一种兼并引物的设计筛选方法与流程
本发明涉及生物信息学分析领域,特别是涉及一种兼并引物的设计筛选方法。
背景技术:
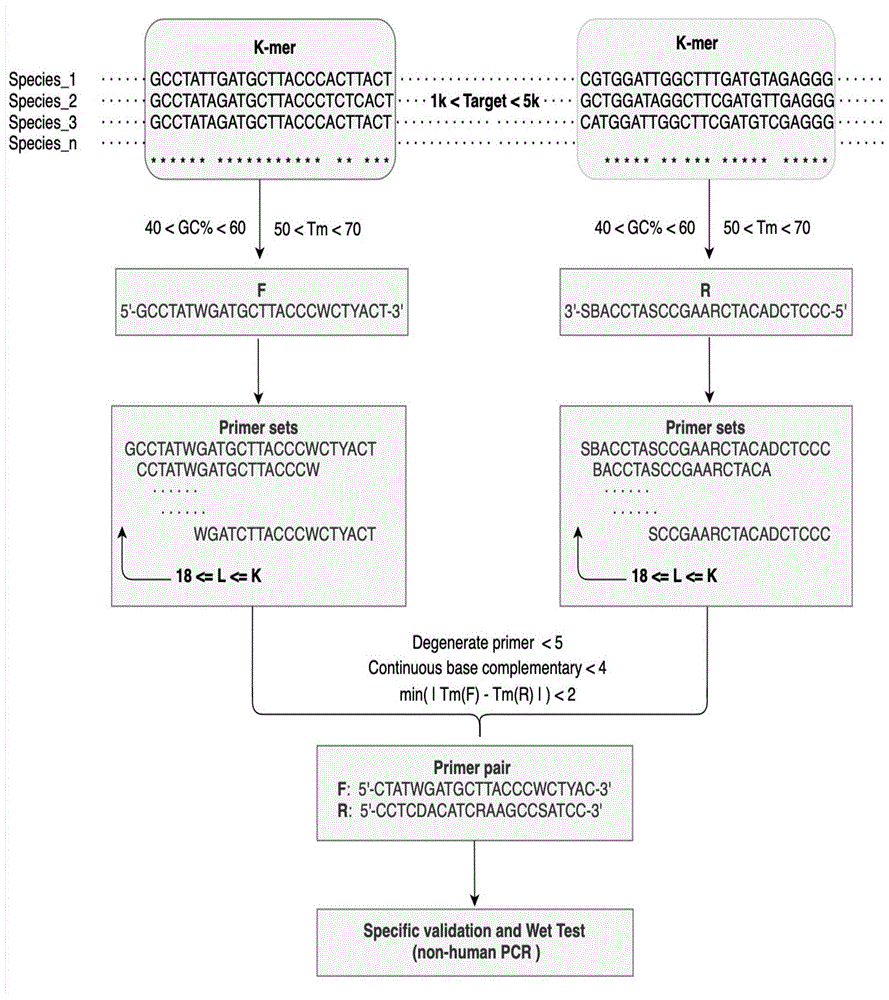
:在临床病原诊断领域,常用检测方法为pcr扩增、16srrna测序和宏基因组测序,其中pcr扩增主要面向靶向扩增检测,16s测序物种区分度不足以作病原鉴定,宏基因组测序在解决大量的宿主dna占比问题上仍有很大的挑战。鉴于常规检测方法中的各种局限和挑战,提供一套临床常见病原菌的靶向检测解决方案尤为重要。病原靶向检测需要富集目标物种特定序列后测序作物种鉴定,达到临床关注病原检测的目的。这就要求富集引物能够覆盖到多种病原物种,且富集序列需具备一定地物种区分度用于后续作物种鉴定分析。靶向富集技术需要在物种序列的保守区设计兼并引物,同时要求富集区间包含一定比例地可变区用于物种区分。靶向富集序列可应用于下一代测序(nextgenerationsequencing,ngs)和纳米孔测序(oxfordnanoporetechnologies,ont)。关于靶向富集引物的设计筛选,现有技术主要根据蛋白保守性domain结构作兼并引物设计,该方法需要预知基因对应地蛋白序列,且引物对组合时具有一定地随意性,比较适用于同属相似物种间的引物设计,而对于非相似物种的引物设计并不适用。目前linux端引物设计经常调用primer3软件,但该软件只适用于提供特定序列设计非兼并引物,并不支持兼并引物设计。为推进靶向富集在临床病原检测中的应用,亟需开发一种靶向富集兼并引物的设计筛选方法。有鉴于此,提出本发明。技术实现要素:本发明要解决的技术问题是提供一种兼并引物的设计筛选方法。考虑到病原靶向检测需要富集目标物种特定序列,然后测序进行物种鉴定,以达到临床病原检测目的,这就要求富集引物能够覆盖到多种病原物种,且富集序列需具备一定地物种区分度用于后续物种鉴定分析。为推进靶向富集在临床病原检测中的应用,本发明通过深入挖掘大量多物种序列数据,从物种基因或全基因组多重序列比对结果入手,惊奇发现在多物种基因多重序列比对下,可通过k-mer方法将所有序列划分为k长度的短序列,便于后续对引物设计区域进行筛选。同时为了设计兼并引物,本发明还开发了一种碱基转码规则及其反向互补规则,在物种个数等于序列数时,保障了引物设计区域的特异性,对该区域的k-mer进行碱基转码形成兼并k-mer,同时记录其位置信息和反向互补序列,进一步对兼并k-mer以不同引物长度l进行划分,便于筛选最优引物对。基于对此的研究发现,本发明构建了一套兼并引物的设计筛选方法,并通过大量多物种兼并引物设计对该方法进行验证,又利用生信、湿实验方法对兼并引物进行特异性验证,结果表明该方法所设计出的兼并引物可以对多物种进行特异性扩增。因此,本发明的第一目的是提供一种兼并引物的设计筛选方法。本发明的第二目的是提供一种兼并引物的设计筛选产品。为实现上述目的,本发明采用如下技术方案:一种兼并引物的设计筛选方法,所述方法包括:1)多重比对:物种序列进行多重比对;2)k-mer划分:多重比对序列进行k-mer划分;3)形成兼并k-mer:划分后的多重比对k-mer以满足基础引物设计条件进行筛选,筛选后的k-mer进行同位置的碱基转码,形成兼并k-mer序列f和其反向互补k-mer序列r;4)形成候选引物集合:以不同引物长度l划分步骤3)后的序列,分别形成候选k-merf引物集合和候选k-merr引物集合;5)组合引物对:对候选k-merf引物集合和候选k-merr引物集合中的引物进行两两引物对组合,筛选最优引物对。在一些实施方式中,所述步骤5)中筛选的条件为:兼并碱基最大个数为0-6,优选为4;f与r引物间连续碱基互补最大个数为4-5,优选为4;兼并引物tm值取值范围为55-70℃,gc含量取值范围40%-60%。在一些实施方式中,所述步骤3)还包括统计碱基转码后k-mer中的兼并碱基占比p,筛选p<p(max)的k-mer序列;在一些优选的实施方式中,所述p(max)取值范围为20%-30%;更优选地为25%。在一些实施方式中,所述步骤3)中基础引物设计条件为:序列不存在gap,gc含量为40%-60%,引物tm为50-70℃。在一些实施方式中,所述步骤3)中碱基转码规则为a→a,t→t,c→c,g→g,a/g→r,c/t→y,a/c→m,g/t→k,c/g→s,a/t→w,a/c/t→h,c/g/t→b,a/c/g→v,a/g/t→d,a/c/g/t→n;转码后的反向互补规则为a→t,t→a,c→g,g→c,r→y,y→r,m→k,k→m,s→s,w→w,h→d,d→h,b→v,v→b,n→n;在一些实施方式中,所述步骤5)中兼并引物tm值计算公式为tm=4×(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)+2×(a+t+w+(r+y+m+k+n)/2+(h+d)×2/3+(b+v)/3),兼并引物gc含量计算公式为gc=(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)/l。在一些实施方式中,所述步骤2)中所述k-mer,k取值范围为20-38;优选为25。在一些实施方式中,所述步骤4)中所述l取值范围为l(min)≤l≤k;l(min)取值范围为18-21,优选为20。在一些实施方式中,所述步骤5)中所述引物对组合取决于目标扩增区间长度t,t的取值范围为80-5k;优选为1-5k。本发明还提供一种兼并引物的设计筛选系统/装置/产品,其特征在于,所述系统/装置/产品包含如下模块:1)多重比对模块:物种序列进行多重比对;2)k-mer划分模块:多重比对序列进行k-mer划分;3)形成兼并k-mer模块:划分后的多重比对k-mer以满足基础引物设计条件进行筛选,筛选后的k-mer进行同位置的碱基转码,形成兼并k-mer序列f和其反向互补k-mer序列r;4)形成候选引物集合模块:以不同引物长度l划分步骤3)后的序列,分别形成候选k-merf引物集合和候选k-merr引物集合;5)组合引物对模块:对候选k-merf引物集合和候选k-merr引物集合中的引物进行两两引物对组合,筛选最优引物对。在一些实施方式中,所述5)组合引物对模块中筛选条件为:兼并碱基最大个数为0-6,优选为4;f与r引物间连续碱基互补最大个数为4-5,优选为4;兼并引物tm值取值范围为55-70℃,gc含量取值范围40%-60%。在一些实施方式中,所述3)形成兼并k-mer模块还包括统计碱基转码后k-mer中的兼并碱基占比p,筛选p<p(max)的k-mer序列;优选地,所述p(max)取值范围为20%-30%,更优选地为25%;在一些实施方式中,所述3)形成兼并k-mer模块中,基础引物设计条件为序列不存在gap,gc含量为40%-60%,引物tm为50-70℃。在一些实施方式中,所述2)k-mer划分模块中k取值范围为20-38,优选为25;在一些实施方式中,所述4)形成候选引物集合模块中l取值范围为l(min)≤l≤k,l(min)取值范围为18-21,优选为20;在一些实施方式中,所述5)组合引物对模块中引物对组合取决于目标扩增区间长度t,t的取值范围为80-5k;优选为1-5k。本发明还提供一种计算机可读介质,其存储有计算机程序,所述计算机程序被处理器执行时,实现上述任一项所述兼并引物的设计筛选方法。本发明还提供一种电子设备,其特征在于,包括处理器以及存储器,所述存储器上存储一条或多条可读指令,所述一条或多条可读指令被所述处理器执行时,实现上述任一项所述兼并引物的设计筛选方法。本发明还提供上述任一所述的兼并引物的设计筛选系统/装置/产品,计算机可读介质或电子设备的应用,其特征在于,所述应用为以下任一应用:1)在多重pcr扩增中的应用;2)在宏基因组病原微生物靶向富集中的应用;3)在宏基因组病原微生物检测中的应用;4)在纳米孔测序检测中的应用。本发明还提供一种通过上述方法设计筛选的针对腺病毒ont靶向富集的引物对,具体序列参见seqidno.5-6。本发明有益的技术效果:1.本发明通过深入挖掘大量多物种序列数据,从物种基因或全基因组多重序列比对结果入手,惊奇发现在多物种基因多重序列比对下,可通过k-mer方法设计筛选兼并引物。2.本发明采用k-mer方法从物种多重序列比对结果中设计兼并引物,克服了第三方软件无法设计兼并引物的缺陷,首次提供一种能够有效应用于多物种靶向富集兼并引物设计的方法;3.本发明通过优化引入兼并引物tm、gc值计算方法,可筛选出tm值差异最小、gc含量适中的引物对作为候选引物,确保了引物对的可用性;4.本发明通过大数据分析优化制定一套适合设计兼并引物的碱基转码、反向互补和引物对筛选规则。附图说明为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1:靶向富集兼并引物设计筛选流程;图2:湿实验pcr产物电泳结果。具体实施方式下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围,并且所述实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。部分术语定义除非在下文中另有定义,本发明具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。如本发明中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由...组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。本发明中的术语“大约”、“大体”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的±10%,优选±5%。在提及单数形式名词时使用的不定冠词或定冠词例如“一个”或“一种”,“所述”,包括该名词的复数形式。此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本发明描述的实施方案能以不同于本发明描述或举例说明的其它顺序实施。以下术语或定义仅仅是为了帮助理解本发明而提供。这些定义不应被理解为具有小于本领域技术人员所理解的范围。本发明中的部分技术术语解释如下:本发明所述的“k-mer”是指将一条序列连续切割,逐个碱基划动得到k个碱基的子字符串,例如reads长度为l,k-mer长度设为k,则产生的k-mers数目为:l-k+1,再例如序列aactgact,设置k为3,则可以将其分割为aac、act、ctg、tga、gac、act共6个k-mers。本发明所述的“碱基转码”是指根据密码子的兼并性将两个或者更多不同的碱基用一个符号代替,例如a/g→r,c/t→y,a/c→m,g/t→k,c/g→s,a/t→w,a/c/t→h,c/g/t→b,a/c/g→v,a/g/t→d,a/c/g/t→n;本发明所述的“位置信息”是指所选k-mer在多重比对结果中的位置。可以理解的是,任何包含上述设计方法的程序、软件、系统等应用都在本发明的保护范围之内。本发明通过附图和如下实施例进一步描述,所述的附图和实施例只是为了例证本发明的特定实施方案,不应理解为以任何方式限制本发明范围之意。除非另外说明,本发明中所公开的实验方法均采用本
技术领域:
常规技术,实施例中所用的试剂和原材料均可由市场购得。实施例1靶向富集兼并引物设计方法的优化开发开发本发明经大量生信分析优化开发如下靶向富集兼并引物的设计筛选方法:1)多重比对:使用mafft软件对物种序列(全基因组或基因,一个物种对应一个id)作多重序列比对,保存为fasta格式;2)k-mer筛选:使用k-mer方法从物种多重序列比对结果中以满足引物设计条件进行过滤筛选。所述k-mer指的是将一条序列连续切割,逐个碱基划动得到的一序列长度为k的核苷酸序列。常规引物所述k取值范围通常为20-38,取值25为最优;所述引物设计条件为不允许存在gap,gc含量满足40%-60%之间,退火温度tm值满足50-70℃之间,其中gc含量计算公式为(c+g)/k,tm值计算公式为4×(c+g)+2×(a+t);3)获得兼并k-mer:筛选与物种个数一致的k-mer多重比对结果进行同位置碱基转码获得兼并k-mer序列(f),同时记录k-mer起始、终止位置信息和反向互补k-mer序列(r)。所述碱基转码规则为a→a,t→t,c→c,g→g,a/g→r,c/t→y,a/c→m,g/t→k,c/g→s,a/t→w,a/c/t→h,c/g/t→b,a/c/g→v,a/g/t→d,a/c/g/t→n;所述序列互补规则为a→t,t→a,c→g,g→c,r→y,y→r,m→k,k→m,s→s,w→w,h→d,d→h,b→v,v→b,n→n。4)兼并k-mer序列过滤:统计碱基转码后k-mer中的兼并碱基占比p,根据占比p是否小于p(max)来判定转码k-mer的保守性,获得保守性k-mer序列。所述p(max)取值范围为20%-30%,以25%最佳。5)获得获选引物集合:根据最小引物长度l(min),分别从对保守性k-mer序列(包含f/和r的k-mer序列),以不同引物长度l,中逐个碱基划分出划动得到所有不同引物长度l的集合,分别获得候选的k-merf引物集合和k-merr引物集合。所述l取值范围为l(min)≤l≤k;l(min)取值范围为18-21,以20最佳。6)组合引物对,获取最优引物对:1.选保守性k-merf引物集合和另一保守性k-merr引物集合进行两两引物对组合,所述组合条件为扩增区间大小t,所述t取值范围为80-5k,优选的为1-5k;2.从引物对组合中筛选出最优引物对,所述组合限定筛选条件为兼并碱基最大个数n1,f与r间连续碱基互补最大个数n2,及兼并引物tm值差异和gc含量,以tm值差异最小的引物对为候选引物。n1取值范围为0-6,以4最佳;n2取值为4-5,以4最佳;tm值取值范围为55-70,且引物对间tm差异不超过2度,其中兼并引物tm值计算公式为tm=4×(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)+2×(a+t+w+(r+y+m+k+n)/2+(h+d)×2/3+(b+v)/3);gc含量取值范围40%-60%,其中兼并引物gc含量计算公式为(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)/l。所述引物不能出现连续4个重复碱基;引物不能出现2个以上连续3个重复碱基;引物对首末端不能出现兼并序列;引物3’末端不能以a碱基结尾。7)双重验证:1)生信验证:根据兼并引物位置提取所选物种对应地碱基序列与nt库比库验证引物的特异性;其中病毒物种的特异性为没有人/细菌/真菌/寄生虫的比对结果,细菌物种的特异性为没有人/病毒/真菌/寄生虫的比对结果,真菌物种的特异性为没有人/细菌/病毒/寄生虫的比对结果。2)湿实验验证:经生信物种特异性验证后,兼并引物进行湿试验靶向富集验证。实施例2腺病毒ont靶向富集引物的具体设计实验1)数据准备从refseq/genbank库筛选靶向12种腺病毒的参考基因组,如下表所示。使用mafft软件对腺病毒全基因组作多重序列比对,并将结果保存为fasta格式。表1靶向12种腺病毒的参考基因组organism_strainassembly_accessionassembly_levelhumanadenovirusb3gca_000880515.1completegenomehumanadenovirus55gca_006401535.1completegenomehumanadenovirus7gcf_000859485.1completegenomehumanmastadenoviruscgcf_000845085.1completegenomehumanadenovirus1gcf_000858645.1completegenomehumanadenovirus2gcf_000859465.1completegenomehumanadenovirus5gcf_000857865.1completegenomehumanmastadenovirusdgcf_000845985.1completegenomehumanmastadenovirusegcf_000859665.1completegenomehumanadenoviruse4gcf_006415355.1completegenomehumanmastadenovirusfgcf_000846685.1completegenomehumanadenovirus52gcf_006448415.1completegenome2)兼并引物设计2.1)对腺病毒全基因组多重序列比对结果切25-mer,以过滤掉gap,同时满足gc含量在40-60%、tm值在50-70之间为条件进行过滤,同时保留25-mer起始/终止位置信息。所述tm值计算公式为tm=4×(c+g)+2×(a+t)。2.2)以同位置25-mer个数和物种数一致为条件进行保守性25-mer筛选,并转化为兼并序列,同时记录k-mer起始、终止位置信息和反向互补k-mer序列(r)。所述转码规则a→at→t,c→c,g→g,a/g→r,c/t→y,a/c→m,g/t→k,c/g→s,a/t→w,a/c/t→h,c/g/t→b,a/c/g→v,a/g/t→d,a/c/g/t→n;所述序列互补规则为a→t,t→a,c→g,g→c,r→yy→r,m→k,k→m,s→s,w→w,h→d,d→h,b→v,v→b,n→n。2.3)以兼并序列个数占比p小于p(max)进行保守性25-mer筛选,p(max)为25%。2.4)对保守性k-mer序列(包含f/和r的k-mer序列),以不同引物长度l,逐个碱基划动得到所有不同引物长度l的集合,分别获得候选的k-merf引物集合和k-merr引物集合。所述l取值范围为l(min)≤l≤k;l(min)取值范围为18-21,以20最佳。按照target扩增区间1k-5k为条件进行25-mer和反向互补序列进行引物对组合,从25-mer和反向互补序列中分别切出各种引物长度l集合,从引物集合间挑选出tm值差异最小的一对作为候选引物,并记录引物gc含量、长度、多重序列比对结果中的位置信息。2.5)按照target扩增区间1k-5k为条件,将k-merf引物和k-merr引物进行引物对组合,再从引物对组合中筛选出最优引物对,所述组合限定筛选条件为兼并碱基引物最大个数n1,f与r和连续间连续碱基互补最大个数n2,及兼并引物tm值差异和gc含量,以n1取值范围为0-6(以4最佳)、n2取值为4-5(以4最佳)、tm值取值范围为55-70℃,且引物对间tm值差异不超过2℃、gc含量取值范围40%-60%的引物对为候选引物。引物对组合时进行仍需满足以下条件:引物对间不能出现连续5个碱基的互补;引物不能出现连续4个重复碱基;引物不能出现2个以上连续3个重复碱基;引物对首末端不能出现兼并序列;引物3’末端不能以a碱基结尾。其中兼并引物tm值的计算公式为tm=4×(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)+2×(a+t+w+(r+y+m+k+n)/2+(h+d)×2/3+(b+v)/3);兼并引物gc含量计算公式为gc=(c+g+s+(r+y+m+k+n)/2+(h+d)/3+(b+v)×2/3)/l。2.6)生信验证及过滤:根据兼并引物位置提取所选物种对应地碱基序列与nt库作blastn-short比对,过滤掉能比对上人、细菌、真菌或寄生虫的兼并引物,以确保候选引物的物种特异性。2.7)按上述方法设计腺病毒兼并引物结果如下表:进一步对引物最适合长度作优化筛选,以计算得到的tm值是否接近60℃来判断最小引物长度为最优。3)结果由上表可知,其中最优的长度为20,因此最终获得引物对序列如下实施例3腺病毒ont靶向富集兼并引物的湿实验验证1)试剂耗材无酶无菌水:thermofisher,nuclease-freewater(notdepc-treated)(货号:am9937);qubit荧光定量仪dna检测试剂盒:qubit1xdsdnahsassaykit(货号:q33231);pcr扩增酶:gxldnapolymerase(r050a)。2)引物验证以腺病毒标准品提取的核酸为模板,同时在pcr反应体系中增加gdna模拟真实的临床样本,分别进行不同的tm值(52℃/55℃/58℃)的筛选,配置体系和反应条件如下表:其中primer1、primer2分别指实施例2中腺病毒兼并引物primerf、primerr。3)pcr产物浓度结果4)pcr产物4200电泳结果以pcr55℃pcr产物为例a1(l):gdnamarkerb1:腺病毒pcr产物模板目的条带2300bp,条带单一且与预期大小一致c1:阴性对照gdnad1:阴性对照zymo4200电泳结果如图2所示。5)结果腺病毒引物,测试了不同的退火温度,该引物在gdna,zymo(8种细菌2种真菌)以及水里不存在非特异性扩增,阳性标准品达到了有效的扩增效果。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。序列表<110>江苏先声医学诊断有限公司北京先声医学检验实验室有限公司江苏先声医疗器械有限公司<120>一种兼并引物的设计筛选方法<160>8<170>siposequencelisting1.0<210>1<211>19<212>dna<213>人工序列(artificialsequence)<400>1tvagbgcvgakgcrtacat19<210>2<211>19<212>dna<213>人工序列(artificialsequence)<400>2ccaccacvytvaactacct19<210>3<211>19<212>dna<213>人工序列(artificialsequence)<400>3tvagbgcvgakgcrtacat19<210>4<211>19<212>dna<213>人工序列(artificialsequence)<400>4ccaccacvytvaactacct19<210>5<211>20<212>dna<213>人工序列(artificialsequence)<400>5gtvagbgcvgakgcrtacat20<210>6<211>20<212>dna<213>人工序列(artificialsequence)<400>6gccaccacvytvaactacct20<210>7<211>21<212>dna<213>人工序列(artificialsequence)<400>7gtvagbgcvgakgcrtacatg21<210>8<211>22<212>dna<213>人工序列(artificialsequence)<400>8gccaccacvytvaactacctbt22当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1