一种炒槐花配方颗粒及其制备方法和质量标准检测方法与流程
本发明属于中药制备及其检测技术领域,尤其涉及一种炒槐花配方颗粒及其制备方法和质量标准检测方法。
背景技术:
炒槐花配方颗粒为豆科植物槐(sophorajaponical.)的干燥花及花蕾制成的配方颗粒。其性味苦,微寒,归肝、大肠经,具有凉血止血,清肝泻火的功效,用于便血,痨血,血痢,崩漏,吐血,衄血,肝热目赤,头痛眩晕。
针对本品的现行制备方法和质量检测尚不完善,会导致药物检测结果的不准确,从而导致药品质量的不可控,同时现行的质量标准中仅包括对部分有效成分的定性检测,没有药物溶剂含量的检查及反应药物内在质量指标含量的测定项,无法在生产中保证产品的质量。
技术实现要素:
本发明目的就是为了弥补已有技术的缺陷,提供一种炒槐花配方颗粒及其制备方法和质量标准检测方法。
为了实现上述的目的,本发明提供以下技术方案:
一种炒槐花配方颗粒的制备方法,取炒槐花饮片加水煎煮二次,加水量分别为炒槐花饮片质量的12倍、10倍,每次1.5h、1h,滤过,合并煎液,于55-60℃下浓缩至相对密度1.10-1.15,喷雾干燥成浸膏粉,加入糊精辅料,混匀,干法制粒,制成颗粒,分装,即得。
一种如上所述的炒槐花配方颗粒的制备方法制备得到的炒槐花配方颗粒。
一种如上所述的炒槐花配方颗粒的质量标准检测方法,对药物定性、鉴别、检查、含量测定的方法进行修订,具体包括:性状检测、鉴别检测、重金属及有害元素检测、有机氯农药残留量检测、浸出物检测、总黄酮含量检测、芦丁含量检测。
进一步的,所述性状检测的性状指标:本品为深黄色至黄褐色颗粒,气微,味微苦涩。
进一步的,所述鉴别检测的方法为:取本品0.2g,研细,加甲醇5ml,密塞,振摇10分钟,滤过,取滤液作为供试品溶液,另取芦丁对照品,加甲醇制成每1ml含4mg的溶液,作为对照品溶液,取按照比例缺炒槐花的阴性样品,按照供试品溶液制备方法制成阴性对照溶液,照薄层色谱法试验,吸取上述三种溶液各10μl,分别点于同一硅胶g薄层板上,按8:1:1以乙酸乙酯-甲酸-水为展开剂,展开,取出,晾干,喷以三氯化铝试液,待乙醇挥干后,置紫外光灯365nm下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,阴性样品在与样品色谱相应的位置上,不显相同颜色的荧光斑点。
进一步的,所述重金属及有害元素检测指标:照铅、镉、砷、汞、铜以原子吸收分光光度法或电感耦合等离子体质谱法测定,铅不得过5mg/kg,镉不得过0.3mg/kg,砷不得过2mg/kg,汞不得过0.2mg/kg。
进一步的,所述有机氯农药残留量检测指标:照农药残留量测定法测定,含总六六六(α-bhc、β-bhc、γ-bhc、δ-bhc之和)不得过0.2mg/kg,总滴滴涕(pp'-dde、pp'-ddd、op'-ddt、pp'-ddt之和)不得过0.2mg/kg,五氯硝基苯不得过0.1mg/kg。
进一步的,所述浸出物检测指标:照醇溶性浸出物测定法项下的热浸法测定,用30%甲醇作溶剂,不得少于70.0%。
进一步的,所述总黄酮含量检测方法如下:
对照品溶液的制备:取芦丁对照品40mg,精密称定,置20ml量瓶中,加甲醇适量,置水浴上微热使溶解,放冷,加甲醇至刻度,摇匀,精密量取10ml,置100ml量瓶中,加水至刻度,摇匀,即得(每1ml中含芦丁0.2mg);
标准曲线的制备:精密量取对照品溶液1ml、2ml、3ml、4ml、5ml与6ml,分别置25ml量瓶中,各加水至6.0ml,加5%亚硝酸钠溶液1ml,混匀,放置6分钟,加10%硝酸铝溶液1ml,摇匀,放置6分钟,加氢氧化钠试液10ml,再加水至刻度,摇匀,放置15分钟,以相应的试剂为空白,照紫外-可见分光光度法,在500nm波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线;
测定法:取本品粉末1g,精密称定,置具塞锥形瓶中,精密加入50%甲醇溶液100ml,密塞,称重,超声提取30min,取出,放冷,称重,用50%甲醇溶液补足减失的重量,摇匀,精密量取10ml,置100ml量瓶中,加水至刻度,摇匀,即得,精密量取3ml,置25ml量瓶中,照标准曲线制备项下的方法,自“加水至6.0ml”起,依法测定吸光度,从标准曲线上读出供试品溶液中含芦丁的浓度,计算,即得;
本品按干燥品计算,每1g含总黄酮以芦丁(c27h30o16)计,不得少于156.0mg。
进一步的,所述芦丁含量检测方法如下:
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以甲醇-1%冰醋酸溶液(32:68)为流动相,检测波长为257nm,柱温为30℃,理论板数按芦丁峰计算应不低于2000;
对照品溶液的制备:取芦丁对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得;
供试品溶液的制备:取本品粉末0.05g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,于25khz频率、250w功率下超声处理30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,即得;
测定法:分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得;
本品按干燥品计算,每1g含芦丁(c27h30o16)槐花不得少于89.0mg。
本发明的优点是:
本发明炒槐花配方颗粒主要含有槐花炭、芦丁、槲皮素、染料木素等化学成分。具有凉血止血,清肝泻火等作用。炒槐花配方颗粒是炒槐花药材饮片经加工制成的配方颗粒,具有用药安全、有效、方便以及易于贮藏和运输等特点,同时本发明优化了制备方法,精确控制了制备参数,保证了药品的质量。与此同时针对本品还提供了行之有效的质量检测方法,全面囊括了炒槐花配方颗粒的各项理化参数,并对该方法进行了方法学验证,实验结果表明,该方法操作方便、可行,准确度高,有利于炒槐花配方颗粒的标准化、规模化、安全化生产制备。
附图说明
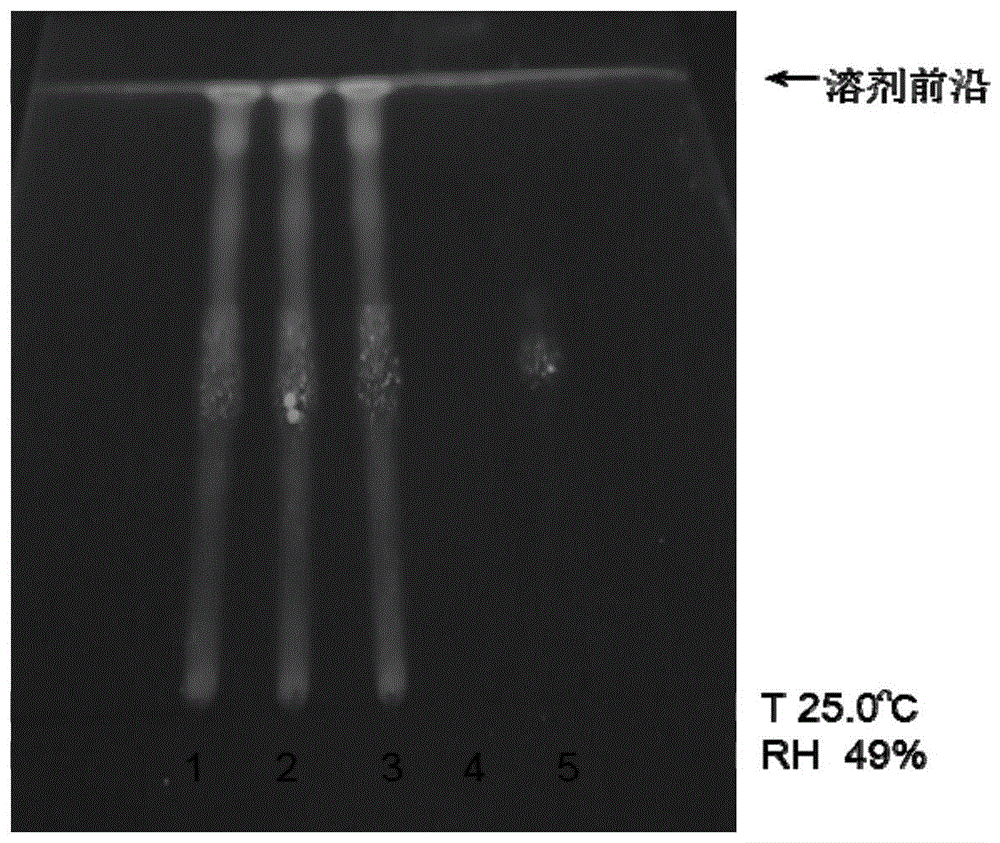
图1所示为炒槐花配方颗粒薄层鉴别色谱图,其中1为炒槐花配方颗粒201711012,2为炒槐花配方颗粒201711022,3为炒槐花配方颗粒201711032,4为阴性样品,5为芦丁对照品。
图2所示为关于铅的不同浓度的标准品吸光度标准曲线图。
图3所示为关于镉的不同浓度的标准品吸光度标准曲线图。
图4所示为关于砷的不同浓度的标准品吸光度标准曲线图。
图5所示为关于汞的不同浓度的标准品吸光度标准曲线图。
图6所示为关于铜的不同浓度的标准品吸光度标准曲线图。
图7所示为不同浓度α-六六六标准品的峰面积标准曲线图。
图8所示为不同浓度β-六六六标准品的峰面积标准曲线图。
图9所示为不同浓度γ-六六六标准品的峰面积标准曲线图。
图10所示为不同浓度δ-六六六标准品的峰面积标准曲线图。
图11所示为不同浓度pp'-dde标准品的峰面积标准曲线图。
图12所示为不同浓度pp'-ddd标准品的峰面积标准曲线图。
图13所示为不同浓度op'-ddt标准品的峰面积标准曲线图。
图14所示为不同浓度pp'-ddt标准品的峰面积标准曲线图。
图15所示为不同浓度五氯硝基苯标准品的峰面积标准曲线图。
图16所示为芦丁对照品溶液标准曲线图。
图17所示为对照品、样品、辅料、空白样品hplc对比图谱。
图18所示为芦丁对照品品溶液标准曲线图。
图19所示为三个不同批号色谱柱的色谱柱耐用性考察。
具体实施方式
以下结合具体的实例对本发明的技术方案做进一步说明:
炒槐花饮片按《中国药典》2015年版一部炒槐花项下炮制标准及饮片炮制通则0213中的净制、炮制标准进行炮制。
取炒槐花饮片2500g加水煎煮二次,加水量分别为12倍、10倍,每次1.5h、1h,滤过,合并煎液,浓缩至相对密度1.10-1.15(55-60℃),喷雾干燥成浸膏粉,加入糊精辅料适量,混匀,干法制粒,制成颗粒1000g,分装,即得。
1、对三批试生产炒槐花配方颗粒样品进行性状检查,结果见表1:
表1三批炒槐花配方颗粒性状描述
根据上述三批生产产品性状检查结果,暂定“本品为深黄色至黄褐色颗粒;气微,味微苦涩”。
2、本实验以芦丁对照品为对照,本方法根据《中华人民共和国药典》2015年版一部槐花药材的薄层鉴别方法,建立了炒槐花配方颗粒的薄层鉴别方法。与此同时对方法的耐用性进行考察,实验结果表明:该炒槐花配方颗粒的薄层鉴别方法稳定、可行。
(1)仪器与试药
仪器:sonydsc-hx400相机、ja2003n千分之一天平(上海佑科科学仪器仪表有限公司)、dhg电热鼓风干燥箱(上海一横科学仪器有限公司)、cary数控超声波清洗器(合肥金尼克机械制造有限公司)、dzkw型水浴锅(上海科恒仪器有限公司)、展开缸、硅胶g薄层板。
试药:
炒槐花配方颗粒(批号:201711012、201711022、201711032)
对照品:芦丁(中国食品药品检定研究院提供,供鉴别用,批号:100080-201610)
乙酸乙酯、甲酸均为分析纯。
(2)溶液的制备
供试品溶液的制备:
取本品粉末0.2g,加甲醇5ml,密塞,振摇10分钟,滤过,取滤液作为供试品溶液。
对照品溶液的制备:
取芦丁对照品,加甲醇制成每1ml含4mg的溶液,作为对照品溶液。
阴性样品溶液的制备:
取按照比例缺炒槐花的阴性样品,按照供试品溶液制备方法制成阴性对照溶液。
(3)薄层色谱条件
薄层板:硅胶g薄层板
点样量:供试品溶液、阴性样品溶液及对照品溶液各10μl
展开剂:乙酸乙酯-甲酸-水(8:1:1)
显色剂:三氯化铝试液
检视:置紫外光灯(365nm)下检视
(4)炒槐花配方颗粒样品薄层鉴别
分别对不同批次的炒槐花配方颗粒样品进行薄层鉴别,实验结果见图1。
采用所述薄层鉴别方法对不同批次炒槐花的配方颗粒进行鉴别,实验结果表明,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,阴性样品在与样品色谱相应的位置上,不显相同颜色的荧光斑点。
3、检查
(1)粒度:照粒度和粒度分布测定法《中国药典》2015年版四部(通则0982第二法双筛分法测定),不能通过一号筛与能通过五号筛的总和,不得超过15%。本品三批检测结果见表2,均符合规定。
(2)水分:中药颗粒剂照水分测定法《中国药典》2015年版四部(通则0832)测定,规定水分不得超过8.0%。本品3批检测结果见表2,均符合规定。
(3)溶化性:根据颗粒剂项下有关的各项规定《中国药典》2015年版四部(通则0104)检查,取供试品10g,加热水200ml,搅拌5分钟,立即观察,应全部溶化,并不得有焦屑等异物。本品3批检测结果见表2,均符合规定。
(4)装量:根据颗粒剂项下有关的各项规定《中国药典》2015年版四部多剂量包装的颗粒剂,照最低装量检查法(通则0942)检查。本品3批检测结果见表2,均符合规定。
表2粒度、水分、溶化性、装量检查结果表
(5)微生物限度检查:根据颗粒剂项下有关的各项规定《中国药典》2015年版四部(通则0104)检查,照微生物限度检查法(中国药典2015年版四部通则1105)检查,规定需氧菌数不得过103cfu/g,霉菌和酵母菌数不得过102cfu/g,大肠埃希菌不得检出,检查结果如表3所示:
表3微生物限度检验结果
4、重金属及有害元素检测
(1)铅
a、仪器与试药
仪器:原子吸收分光光度计(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、微波消解仪(南京先欧仪器制造有限公司)、赶酸器(北京莱伯泰科仪器股份有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:铅单元素标准溶液(中国计量科学研究院,批号:gbw08619-13115)硝酸(优级纯)、双氧水。
b、溶液的制备
供式品溶液的制备:取50g样品粉碎,存于塑料袋中。称取供试品0.5g于消解罐内罐中,加入浓硝酸8ml,双氧水2ml,盖上盖子,放置过夜。次日将装有供式品的内罐套上外罐,放入微波消解仪中。按照微波消解仪的操作规程操作,消解完毕,取出,将消解罐内罐放电热板上加热驱酸,至无黄烟溢出,取下,放凉。用硝酸(2.0%)定量转移并定容至50ml;同时做试剂空白。
铅标准使用液的制备:吸取1.0ml铅标准溶液,置于1000ml容量瓶中,加2%硝酸稀释至刻度。如此多次稀释成每毫升含10.0、20.0、30.0、40.0、50.0ng铅的标准吸收液。
仪器条件:波长283.3nm,狭缝0.5nm,灯电流10ma,干燥温度100℃持续10s,灰化温度600℃持续20s,原子化温度2100℃持续3s,背景校正为氘灯。
标准曲线绘制:吸取上面配置的铅标准使用液10.0、20.0、30.0、40.0、50.0ng/ml各10ul,注入石墨炉,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。
试样测定:分别吸取样液和空白液各10ul,注入石墨炉,测得其吸光值,代入标准系列的一元线性回归方程中求得样液中铅的含量。
c、方法学考察
线性关系考察
吸取上面配置的铅标准使用液10.0、20.0、30.0、40.0、50.0μg/l各10μl,注入石墨炉,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。结果见表4及图2。
表4不同浓度标准品的吸光度
d、炒槐花配方颗粒样品铅的测定
取炒槐花配方颗粒3批,照铅测定项下测定,结果见表5。
表5样品铅的测定结果
(2)镉
a、仪器与试药
仪器:原子吸收分光光度计(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、微波消解仪(南京先欧仪器制造有限公司)、赶酸器(北京莱伯泰科仪器股份有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:镉单元素标准溶液(国家有色金属及电子材料分析测试中心,批号:gsb04-1721-2004:163041-1)
硝酸(优级纯)、双氧水。
b、溶液的制备
供式品溶液的制备:取50g样品粉碎,存于塑料袋中。称取供试品0.5g于消解罐内罐中,加入浓硝酸8ml,双氧水2ml,盖上盖子,放置过夜。次日将装有供式品的内罐套上外罐,放入微波消解仪中。按照微波消解仪的操作规程操作,消解完毕,取出,将消解罐内罐放电热板上加热驱酸,至无黄烟溢出,取下,放凉。用硝酸(2.0%)定量转移并定容至50ml;同时做试剂空白。
镉标准使用液的制备:吸取1.0ml镉标准溶液,置于1000ml容量瓶中,加2%硝酸稀释至刻度。如此多次稀释成每毫升含0.5、1.0、1.5、2.0、2.5ng镉的标准吸收液。
仪器条件:波长228.8nm,狭缝0.5nm,灯电流4ma,干燥温度100℃持续10s,灰化温度600℃持续20s,原子化温度1800℃持续3s,背景校正为氘灯。
标准曲线绘制:吸取上面配置的镉标准使用液0.5、1.0、1.5、2.0、2.5μg/l各10ul,注入石墨炉,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。
试样测定:分别吸取供式液和空白液各10ul,注入石墨炉,测得其吸光值,代入标准系列的一元线性回归方程中求得样液中镉的含量。
c、方法学考察
线性关系考察
吸取上面配置的镉标准使用液0.5、1.0、1.5、2.0、2.μg/l各10ul,注入石墨炉,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。结果见表6及图3。
表6不同浓度标准品的吸光度
d、炒槐花配方颗粒样品镉的测定
取炒槐花配方颗粒3批,照试样测定项下测定,结果见表7。
表7样品镉的测定结果
(3)砷
a、仪器与试药
仪器:原子吸收分光光度计(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、微波消解仪(南京先欧仪器制造有限公司)、赶酸器(北京莱伯泰科仪器股份有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:砷单元素标准溶液(中国计量科学研究院,批号:gbw08611-15083)硝酸(优级纯)、双氧水、硼氢化钾、氢氧化钾。
b、溶液的制备
供式品溶液的制备:取50g样品粉碎,存于塑料袋中。称取供试品0.5g于消解罐内罐中,加入浓硝酸8ml,双氧水2ml,盖上盖子,放置过夜。次日将装有供式品的内罐套上外罐,放入微波消解仪中。按照微波消解仪的操作规程操作,消解完毕,取出,将消解罐内罐放电热板上加热驱酸,至无黄烟溢出,取下,放凉。用硝酸(2.0%)定量转移并定容至50ml;同时做试剂空白。
砷标准使用液的制备:吸取1.0ml砷标准溶液,置于1000ml容量瓶中,加2%硝酸稀释至刻度。如此多次稀释成每毫升含5.0、10.0、15.0、20.0、25.0ng砷的标准吸收液。
仪器条件:波长193.7nm,狭缝0.5nm,灯电流10ma,氢化物发生装置。
标准曲线绘制:吸取上面配置的砷标准使用液5.0、10.0、15.0、20.0、25.0μg/l,注入氢化物发生装置,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。
试样测定:分别吸取样液和空白液注入氢化物发生装置,测得其吸光值,代入标准系列的一元线性回归方程中求得样液中砷的含量。
c、方法学考察
线性关系考察
吸取上面配置的砷标准使用液5.0、10.0、15.0、20.0、25.0μg/l,注入氢化物发生装置,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。结果见表8及图4。
表8不同浓度标准品的吸光度
d、炒槐花配方颗粒样品砷的测定
取炒槐花配方颗粒3批,照试样测定项下测定,结果见表9。
表9样品砷的测定结果
(4)汞
a、仪器与试药
仪器:原子吸收分光光度计(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、微波消解仪(南京先欧仪器制造有限公司)、赶酸器(北京莱伯泰科仪器股份有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:汞单元素标准溶液(中国计量科学研究院,批号:gbw08617-15034)硝酸(优级纯)、双氧水、硼氢化钾、氢氧化钾。
b、溶液的制备
供式品溶液的制备:取50g样品粉碎,存于塑料袋中。称取约0.4g试样于消解罐内罐中,加入浓硝酸8ml,双氧水2ml,盖上盖子,放置过夜。次日将装有供试品的内罐套上外罐,放入微波消解仪中。按照微波消解仪的操作规程操作,消解完毕,取出,将消解罐内罐放电热板上加热驱酸,至无黄烟溢出,并继续浓缩至2-3ml,放冷,加20%硫酸溶液2ml、5%高锰酸钾溶液0.5ml,摇匀,滴加5%盐酸羟胺溶液至紫红色恰消失,转入50ml量瓶中,用水洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,必要时离心,取上清液,即得。同法同时制备试剂空白溶液。
汞标准使用液的制备:精密量取汞单元素标准溶液适量,用2%硝酸溶液稀释,制成每1ml含汞(hg)0.1μg的溶液,即得(0-5℃贮存)。分别精密量取汞标准贮备液0ml、1.0ml、2.0ml、3.0ml、4.0ml、5.0ml,置50ml量瓶中,加20%硫酸溶液10ml、5%高锰酸钾溶液0.5ml,摇匀,滴加5%盐酸羟胺溶液至紫红色恰消失,用水稀释至刻度,摇匀如此多次稀释成每毫升含2.0、4.0、6.0、8.0、10.0ng汞的标准吸收液。
仪器条件:波长253.6nm,狭缝0.5nm,灯电流4ma,氢化物发生装置。
标准曲线绘制:吸取上面配置的汞标准使用液2.0、4.0、6.0、8.0、10.0μg/l,注入氢化物发生装置,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。
试样测定:分别吸取样液和空白液注入氢化物发生装置,测得其吸光值,代入标准系列的一元线性回归方程中求得样液中汞的含量。
c、方法学考察
线性关系考察
吸取上面配置的汞标准使用液2.0、4.0、6.0、8.0、10.0μg/l,注入氢化物发生装置,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。结果见表10及图5。
表10不同浓度标准品的吸光度
d、炒槐花配方颗粒样品汞的测定
取炒槐花配方颗粒3批,照试样测定项下测定,结果见表11。
表11样品汞的测定结果
(5)铜
a、仪器与试药
仪器:原子吸收分光光度计(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、微波消解仪(南京先欧仪器制造有限公司)、赶酸器(北京莱伯泰科仪器股份有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:铜单元素标准溶液(中国计量科学研究院,批号:gbw08615-16032)硝酸(优级纯)、双氧水。
b、溶液的制备
供式品溶液的制备:取50g样品粉碎,存于塑料袋中。称取0.5g试样于消解罐内罐中,加入浓硝酸8ml,双氧水2ml,盖上盖子,放置过夜。次日将装有供式品的内罐套上外罐,放入微波消解仪中。按照微波消解仪的操作规程操作,消解完毕,取出,将消解罐内罐放电热板上加热驱酸,至无黄烟溢出,取下,放凉。用硝酸(2.0%)定量转移并定容至50ml;同时做试剂空白。
铜标准使用液的制备:吸取1.0ml铜标准溶液,置于1000ml容量瓶中,加2%硝酸稀释至刻度。如此多次稀释成每毫升含0.2、0.4、0.6、0.8、1.0μg铜的标准吸收液。
仪器条件:波长324.7nm,狭缝0.2nm,灯电流4ma,火焰检测器检测。
标准曲线绘制:吸取上面配置的铜标准使用液0.2、0.4、0.6、0.8、1.0μg/ml,注入火焰检测器,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。
试样测定:分别吸取样液和空白液注入火焰检测器,测得其吸光值,代入标准系列的一元线性回归方程中求得样液中铜的含量。
c、方法学考察
线性关系考察
吸取上面配置的铜标准使用液0.2、0.4、0.6、0.8、1.0μg/ml,注入火焰检测器,测得吸光值并求得吸光值与浓度关系的一元线性回归方程。结果见表12及图6。
表12不同浓度标准品的吸光度
d、炒槐花配方颗粒样品铜的测定
取炒槐花配方颗粒3批,照试样测定项下测定,结果见表13。
表13样品铜的测定结果
根据上述3批样品测定结果,炒槐花配方颗粒中铅的含量为0.841-0.915mg/kg考虑到不同批次之间的差异,暂定本品每1kg含铅不得过5mg,镉的含量为0.017-0.026mg/kg考虑到不同批次之间的差异,暂定本品每1kg含镉不得过0.3mg,砷的含量为0.366-0.417mg/kg考虑到不同批次之间的差异,暂定本品每1kg含砷不得过2mg,汞的含量为0mg/kg考虑到不同批次之间的差异,暂定本品每1kg含汞不得过0.2mg,铜的含量为5.2-5.3mg/kg考虑到不同批次之间的差异,暂定本品每1kg含铜不得过20mg。
5、有机氯农药残留量检测
照农药残留量测定法(通则2341有机氯类农药残留量测定法-第一法)测定。
(1)仪器与试药
仪器:7890b气相色谱仪(安捷伦科技有限公司)、bsa224s万分之一天平(赛多利斯科学仪器有限公司)、ika旋转蒸发仪(艾卡(广州)仪器设备有限公司)、zhp-y2102f双层恒温摇床(江苏盛蓝仪器制造有限公司)、tg16-ws台式高速离心机(湖南湘仪实验室仪器开发有限公司)
试药:炒槐花配方颗粒(批号:201711011、201711021、201711031);
标准品:α-六六六标准溶液(中国食品药品检定研究院,批号:gsb05-2276-2008)
β-六六六标准溶液(中国食品药品检定研究院,批号:gsb05-2277-2008)
γ-六六六标准溶液(中国食品药品检定研究院,批号:gsb05-2278-2008)
δ-六六六标准溶液(中国食品药品检定研究院,批号:gsb05-2279-2008)
pp'-dde标准溶液(中国食品药品检定研究院,批号:gsb05-2280-2008)
op'-ddt标准溶液(中国食品药品检定研究院,批号:gsb05-2281-2008)
pp'-ddd标准溶液(中国食品药品检定研究院,批号:gsb05-2282-2008)
pp'-ddt标准溶液(中国食品药品检定研究院,批号:gsb05-2283-2008)
五氯硝基苯标准溶液(中国食品药品检定研究院,批号:gsb05-1845-2008)
石油醚(60-90℃)、丙酮、二氯甲烷、氯化钠、无水硫酸钠、均为分析纯。
(2)溶液的制备
对照品贮备溶液的制备:精密称取六六六(bhc)(α-bhc,β-bhc,γ-bhc,δ-bhc)、滴滴涕(ddt)(pp'-dde,pp'-ddd,op'-ddt,pp'-ddt)及五氯硝基苯(pcnb)农药对照品适量,用石油醚(60-90℃)分别制成每1ml约含10μg的溶液,即得。
混合对照品储备溶液制备:精密量取上述各对照品贮备液0.5ml,置10ml量瓶中,用石油醚(60-90℃)稀释至刻度,摇匀,即得。
混合对照品溶液的制备:精密量取上述混合对照品贮备液,用石油醚(60-90℃)制成每1l分别含10μg、20μg、40μg、60μg、80μg的溶液,即得。
供式品溶液的制备:取供试品,粉碎,取约2g,精密称定,置100ml具塞锥形瓶中,加水20ml浸泡过夜,精密加丙酮40ml,称定重量,超声处理30分钟,放冷,再称定重量,用丙酮补足减失的重量,再加氯化钠约6g,精密加二氯甲烷30ml,称定重量,超声15分钟,再称定重量,用二氯甲烷补足减失的重量,静置(使分层),将有机相迅速移入装有适量无水硫酸钠的100ml具塞锥形瓶中,放置4小时。精密量取35ml,于40℃水浴上减压浓缩至近干,加少量石油醚(60-90℃)如前反复操作至二氯甲烷及丙酮除净,用石油醚(60-90℃)溶解并转移至10ml具塞刻度离心管中,加石油醚(60-90℃)精密稀释至5ml,小心加入硫酸1ml,振摇1分钟,离心(3000转/分钟)10分钟,精密量取上清液2ml,置具刻度的浓缩瓶(见图)中,连接旋转蒸发器,40℃下(或用氮气)将溶液浓缩至适量,精密稀释至1ml,即得。
测定法:分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,注入气相色谱仪,按外标法计算供试品中9种有机氯农药残留量。
(3)色谱条件:以db-1701石英毛细管(30m×0.32mm×0.25μm),ecd电子捕获检测器。进样口温度230℃,检测器温度300℃,不分流进样。程序升温:初始70℃保持1分钟,每分钟7℃升至240℃,保持17分钟。
(4)方法学考察
线性关系考察
吸取上面配置的9种混合标准使用液10、20、40、60、80μg/l,注入气相色谱仪,测得峰面积并求得峰面积与浓度关系的一元线性回归方程。结果见表14-22及图7-15。
表14不同浓度α-六六六标准品的峰面积
表15不同浓度β-六六六标准品的峰面积
表16不同浓度γ-六六六标准品的峰面积
表17不同浓度δ-六六六标准品的峰面积
表18不同浓度pp'-dde标准品的峰面积
表19不同浓度pp'-ddd标准品的峰面积
表20不同浓度op'-ddt标准品的峰面积
表21不同浓度pp'-ddt标准品的峰面积
表22不同浓度五氯硝基苯标准品的峰面积
(5)炒槐花配方颗粒样品9种有机氯类农药残留量测定
取炒槐花配方颗粒3批,照试样测定项下测定,结果见表23。
表23样品9种有机氯类农药残留的测定结果
根据上述3批样品测定结果,炒槐花配方颗粒中六六六的含量为0-0mg/kg考虑到不同批次之间的差异,暂定本品每1kg含六六六不得过0.2mg,滴滴涕的含量为0-0.037mg/kg考虑到不同批次之间的差异,暂定本品每1kg含滴滴涕不得过0.2mg,五氯硝基苯的含量为0-0mg/kg,考虑到不同批次之间的差异,暂定本品每1kg含五氯硝基苯不得过0.1mg。
6、浸出物检测
照醇溶性浸出物测定法(通则2201)项下的热浸法,用30%甲醇作溶剂,不得少于70.0%。本品3批检测结果见表24,均符合规定。
表24浸出物检验结果
7、总黄酮含量检测
照紫外-可见分光光度法(通则0401)测定。
(1)仪器与试药
紫外分光光度计:tu-1810,北京普析通用仪器有限责任公司;
电子分析天平:bsa224s(万分之一),赛多利斯科学仪器有限公司;mcm36(百万分之一),赛多利斯科学仪器有限公司;
甲醇:分析纯,国药集团化学试剂有限公司;
亚硝酸钠:分析纯,天津市科密欧化学试剂有限公司
硝酸铝:分析纯,天津市科密欧化学试剂有限公司
氢氧化钠:分析纯,国药集团化学试剂有限公司
水:娃哈哈纯净水,杭州娃哈哈集团;
芦丁对照品(批号:100080-201811,纯度92.4%),购自中国食品药品检定研究院。
(2)参照《中国药典》2015年版一部中炒槐花【含量测定】项下总黄酮测定条件进行实验,对其进行提取方式、提取溶剂考察,初步确定以下方法,并对其进行方法学的考察。
对照品溶液的制备:取芦丁对照品40mg,精密称定,置20ml量瓶中,加甲醇适量,置水浴上微热使溶解,放冷,加甲醇至刻度,摇匀。精密量取10ml,置100ml量瓶中,加水至刻度,摇匀,即得。
供试品溶液的制备:取本品粉末约1g,精密称定,置具塞锥形瓶中,精密加入50%甲醇溶液100ml,密塞,称重,超声提取30min,取出,放冷,称重,用50%甲醇溶液补足减失的重量,摇匀。精密量取10ml,置100ml量瓶中,加水至刻度,摇匀,即得。
标准曲线的制备:精密量取对照品溶液1ml、2ml、3ml、4ml、5ml、6ml分别置25ml量瓶中,各加水至6ml,加5%亚硝酸钠溶液1ml,摇匀,放置6分钟,加10%硝酸铝溶液1ml,摇匀,放置6分钟,加氢氧化钠试液10ml,再加水至刻度,摇匀,放置15分钟,以相应的试剂为空白,照紫外-可见分光光度法(通则0410),在500nm波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
测定法:精密量取供试品溶液3ml,置25ml量瓶中,照标准曲线制备项下方法,自“加水至6.0ml”起,依法测定吸光度,从标准曲线上读出供试品溶液中芦丁的浓度(μg/ml),计算,即得。
a、供试品溶液制备方法考察
提取方式考察
取本品(批号:201711011)约1g(共2组),精密称定,分别置具塞锥形瓶中,精密加入甲醇溶液100ml,密塞,称重,分别超声30min、回流提取30min,取出,放冷,称重,用甲醇溶液补足减失的重量,摇匀,精密量取10ml,置100ml量瓶中,加水至刻度,摇匀。精密量取3ml,置25ml量瓶中,照标准曲线制备项下方法,自“加水至6ml”起,依法测定吸光度,从标准曲线上读出供试品溶液中芦丁的浓度(μg/ml),计算,即得,结果见表25。
表25提取方式考察结果
结果表明,本品每克取样量下,超声提取方式溶液中芦丁含量明显高于回流提取方式,故确定提取方式为超声处理。
b、提取溶剂考察
取本品(批号:201711011)约1g(共3组),精密称定,分别置具塞锥形瓶中,分别精密加入水100ml、50%甲醇溶液100ml、甲醇100ml,超声处理30分钟,放冷,摇匀。精密量取10ml,置100ml量瓶中,加水至刻度,摇匀。精密量取3ml,置25ml量瓶中,照标准曲线制备项下方法,自“加水至6ml”起,依法测定吸光度,从标准曲线上读出供试品溶液中芦丁的浓度(μg/ml),计算,即得,结果见表26。
表26提取溶剂考察结果
结果表明,本品每克取样量下,以50%甲醇为提取溶剂时,芦丁含量明显高于其他溶剂,故确定提取溶剂为50%甲醇。
(3)方法学考察
a、专属性试验
炒槐花配方颗粒中含有芦丁,为考察辅料是否干扰芦丁的测定,按处方比例称取辅料同法制成辅料样品溶液,记录数据。结果表明,辅料对芦丁的测定无干扰,以本法同时测定本品中芦丁的含量具有专属性(见表27)。
表27辅料与样品芦丁含量
b、线性关系的考察
精密称量芦丁对照品43.540mg(100080-201811,92.4%)置20ml量瓶,加甲醇适量,置水浴上微热使溶解,加甲醇至刻度线,摇匀。精密量取10ml,置100ml量瓶中,加水至刻度,摇匀,即得对照品溶液。
标准曲线的制备:精密量取对照品溶液1ml、2ml、3ml、4ml、5ml、6ml分别置25ml量瓶中,各加水至6ml,加5%亚硝酸钠溶液1ml,混匀,放置6分钟,加10%硝酸铝溶液1ml,摇匀,放置6分钟,加氢氧化钠试液10ml,再加水至刻度,摇匀,放置15分钟,以相应的试剂为空白,照紫外-可见分光光度法(通则0410),在500nm波长处测定吸光度,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。
y=0.01171x-0.0005,相关系数:r2=0.9997
结果表明,芦丁在8.0480ug/ml~48.2880ug/ml范围内线性良好。
表28芦丁线性关系
c、稳定性试验
取本品(批号:201711011),研细,取约1g,精密称定,按正文供试品溶液的制备和测定法项下操作,分别在制备后放置0、1、2、4、6、8h进样测定,测定结果见表29。
表29稳定性试验结果
结果表明,在8小时内测定溶液含量的rsd>2.0%,供试品溶液在0-8h内稳定性不好,样品处理完后,应立即上机,快速测量。
d、重复性试验
取本品(批号:201711011),研细,取约1g(共6份),精密称定,按正文供试品溶液制备和测定法项下进行操作。结果见表30。
表30重复性试验结果(n=6)
结果表明,rsd<2.0%,说明本方法的重复性良好。
e、加样回收率试验
采用加样回收法,取本品(批号:201711011),研细,取约50mg(共6份),精密称定,分别添加芦丁对照品粉末约12.48mg(批号:100080-201811,纯度92.4%),下表中对照品加入量已扣除纯度,按供试品溶液制备方法提取处理。结果见表31。
根据炒槐花配方颗粒(批号:201711011)中芦丁含量为230.67mg/g。
表31芦丁准确度试验结果(n=6)
结果,平均回收率为101.07%,rsd<2.0%,表明本方法准确度好。
(4)炒槐花配方颗粒样品的含量测定
取炒槐花配方颗粒3批,照标准含量测定项下测芦丁的含量,结果见表32。
表32样品的含量测定结果
(5)含量限度的制定
由于配方颗粒在生产中加入了辅料,故在计算中需扣除辅料量,再结合标准汤剂中含量限度范围与出膏率,暂定本品按干燥品计算,每克含芦丁(c27h30o16)不得少于156.0mg。
8、芦丁含量检测
照高效液相色谱法(通则0512)测定。
(1)仪器与试药
高效液相色谱仪:agilentvwd,安捷伦科技有限公司;
色谱柱:agilentc18(4.6*250mm,5μm),lnb17409;agilentc18(4.6*250mm,5μm),lnb18123;agilentc18(4.6*250mm,5μm),lnb18102
电子分析天平:bsa224s(万分之一),赛多利斯科学仪器有限公司;mcm36(百万分之一),赛多利斯科学仪器有限公司;bt25s(十万分之一),赛多利斯科学仪器有限公司;
甲醇:色谱纯,tedia(天地)科技有限公司;
冰醋酸:分析纯,天津市大茂化学试剂厂;
水:娃哈哈纯净水,杭州娃哈哈集团;
芦丁对照品(批号:100080-201811,纯度91.7%),购自中国食品药品检定研究院。
(2)含量测定方法的初步确定
参照《中国药典》2015年版一部中炒槐花【含量测定】项下条件进行实验,所得图谱基线平稳,峰形、分离度良好,故沿用炒槐花含量测定方法,并进行方法的考察。
色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇-1%冰醋酸溶液(32:68)为流动相;检测波长为257nm;柱温为30℃。理论板数按芦丁峰计算应不低于2000。
对照品溶液的制备取芦丁对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得。
供试品溶液的制备取本品粉末0.05g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,称定重量,超声处理(功率250w,频率25khz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,即得。
测定法分别精密吸取对照品溶液与供试品溶液各5μl,注入液相色谱仪,测定,即得。
供试品溶液制备方法考察
a、提取方式考察
取本品(批号:201711011)约0.05g,精密称定,分别置具塞锥形瓶中,精密加入甲醇50ml,密塞,称重,分别超声30分钟、回流提取30分钟,取出,放冷,称重,用甲醇补足减失的重量,摇匀,滤过,即得。结果见表33。
表33提取方式考察结果
结果表明,不同提取方式对应的每克取样量下超声提取芦丁峰面积大于回流提取,故确定提取方式为超声处理。
b、提取溶剂考察
取本品(批号:201711011)0.05g(共3组),精密称定,置具塞锥形瓶,分别精密加入甲醇50ml、50%甲醇溶液50ml、水50ml,称定重量,超声30分钟,取出,放冷,称重,以对应溶剂补足减失的重量,摇匀,微孔滤膜滤过,即得。结果见表34。
表34炒槐花配方颗粒提取溶剂
由此可知,甲醇为提取溶剂时,每克取样量下芦丁峰面积最高,综合考虑,确定提取溶剂为甲醇。
因此确定供试品制备方法为:取本品粉末约0.05g,精密称定,置具塞锥形瓶中,精密加入甲醇50ml,密塞,称定重量,超声处理(功率250w,频率25khz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
(3)方法学考察
a、专属性试验
炒槐花配方颗粒中含有芦丁,为考察辅料是否干扰芦丁的测定,按处方比例称取辅料同法制成辅料样品溶液,记录色谱图。结果表明,辅料糊精溶剂色谱中在与芦丁相应的保留时间没有色谱峰,表明辅料对芦丁的测定无干扰,以本法同时测定本品中芦丁的含量具有专属性(见图17)。
b、线性关系的考察
精密称取芦丁对照品7.205mg(100080-201811,91.7%)置25ml容量瓶中,加甲醇适量,超声溶解,加甲醇至刻度线,摇匀,作为储备液。分别精密量取上述储备液1ml、2ml、2.5ml至10ml的容量瓶中、1ml至2ml的容量瓶中,加甲醇至刻度线,混匀,即得对照品溶液。
分别精密吸取对照品溶液5μl,注入液相色谱仪,照液相色谱法(2015年版中国药典四部通则0512)测定。以浓度(mg/ml)为横坐标,峰面积为纵坐标,绘制标准曲线。芦丁回归方程:
y=10571x+27.281,相关系数:r2=1
结果表明,芦丁在0.0264mg/ml~0.264mg/ml范围内线性良好。
表35芦丁线性关系
c、仪器精密度试验
精密吸取芦丁对照品溶液(浓度为0.132mg/ml)5μl,按正文【含量测定】项下的色谱条件和测定法项下进行操作,连续进样6次,测定结果见表36。
表36仪器精密度试验结果(n=6)
结果表明,rsd<2.0%,仪器精密度良好。
d、稳定性试验
取本品(批号:201711011),研细,取约0.05g,精密称定,按正文供试品溶液的制备和测定法项下操作,分别在制备后放置0、2、4、8、12、18、24h进样测定,测定结果见表37。
表37稳定性试验结果
结果表明,rsd<2.0%,供试品溶液在24h内稳定性良好。
e、耐用性验证
取样品溶液,分别连接agilentc18(4.6*250mm,5μm),lnb17409;agilentc18(4.6*250mm,5μm),lnb18123;agilentc18(4.6*250mm,5μm),lnb18102;三个不同批号色谱柱,在同一色谱条件下对同一炒槐花配方颗粒样品进行进样,色谱峰型良好,分离度良好,结果表明该色谱条件具有普适性。
f、重复性试验
取本品(批号:201711011),研细,取约0.05g(共6份),精密称定,按正文供试品溶液制备和测定法项下进行操作。结果见表38。
表38重复性试验结果(n=6)
结果表明,rsd<2.0%,说明本方法的重复性良好。
g、加样回收率试验
采用加样回收法,取本品(批号:201711011),研细,取约0.025g(共6份),精密称定,分别添加对照品粉末约3mg(批号:100080-201811,纯度91.7%),按供试品溶液制备方法提取处理。结果见表39。
根据炒槐花配方颗粒(批号:201711011)中芦丁含量为122.02mg/g。
表39芦丁准确度试验结果(n=6)
结果,平均回收率为99.21%,rsd<2.0%,表明本方法准确度较好。
(4)配方颗粒样品的含量测定
取炒槐花配方颗粒3批,照标准含量测定项下测芦丁的含量,结果见表40。
表40样品的含量测定结果
(5)含量限度的制定
由于配方颗粒在生产中加入了辅料,故在计算中需扣除辅料量,再结合标准汤剂中含量限度范围与出膏率,暂定本品按干燥品计算,每克含芦丁(c27h30016)不得少于89.0mg。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!