(m,2h),2.12-2.10(m,2h),1.13(dd,3h)。
1.4:目标化合物2-8的合成
目标化合物2:参考化合物1方法,以m-2和曲氟脲苷合成。1hnmr(400mhz,dmso-d6)δ(ppm)7.40–7.33(m,2h),7.22–7.13(m,3h),6.65(dd,1h),6.10-6.07(t,1h),5.27-5.26(d,1h),5.22-5.20(d,1h),4.96(m,1h),4.26-4.24(t,1h),4.00–3.85(m,2h),3.83-3.82(d,1h),3.66-3.65(d,1h),3.60-3.59(d,1h),3.66-3.65(d,1h),3.45–3.30(m,2h),2.21-2.19(t,2h),1.12(dd,3h).
目标化合物3:参考化合物1方法,以m-1、二氯化磷酸1-萘酯和5-氟脱氧尿苷合成。1hnmr(400mhz,dmso-d6)δ(ppm)8.22-8.21(t,1h),8.22–8.19(m,1h),7.93–7.90(m,1h),7.76–7.72(m,1h),7.58–7.39(m,4h),6.66(dd,1h),6.15-6.11(m,1h),5.25-5.10(m,2h),4.97(m,1h),4.25-4.22(m,1h),4.01–3.87(m,2h),3.80-3.77(m,1h),3.65-3.55(m,2h),3.47–3.31(m,2h),2.12-2.10(m,2h),1.13(dd,3h)。
目标化合物4:参考化合物1方法,以m-1、二氯化磷酸1-萘酯和曲氟脲苷合成。1hnmr(400mhz,dmso-d6)δ(ppm)8.21–8.17(m,1h),7.91–7.89(m,1h),7.74–7.70(m,1h),7.56–7.36(m,4h),6.65(dd,1h),6.10-6.07(t,1h),5.27-5.26(d,1h),5.22-5.20(d,1h),4.96(m,1h),4.26-4.24(t,1h),4.00–3.85(m,2h),3.83-3.82(d,1h),3.66-3.65(d,1h),3.60-3.59(d,1h),3.66-3.65(d,1h),3.45–3.30(m,2h),2.21-2.19(t,2h),1.12(dd,3h).
实施例2:目标化合物正常状态、预先去除自由基状态下对肿瘤细胞增殖体外抑制作用研究取对数生长期肿瘤细胞,加入0.25%胰酶消化3min,用含10%小牛血清rpmi-1640悬浮细胞,计数,调细胞浓度为1×105个/ml,以100μl/孔接种于top-count专用96孔细胞培养板中,37℃,5%co2孵育24h。然后将细胞分为实验组和对照组,实验组加入目标化合物溶液(0.001μg/ml,0.01μg/ml,0.1μg/ml,1μg/ml,10μg/ml),每一浓度均为四复孔,且每孔体积均补足200μl。预先去除自由基组细胞先用自由基清除剂n-乙酰半胱胺酸(20mmol/l)预处理1小时。各组加样后分别继续培养48h,于培养结束前,每孔分别加入3h-tdr3×105bq,用top-count测定各孔cpm(countperminute)值。计算各实验组药物对细胞增殖的半数抑制浓度(medianinhibitionconcentration,ec50)。同法测定药物对人正常细胞外周血单核细胞pbmc增殖的半数抑制浓度(medianinhibitionconcentration,ec50)。
表1目标化合物对肿瘤细胞增殖(48小时)的半数抑制浓度(ec50,μmol/l)
以上实验结果显示:5-氟脲嘧啶、5-氟脱氧尿苷、曲氟脲苷或nuc-3373在正常状态、预先去除自由基下对肿瘤细胞增殖体外抑制作用没有显著差异,本发明的实施例化合物(1-4)在正常状态、预先去除自由基下的对肿瘤细胞增殖体外抑制作用具有显著差异(26-43倍)。提示本发明的实施例化合物对富含自由基的肿瘤细胞具有更强的细胞毒性。本发明的目标化合物对肿瘤细胞增殖体外抑制作用显著强于5-氟脲嘧啶、5-氟脱氧尿苷、曲氟脲苷,和nuc-3373相当,但是其对人正常细胞外周血单核细胞pbmc增殖的半数抑制浓度显著大于nuc-3373,提示其具有更好的安全性。
实施例3:目标化合物1对人结肠癌细胞ht-29肿瘤的生长抑制作用
取对数生长期的人结肠癌细胞ht-29细胞,以1×107个细胞·0.2ml-1·只-1的浓度,接种于裸鼠右侧腋窝皮下,第14天选取移植瘤体积≥500mm3的裸鼠,随机分为阴性对照组、低剂量组(化合物001,0.12mmol/kg)、中剂量组(化合物001,0.23mmol/kg)、高剂量组(化合物001,0.46mmol/kg)、5-氟脲嘧啶组(0.23mmol/kg)、nuc-3373组(0.23mmol/kg)。隔天进行腹腔药物注射。注射药物开始后每4天使用游标卡尺测量肿瘤的长径、短径,计算肿瘤体积。给药第21天处死荷瘤小鼠,称量每只裸鼠重量及移植瘤重量(g)。
抑制效果见图1,第21天裸鼠重量及移植瘤重量见表2:给药后,各组均显示出显著的抑制肿瘤生长作用,所有试验组裸小鼠体重均小于阴性对照组,目标化合物组祼鼠体重均高于nuc-3373组和5-氟脲嘧啶组组。目标化合物1中剂量组抗肿瘤作用优于nuc-3373组和5-氟脲嘧啶组,目标化合物1中剂量小鼠体重明显高于nuc-3373组和5-氟脲嘧啶组,。提示目标化合物1比nuc-3373和5-氟脲嘧啶更有效、更安全。
表2目标化合物1对祼鼠ht-29结肠癌移植瘤的生长抑制作用(以5-氟尿嘧啶为对照)。
以上实例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人是能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所做的等效变换或修饰,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!