一种在胃肠道内稳定的口服纳米粒子及其制备方法和应用
1.本发明是涉及一种在胃肠道内稳定的口服纳米粒子及其制备方法和应用,属于药用纳米材料技术领域。
背景技术:
2.难溶性药物因溶解度小,难以达到治疗所需的血药浓度和体内消除速度过快等因素导致其口服生物利用度较低。而口服纳米粒子可以改善难溶性药物的溶解性,是提高难溶性药物口服生物利用度的有效方法之一,但口服纳米粒子进入胃肠道时将面临如胃液和肠液的ph值、酶、盐类物质等复杂而严峻的生理环境的破坏影响,因此口服纳米粒子能在胃肠道生理环境下保持良好稳定性是其更好发挥潜在作用如黏液层穿越、口服吸收及提高生物利用度的前提。
3.虽然利用亲水性聚合物如透明质酸、壳聚糖及其衍生物等可以提高纳米粒子在体外的物理稳定性,但在胃肠道内,因受胃液和肠液中有机物和离子的影响,含上述聚阳离子或聚阴离子稳定剂的纳米粒子很容易发生聚集从而导致纳米粒子粒径增大,进而会影响纳米粒子在胃肠道内的稳定性和口服吸收及生物利用度。因此,本领域亟需研发在胃肠道内稳定的口服纳米粒子,以利于其负载难溶药物穿越黏液层实现细胞顺利摄取,进而真正达到提高难溶药物口服生物利用度的目的。
技术实现要素:
4.针对现有技术存在的上述问题和需求,本发明的目的是提供一种在胃肠道内稳定的口服纳米粒子及其制备方法和应用。
5.为实现上述发明目的,本发明采用的技术方案如下:
6.一种在胃肠道内稳定的口服纳米粒子,是由聚乳酸-羟基乙酸共聚物(plga)与槐糖脂和二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000(dspe-peg2000)依次按质量比为:1:(1~4):(0.02~0.10)采用改良的纳米沉淀法制备而得的平均粒径小于200nm的纳米粒子。
7.一种制备在胃肠道内稳定的口服纳米粒子的方法,为改良的纳米沉淀法,具体包括如下步骤:
8.a)取聚乳酸-羟基乙酸共聚物(plga)溶于有机溶剂中,制得溶液a;
9.b)取槐糖酯溶于去离子水中,制得溶液b;
10.c)取二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000溶于无水乙醇中,制得溶液c;
11.d)将溶液c加入已预热到60~70℃的溶液b中,搅拌,制得混合溶液d;
12.e)将溶液a滴加或倒入到混合溶液d中,搅拌,制得混合溶液e;
13.f)使混合溶液e旋转蒸发挥去有机溶剂,即得均匀的胶体分散液。
14.一种优选方案,所述方法还包括如下步骤:
15.g)向步骤f)所得胶体分散液中加入冻干保护剂进行冷冻干燥。
16.一种优选方案,所述聚乳酸-羟基乙酸共聚物(plga)选用乳酸与乙醇酸的摩尔比
值为50:50或75:25的酯封端规格。
17.一种优选方案,步骤a)中的有机溶剂选用乙腈或丙酮。
18.一种优选方案,溶液a中的聚乳酸-羟基乙酸共聚物的浓度为2~6mg
·
ml-1
。
19.一种优选方案,溶液b中的槐糖酯的浓度为2~10mg
·
ml-1
。
20.一种优选方案,溶液c中的二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000的浓度为8~12mg
·
ml-1
。
21.一种优选方案,混合溶液d中的槐糖酯与二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000的质量比为(25~50):1。
22.一种优选方案,混合溶液e中的有机溶剂与去离子水的体积比为1:1~1:5。
23.一种优选方案,所述冻干保护剂为甘露醇。
24.一种优选方案,冷冻干燥工艺为:向步骤f)所得胶体分散液中加入冻干保护剂,添加含量为8~12wt%,先在-90℃~-70℃预冻1~3小时,然后在-50℃~-40℃及真空度<100pa下冻干45~50小时。
25.上述的在胃肠道内稳定的口服纳米粒子可用作口服难溶药物(如水飞蓟素)的载体。
26.与现有技术相比,本发明具有如下有益技术效果:
27.1)因本发明所述的口服纳米粒子,是由聚乳酸-羟基乙酸共聚物(plga)与槐糖脂和二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000(dspe-peg2000)制备而得,因此具有可生物降解、无毒、生物相容性好的优点;
28.2)实验证明:本发明所述的口服纳米粒子在胃肠道中具有非常好的稳定性,可在模拟胃肠液中保持24小时以上不发生粒径增大的团聚现象,而由单一dspe-peg2000包覆的plga纳米粒及由磷脂lipoid s100与槐糖脂共包覆的plga纳米粒均在模拟胃肠液中保持2小时就发生了粒径骤增的团聚现象,进一步说明本发明采用槐糖脂和二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000共同包覆plga制备纳米粒子,产生了出乎意料的技术效果,即:获得了在胃肠道中非常稳定的口服纳米粒子;
29.3)实验还证明:采用本发明所述的口服纳米粒子包载口服难溶药物水飞蓟素时,与市售药物益肝灵相比,可使相对生物利用度达到236.89%,使口服难溶药物水飞蓟素的口服生物利用度得到了显著提高;
30.4)另外,本发明所述的口服纳米粒子的制备工艺简单,将油相滴加或倒入水相均可,无需调ph和后续离心或滤过,所得的纳米粒粒径较小(平均粒径《200nm),易于实现工业化;
31.总之,本发明相对于现有技术,不仅产生了出乎意料的技术效果,而且获得了显著性进步,对解决口服难溶药物的口服生物利用度低的难题具有重要价值。
附图说明
32.图1为各组纳米粒子在模拟胃液中的粒径变化,其中:spdns组为本发明实施例4所获得的纳米粒子,pdns组为对比例1所获得的纳米粒子,sp
l
ns组为对比例2所获得的纳米粒子;
33.图2为各组纳米粒子在模拟肠液中的粒径变化,其中:spdns组为本发明实施例4所
获得的纳米粒子,pdns组为对比例1所获得的纳米粒子,sp
l
ns组为对比例2所获得的纳米粒子。
34.图3为本发明所述纳米粒子(spdns)和市售益肝灵片(ygl)的血药浓度-时间曲线图。
35.图4为图3的局部放大图。
具体实施方式
36.下面结合实施例和对比例对本发明技术方案做进一步详细、完整地说明。
37.实施例1
38.a)称取65.4mg plga(75:25),溶于16ml乙腈中,得到plga的浓度为4.1mg
·
ml-1
的溶液a;
39.b)称取236.6mg槐糖脂溶于47ml去离子水中,制得槐糖酯的浓度为5.0mg
·
ml-1
的溶液b;
40.c)称取5.1mg dspe-peg2000溶于500μl无水乙醇中,制得dspe-peg2000的浓度为10.2mg
·
ml-1
的溶液c;
41.d)取150μl溶液c加入已预热到65℃的15ml溶液b中,于100r
·
min-1
搅拌30min,制得混合溶液d,即:水相,其中:槐糖酯与dspe-peg2000的质量比为50:1;
42.e)称取约8.8mg水飞蓟素溶于5ml溶液a中,得油相;然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液e;
43.f)使混合溶液e于40℃水浴下旋转蒸发挥去有机溶剂,即得均匀的胶体分散液(对所得样品溶液稀释2倍后采用激光笔照射,可产生明显的丁达尔效应);
44.g)向步骤f)所得胶体分散液中加入10%的甘露醇进行冷冻干燥:先在-80℃预冻1小时,然后在-50℃~-40℃及真空度<100pa下冻干48小时,即得本发明所述的口服纳米粒子,简记为:spdns。
45.重复上述步骤,共制备3份平行样品,对所得3份平行样品进行如下检测:
46.采用粒径分析仪测定纳米粒的粒径和zeta电位;
47.采用超滤离心法测定纳米粒的包封率(ee)和载药量(dl);
48.检测结果详见表1所示。
49.表1
50.样品编号size/nmpdizeta potential/mvee/%dl/%1173.6
±
2.50.128
±
0.020-0.563
±
2.07392.002.602162.4
±
1.20.156
±
0.0110.264
±
2.49192.792.723159.3
±
0.70.170
±
0.0091.499
±
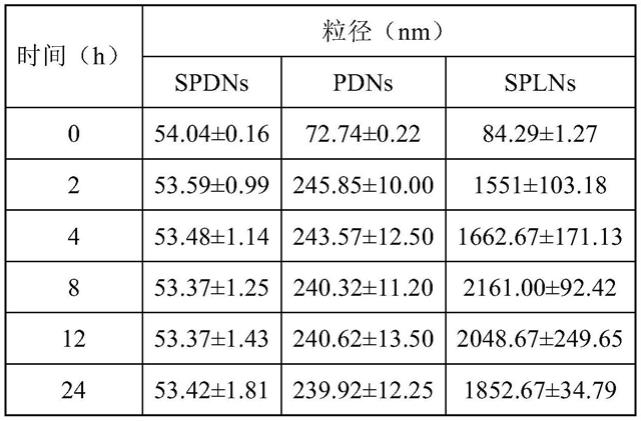
1.20997.002.81
51.由表1所示结果可见:采用本实施例方法可获得分布均匀的粒径小于200nm的纳米粒子,且所述纳米粒子对难溶药物的包封率(ee)高于92%,载药量(dl)大于2.5%。
52.实施例2
53.a)称取80.4mg plga(50:50),溶于16ml丙酮中,得到plga的浓度为5.0mg
·
ml-1
的溶液a;
54.b)称取152.9mg槐糖脂溶于30ml去离子水中,制得槐糖酯的浓度为5.1mg
·
ml-1
的
溶液b;
55.c)称取14.2mg dspe-peg2000溶于1.4ml无水乙醇中,制得dspe-peg2000的浓度为10.1mg
·
ml-1
的溶液c;
56.d)取200μl溶液c加入已预热到65℃的10ml溶液b中,于100r
·
min-1
搅拌30min,制得混合溶液d,即:水相,其中:槐糖酯与dspe-peg2000的质量比为25:1;
57.e)称取约9.4mg水飞蓟素溶于5ml溶液a中,得油相;然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液e;
58.f)使混合溶液e于40℃水浴下旋转蒸发挥去有机溶剂,即得均匀的胶体分散液(对所得样品溶液采用激光笔照射,可产生明显的丁达尔效应);
59.g)向步骤f)所得胶体分散液中加入10%的甘露醇进行冷冻干燥:先在-80℃预冻1小时,然后在-50℃~-40℃及真空度<100pa下冻干48小时,即得本发明所述的口服纳米粒子,简记为:spdns。
60.重复上述步骤,共制备3份平行样品,对所得3份平行样品参照实施例1进行粒径、粒径分布、zeta电位、包封率(ee)和载药量(dl)进行检测。
61.检测结果详见表2所示。
62.表2
63.样品编号size/nmpdizeta potential/mvee/%dl/%168.8
±
0.40.091
±
0.0221.780
±
0.62494.573.24265.8
±
0.40.130
±
0.0130.472
±
0.21294.473.31364.9
±
0.50.117
±
0.009-0.387
±
1.34895.473.58
64.由表2所示结果可见:采用本实施例方法可获得分布均匀的粒径小于100nm的纳米粒子,且所述纳米粒子对难溶药物的包封率(ee)高于94%,载药量(dl)大于3.0%。
65.实施例3
66.a)称取52.7mg plga(50:50),溶于17ml乙腈中,得到plga的浓度为3.1mg
·
ml-1
的溶液a;
67.b)称取112.1mg槐糖脂溶于28ml去离子水中,制得槐糖酯的浓度为4.0mg
·
ml-1
的溶液b;
68.c)称取3.1mg dspe-peg2000溶于308μl无水乙醇中,制得dspe-peg2000的浓度为10.1mg
·
ml-1
的溶液c;
69.d)取80μl溶液c加入已预热到65℃的8ml溶液b中,于100r
·
min-1
搅拌30min,制得混合溶液d,即:水相,其中:槐糖酯与dspe-peg2000的质量比为40:1;
70.e)称取约5.6mg水飞蓟素溶于5ml溶液a中,得油相;然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液e;
71.f)使混合溶液e于40℃水浴下旋转蒸发挥去有机溶剂,即得均匀的胶体分散液(对所得样品溶液采用激光笔照射,可产生明显的丁达尔效应);
72.g)向步骤f)所得胶体分散液中加入10%的甘露醇进行冷冻干燥:先在-80℃预冻1小时,然后在-50℃~-40℃及真空度<100pa下冻干48小时,即得本发明所述的口服纳米粒子,简记为:spdns。
73.重复上述步骤,共制备3份平行样品,对所得3份平行样品参照实施例1进行粒径、
粒径分布、zeta电位、包封率(ee)和载药量(dl)进行检测。
74.检测结果详见表3所示。
75.表3
76.样品编号size/nmpdizeta potential/mvee/%dl/%1120.6
±
1.30.174
±
0.0252.343
±
0.52694.023.172118.6
±
0.90.282
±
0.0050.609
±
0.81996.463.273124.3
±
0.60.242
±
0.0070.022
±
2.16195.913.39
77.由表3所示结果可见:采用本实施例方法可获得分布均匀的粒径小于150nm的纳米粒子,且所述纳米粒子对难溶药物的包封率(ee)高于94%,载药量(dl)大于3.0%。
78.实施例4
79.a)称取103.7mg plga(50:50),溶于20ml乙腈中,得到plga的浓度为5.2mg
·
ml-1
的溶液a;
80.b)称取56.4mg槐糖脂溶于11ml去离子水中,制得槐糖酯的浓度为5.1mg
·
ml-1
的溶液b;
81.c)称取9.1mg dspe-peg2000溶于900μl无水乙醇中,制得dspe-peg2000的浓度为10.1mg
·
ml-1
的溶液c;
82.d)取200μl溶液c加入已预热到65℃的10ml溶液b中,于100r
·
min-1
搅拌30min,制得混合溶液d,即:水相,其中:槐糖酯与dspe-peg2000的质量比为25:1;
83.e)取5ml溶液a作为油相,然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液e;
84.f)使混合溶液e于40℃水浴下旋转蒸发挥去有机溶剂,得到本发明所述的纳米粒子,简记为:spdns。
85.为了体现本发明的纳米粒在胃肠道中的物理稳定性优势,将如下对比例1和对比例2所示的纳米粒分别作为参比纳米粒。
86.对比例1
87.1)称取103.7mg plga(50:50),溶于20ml乙腈中,得到plga的浓度为5.2mg
·
ml-1
的乙腈溶液;
88.2)称取9.1mg dspe-peg2000溶于900μl无水乙醇中,制得dspe-peg2000的浓度为10.1mg
·
ml-1
的无水乙醇溶液;
89.3)取200μl dspe-peg2000的浓度为10.1mg
·
ml-1
的无水乙醇溶液加入已预热到65℃的10ml去离子水中,于100r
·
min-1
搅拌30min,得到水相;
90.4)取5ml plga的浓度为5.2mg
·
ml-1
的乙腈溶液作为油相,然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液;然后使混合溶液于40℃水浴下旋转蒸发挥去有机溶剂,得到该对比例所述的纳米粒子,简记为:pdns。
91.对比例2
92.a)称取103.7mg plga(50:50),溶于20ml乙腈中,得到plga的浓度为5.2mg
·
ml-1
的溶液a;
93.b)称取52.3mg槐糖脂溶于10ml去离子水中,制得槐糖酯的浓度为5.2mg
·
ml-1
的溶液b;
94.c)称取5.0mg lipoid s100溶于500μl无水乙醇中,制得lipoid s100的浓度为10mg
·
ml-1
的溶液c;
95.d)取200μl溶液c加入已预热到65℃的10ml溶液b中,于100r
·
min-1
搅拌30min,制得混合溶液d,即:水相,其中:槐糖酯与lipoid s100的质量比为25:1;
96.e)取5ml溶液a作为油相,然后将油相倒入到水相中,于300r
·
min-1
搅拌2h,制得混合溶液e;
97.f)使混合溶液e于40℃水浴下旋转蒸发挥去有机溶剂,得到该对比例所述的纳米粒子,简记为:sp
l
ns。
98.对上述实施例和对比例所获得的纳米粒进行在胃肠道中的物理稳定性考察:
99.参照2020版《中国药典》第四部通则配制模拟胃液和模拟肠液,均不含酶,具体方法如下:
100.①
模拟胃液:取浓盐酸23.4ml,用去离子水定容配制成100ml稀盐酸,然后取16.4ml稀盐酸用去离子水定容配制成1000ml,即得模拟胃液;
101.②
模拟肠液:称取6.8g磷酸二氢钾溶于500ml去离子水中,然后用0.1m氢氧化钠溶液调至ph=6.8,再用去离子水定容配制成1000ml,即得模拟肠液;
102.取上述spdns纳米粒,用介质稀释10倍,然后于37℃恒温振荡,再测定0h,2h,4h,8h,12h,24h各时间点的纳米粒子的粒径。详细结果见表4和表5所示。
103.表4各组纳米粒子在模拟胃液中的粒径(n=3)
[0104][0105]
表5各组纳米粒子在模拟肠液中的粒径(n=3)
[0106][0107]
另外,图1为各组纳米粒子在模拟胃液中的粒径变化,其中:spdns组为本发明实施例4所获得的纳米粒子,pdns组为对比例1所获得的纳米粒子,sp
l
ns组为对比例2所获得的纳米粒子;图2为各组纳米粒子在模拟肠液中的粒径变化,其中:spdns组为本发明实施例4所获得的纳米粒子,pdns组为对比例1所获得的纳米粒子,sp
l
ns组为对比例2所获得的纳米粒子。
[0108]
结合表4和表5及图1和图2可见:本发明所获得的口服纳米粒子在胃肠道中具有非常好的稳定性,可在模拟胃肠液中保持24小时以上不发生粒径增大的团聚现象,而由单一dspe-peg2000包覆的plga纳米粒(即:对比例1所获得的pdns)及由磷脂lipoid s100与槐糖脂共包覆的plga纳米粒(即:对比例2所获得的sp
l
ns)均在模拟胃肠液中保持2小时就发生了粒径骤增的团聚现象,进一步说明本发明采用槐糖脂和二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000共同包覆plga制备纳米粒子,产生了出乎意料的技术效果,即:获得了在胃肠道中非常稳定的口服纳米粒子。
[0109]
实施例5载水飞蓟素的spdns在大鼠体内的药动学考察
[0110]
对雌性sd大鼠(200
±
20g,清洁级)口服灌胃给予实施例3制备的载水飞蓟素spdns,给药剂量为20mg/kg;并以市售益肝灵片(研碎后用peg 400:水=1:1分散,后续简称为ygl)为参比制剂,给药剂量为20mg/kg;给药后,分别于0.5h、1h、1.5h、2h、3h、4h、6h、10h、24h和48h经大鼠眼球后静脉丛取血,用肝素处理过的离心管收集,于3500r
·
min-1
离心10min,分离血浆,利用高效液相色谱法(参照文献j nanobiotechnol(2020)18:83)测定血浆中的药物浓度。
[0111]
将血药浓度数据经过das 2.0软件处理,得药动学参数(见表6所示)。
[0112]
将slb的血药浓度对时间作图,绘制血药浓度-时间曲线(见图3和图4所示)。
[0113]
将药动学参数用spss 21.0软件进行统计学分析,p《0.05表示有统计学差异。
[0114]
表6 slb在spdns和ygl组的药动学参数
[0115]
药动学参数单位spdns组ygl组t
1/2z
h29.71
±
32.408.07
±
6.46t
max
h0.90
±
0.420.90
±
0.42c
max
μg/l1090.00
±
302.61*572.12
±
122.64
auc
(0-t)
μg/l
·
h9128.76
±
3816.22*3853.66
±
2339.19auc
(0-∞)
μg/l
·
h13884.81
±
8953.13*4114.66
±
2399.78mrt
(0-t)
h14.54
±
4.679.64
±
6.43mrt
(0-∞)
h44.10
±
36.9616.05
±
12.38
[0116]
*p《0.05,表示与ygl相比有显著性差异。
[0117]
由表6所示结果可见:与市售药益肝灵相比,spdns中slb在大鼠体内的平均滞留时间明显延长,spdns的c
max
是市售药益肝灵的1.91倍。口服给spdns和益肝灵后,两者的auc
(0-t)
分别为9128.76μg
·
h/l和3853.66μg
·
h/l,以益肝灵为参比制剂,根据如下公式计算spdns的相对生物利用度:
[0118][0119]
结果表明,与益肝灵相比,spdns的相对生物利用度为236.89%。
[0120]
另外,结合图3和图4还可知,slb在spdns组的各时间点的平均血药浓度均高于同等条件下的ygl组,说明本发明所述的口服纳米粒子spdns可显著提高口服难溶药物水飞蓟素的口服生物利用度,对解决口服难溶药物的口服生物利用度低的难题具有重要价值。
[0121]
最后需要在此指出的是:以上仅是本发明的部分优选实施例,不能理解为对本发明保护范围的限制,本领域的技术人员根据本发明的上述内容做出的一些非本质的改进和调整均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1