一种全氟化碳硅质体及其制备方法和应用与流程
1.本发明属于生物医用材料领域,具体涉及一种硅质体包载的全氟化碳制剂及其制备方法和应用。
背景技术:
2.肿瘤乏氧是实体瘤最普遍的特征之一。乏氧是肿瘤微环境的一种恶性特征,它和肿瘤对各种疗法的抵抗性(包括化疗,放疗和光动力治疗)密切相关。特别是对于化疗的耐药,乏氧可能导致肿瘤的多药耐药(multidrug resistance),最终可能导致化疗失败。在多药耐药的发生过程中,肿瘤乏氧首先会提高肿瘤细胞的乏氧诱导因子
‑
1α(hif
‑
1α)的相对表达水平,从而提高多药耐药基因1(mdr1)的转录水平,最终增加p
‑
糖蛋白(p
‑
gp)的蛋白表达水平,而p
‑
gp作为多种化疗药物的“外排泵”,可以将进入肿瘤细胞内的药物泵回胞外。因此,改善乏氧是增强化疗疗效的有效方法。肿瘤转移是大多数癌症患者死亡的主要原因。据报道,乏氧可以促进肿瘤细胞的上皮
‑
间质转化(epithelial
‑
mesenchymal transition),加速肿瘤转移。因此,改善乏氧可以同时增加化疗的疗效、抑制肿瘤的转移。
3.目前,研究人员已经探索了很多方法来改善肿瘤乏氧。高压氧疗法可直接增加全身血液中的氧气浓度,尽管在临床上已被使用,但它缺少肿瘤特异性,增加了放疗、化疗对正常组织的损伤,并且存在高氧中毒的风险。现在迫切需要开发肿瘤特异性的氧气递送系统。在不同种类的氧气递送材料中,全氟化碳(perfluocarbon)由于其较高的氧气溶解度和较好的生物相容性而具有很好的优势。全氟化碳已在临床中被广泛用作超声成像对比剂、预防缺血和再灌注损伤,并且有望作为人工血液。根据报道,基于全氟化碳的纳米液滴可以缓解肿瘤乏氧,并增强放射疗法和光动力疗法的疗效。
4.尽管全氟化碳的氧气溶解度很高,但是,它释放氧气的速度效率较低。血红蛋白在低氧条件下,依靠其四个亚基之间的协同效应迅速释放氧气分子,与此不同,全氟化碳仅依靠氧气的顺浓度梯度自由扩散来释放氧气。因此,为了增强全氟化碳释放氧气的效率,很有必要依靠外部刺激触发氧气的释放。以往的报道已经利用近红外光或超声波触发氧气的释放,但是,因为近红外光的组织穿透性有限,临床应用受到限制。普通超声的组织穿透性高,但是超声波不能聚焦,不能定点控制氧气的释放,控释不够精确,肿瘤附近的正常组织也会受到超声处理。
5.全氟化碳不仅可以作为氧气递送的载体,还可以作为药物递送系统(drug deliverysystem)。小分子的化疗药物往往治疗效果有限,并且有严重的副作用,研究人员长期以来一直在关注智能药物输送系统的开发,实现化疗药物的增效、减毒。全氟化碳可以实现超声成像监测下的药物递送,以及超声触发的药物释放,因此,基于全氟化碳的药物递送系统具有独特的优势。
6.良好的结构稳定性和载药稳定性对于药物递送系统至关重要,因为它可以避免递送系统在血液循环系统中过早地释放药物,从而减少药物的副作用。文献已经报道的全氟化碳纳米材料的包裹剂主要包括磷脂或其他表面活性剂,白蛋白,红细胞膜,介孔纳米材
料,如bi2se和sio2。这些包裹剂具有有限的载药稳定性,并且合成较为复杂。开发合成过程简单、稳定性较高的全氟化碳药物递送系统很有必要。
技术实现要素:
7.本发明的目的之一是以硅质体作为氟化碳的包裹剂,提高氟化碳药物递送系统的结构稳定性和载药稳定性,减少药物在血液循环中提前泄漏,降低药物的副作用。
8.本发明的目的之二是利用高强度聚焦超声(hifu)触发氟化碳制剂释放氧气和药物,实现高强度聚焦超声刺激响应型的氧气和药物控释,提高药物在肿瘤的富集量,增加药物的药效。
9.本发明的目的之三是利用硅质体包载的氟化碳制剂,向肿瘤靶向递送氧气,改善肿瘤的乏氧,缓解肿瘤的多药耐药和上皮
‑
间质转化,提高化疗的药效,减少肿瘤的转移。
10.本发明的目的是通过以下技术方案实现的:
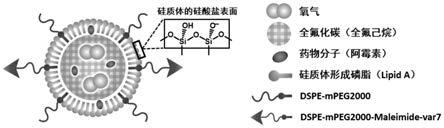
11.一种包载氟化碳类化合物的硅质体,所述硅质体是具有核壳结构的粒子,所述壳是由形成硅质体脂质和不含si的磷脂成分组成的单层脂质分子层,所述形成硅质体脂质在所述壳的外表面形成
‑
si
‑
o
‑
si
‑
形式的硅酸盐网络结构;所述核含有氟化碳类化合物。
12.进一步的,根据本发明,以单层脂质分子层的摩尔总量为基准,所述形成硅质体脂质的摩尔百分比是50%
‑
100%,不含si的磷脂成分的摩尔百分比是0%
‑
50%。在本发明的一些实施方式中,形成硅质体脂质的摩尔百分比例如是94~95%,60%,50%,以及余量的不含si的磷脂成分。
13.进一步的,根据本发明,所述氟化碳类化合物是指有机化合物中与碳相连的氢被氟全部或部分取代后形成的化合物。所述有机化合物包括但不限于烃类、醇类、酯类、醚类。所述烃类例如为c3
‑
10的饱和或不饱和的链状或环状烃。所述醇类例如为c1
‑
10一元醇或多元醇。所述酯类例如为c2
‑
10的酯。所述醚类例如为c2
‑
30的链状或环状的一元醚或多元醚。在本发明的一些实施方式中,所述氟化碳类化合物选自全氟丙烷、全氟戊烷、全氟己烷、全氟溴辛烷、全氟冠醚组成的组。根据本发明,优选常压下沸点为40
‑
80℃的氟化碳类化合物,所述化合物包括但不限于全氟己烷、全氟庚烷、十二氟己烷、一溴十二氟己烷。本发明的发明人通过研究发现,在上述沸点范围内的氟化碳类化合物,才能满足靶向肿瘤给药、恰当的药物包封率和载药量、携带氧气,并能在hifu指导下的释药。
14.进一步的,根据本发明,所述形成硅质体脂质包括无机前体、连接基团和疏水性尾部三部分,其具有如下结构式:a
m
ch
(3
‑
m)
—ch2—y—(ch2)
p
—z—r
q
,
15.其中,a为:(r1o)3si—(ch2)3—n—c(o)—x—(ch2)
n
—;r1为烷基,优选为c1
‑
10烷基,更优选为甲基、乙基;x为
‑
o
‑
或键;n为0
‑
10的整数,例如0、1、2、3、4、5、6、7、8、9、或10,优选为0、1、2;
16.m为1
‑
3的整数;当m为2或3时,所述的2个或3个a可以相同也可以不同;
17.y为—ch2—、—o—c(o)—或者—c(o)—o—;
18.p为0
‑
10的整数,例如0、1、2、3、4、5、6、7、8、9或10,优选为0、1、2;
19.z为—c(o)—nh(2‑
q
)—或者—ch(3‑
q
)—;
20.r为c原子数大于5的烷基,优选为c7
‑
20的直链饱和烷基;
21.当z为—c(o)—nh(2‑
q
)—时,q为1
‑
2的整数;当z为—ch(3‑
q)—时,q为1
‑
3的整数;
当q为2或3时,所述的2个或3个r可以相同也可以不同。
22.在本发明的一些实施方式中,所述形成硅质体脂质选自如下所示复合脂质a
‑
d组成的组。四种复合脂质的合成步骤如文献(chem.eur.j.2013,19,16113
–
16121)所述。
[0023][0024]
进一步的,根据本发明,所述硅质体表面的硅酸盐网络结构可以由形成硅质体脂质通过原位溶胶
‑
凝胶反应(in situ sol
‑
gel reaction)形成,反应机理例如为:
[0025]
‑
si
‑
och2ch3+h2o
→‑
si
‑
oh+ch3ch2oh
[0026]2‑
si
‑
oh
→‑
si
‑
o
‑
si
‑
+h2o
[0027]
进一步的,根据本发明,所述的不含si的磷脂成分可以选自本领域已知的不含si的磷脂成分,包括但不限于:可以延长血液循环时间的二硬脂酰基磷脂酰乙醇胺的聚乙二醇衍生物dspe
‑
mpeg
1000~20000
,例如dspe
‑
mpeg2000(二硬脂酰基磷脂酰乙醇胺
‑
聚乙二醇2000)和dspe
‑
mpeg5000(二硬脂酰基磷脂酰乙醇胺
‑
聚乙二醇5000);可以用来连接靶向分子的被马来酰亚胺改性的二硬脂酰磷脂酰乙醇胺或被马来酰亚胺改性的二硬脂酰基磷脂酰乙醇胺的聚乙二醇衍生物dspe
‑
mpeg
1000~20000
,例如dspe
‑
maleimide(二硬脂酰磷脂酰乙醇胺改性马来酰亚胺)、dspe
‑
mpeg
2000
‑
maleimide(二硬脂酰基磷脂酰乙醇胺
‑
聚乙二醇2000
‑
马来酰亚胺);可以调整硅质体相变温度的二棕榈酰磷脂酰胆碱(dppc)和1
‑
肉豆蔻酰基
‑2‑
硬脂酰基卵磷脂(mspc);氢化大豆卵磷脂(hspc);二硬脂酰基磷脂酰胆碱(dspc)。所述不含si的磷脂成分,可以是一种或多种不含si的磷脂的混合物。
[0028]
进一步的,根据本发明,所述硅质体的表面可以修饰肿瘤靶向分子,所述肿瘤靶向分子选自可以靶向肿瘤的抗体、多肽、适配体、叶酸或叶酸衍生物中的任意一种。在本发明的一个实施方式中,所述肿瘤靶向分子是var7多肽(参考文献:mol.pharmaceutics 2014,11,2896
‑
2905),其氨基酸序列为aceeqnpwarylewlfptetlllel。在本发明的一些实施方式中,所述肿瘤靶向分子是叶酸衍生物,例如巯基
‑
聚乙二醇
2000~6000
‑
叶酸,具体为巯基
‑
聚乙
二醇
5000
‑
叶酸。
[0029]
进一步的,根据本发明,所述硅质体的粒径为10nm
‑
1μm。
[0030]
进一步的,根据本发明,所述硅质体的核可以包载小分子药物。所述小分子药物是水不溶性或疏水性药物,包括但不限于化疗药物、蛋白激酶抑制剂、光动力药物的一种或多种。在本发明的一些实施方式中,所述小分子药物选自阿霉素、顺铂、紫杉醇、伊立替康、索拉菲尼、吉非替尼、卟吩姆钠、维替泊芬、吲哚箐绿组成的组。
[0031]
进一步的,根据本发明,所述硅质体的核可以携带氧气。所述硅质体携带氧气的能力是通过氟化碳类化合物对氧气的溶解实现的。在15
‑
40℃、氧气分压为0
‑
101325pa的环境中,当所述硅质体对氧气的吸附达到平衡时,每毫升氟化碳类化合物可以溶解0
‑
2毫升氧气。当所述硅质体所处环境的氧气分压升高(或降低)时,所述硅质体吸附氧气的量会相应地升高(或降低)。当所述硅质体经过静脉注射进入人体后,从a区被转运到b区时,若b区的氧气浓度高于a区,硅质体制剂将在b区吸附氧气;若b区的氧气浓度低于a区,硅质体将在b区释放氧气。正常人体静脉血氧含量为110
‑
180ml/l,动脉血氧含量为150
‑
230ml/l,静脉血液氧气浓度低于动脉血液氧气浓度,并且,动脉血液氧气浓度高于肿瘤血管外的肿瘤组织(包括肿瘤细胞和肿瘤细胞外间质)。当所述硅质体经过静脉注射进入人体后,从静脉血液进入动脉血液时,硅质体将吸附氧气;从动脉血液进入肿瘤血管外的肿瘤组织时,硅质体将释放氧气。因此,所述硅质体可以将动脉血液中的氧气携带到肿瘤组织。
[0032]
进一步的,根据本发明,所述硅质体具有很好的稳定性:经过0
‑
100μm的表面活性剂triton
‑
100的处理后,所述硅质体的药物释放率小于20%。所述硅质体相比于由普通磷脂组成的包载氟化碳类化合物的脂质体具有更高的稳定性:经过10
‑
100μm的表面活性剂triton
‑
100的处理后,所述硅质体的药物释放率比脂质体低30
‑
90%。
[0033]
进一步的,根据本发明,所述硅质体可以响应高强度聚焦超声的刺激,实现高强度聚焦超声控制的药物释放和氧气释放:将所述硅质体使用超声强度(脉宽调制)为1%
‑
10%的高强度聚焦超声处理0.5
‑
5分钟,所述硅质体的氧气释放率可达到5%
‑
100%,所述硅质体的药物释放率可达到5%
‑
100%。
[0034][0035]
进一步的,根据本发明,所述硅质体还具有增强超声成像的能力。
[0036]
本发明进一步提供所述硅质体的制备方法。
[0037]
一种制备前述硅质体的方法,其包括如下步骤:将氟化碳类化合物、形成硅质体脂质和不含si的磷脂成分,或进一步包含的药物,加入缓冲溶液或水中,均质化,再通过高压挤出器获得粒径分布均匀的制剂。
[0038]
进一步的,根据本发明,在加入缓冲溶液或水之前,氟化碳类化合物、形成硅质体脂质和不含si的磷脂成分,或进一步包含的药物,可以先溶解在有机溶剂中。所述有机溶剂可以是醇类(例如乙醇,丙醇等)、二甲基亚砜等。
[0039]
进一步的,根据本发明,所述缓冲溶液可以是pbs缓冲液、tris
‑
hcl缓冲液等。所述
缓冲溶液的ph值可以是5.0
‑
8.0。
[0040]
本发明所述硅质体还可通过另一种方法制备获得。所述制备方法包括如下步骤:1)将形成硅质体脂质在酸性醇溶液中水解;2)将不含si的磷脂成分,或者进一步包含的药物,溶解在有机溶剂中,加入步骤1)的水解液中形成混合物;3)挥干步骤2)的混合物得到磷脂薄膜,将所述薄膜水化,再进行均质化;氟化碳类化合物在水化操作时或在均质化操作时加入。
[0041]
进一步的,根据本发明,所述酸性醇溶液是将醇溶解在酸的水性稀溶液中制备得到的。所述醇可以是乙醇,丙醇,异丙醇等;所述酸可以是盐酸,硫酸,硝酸等。
[0042]
进一步的,根据本发明,所述有机溶剂可以是醇类(例如乙醇等)、二氯甲烷、三氯甲烷、二甲基甲酰胺等。
[0043]
进一步的,根据本发明,所述水化可以使用水、pbs缓冲液、tris
‑
hcl缓冲液等。
[0044]
根据本发明,均质化后通过离心得到硅质体制剂。也可以,在均质化后进一步通过高压挤出器得到粒径更均匀的硅质体制剂。
[0045]
根据本发明,在得到硅质体的制剂后,用肿瘤靶向分子修饰所述制剂。
[0046]
根据本发明,在得到硅质体的制剂后,去掉未包封入硅质体的物质,例如可以通过洗涤、过滤和/或色谱柱等方式。
[0047]
根据本发明,如果含有药物,在制备所述硅质体时,药物和磷脂成分(形成硅质体脂质和不含si的磷脂的总和)的投料量的质量比优选为1:10~1:2,该比例会影响硅质体的载药量和包封率,在本发明优选的质量比范围内,硅质体的载药量和包封率最佳。
[0048]
本发明进一步提供所述硅质体的制药用途。
[0049]
本发明所述的硅质体在制备递送氧气的载体或药物中的应用。基于所述的应用,本发明的硅质体可以制备成用于改善肿瘤乏氧的药物。
[0050]
本发明所述的硅质体在制备递送药物的载体中的应用。基于所述的应用,本发明的硅质体可以制备成靶向肿瘤的药物,其中包封肿瘤治疗药物,可以实现靶向肿瘤同时给予氧气和药物,改善肿瘤乏氧,提高药物治疗效果,降低肿瘤转移发生率。本发明所述的硅质体在制备肿瘤治疗药物中的应用。
[0051]
本发明所述的硅质体在制备和hifu联合治疗肿瘤的药物中的应用。所述药物能够改善肿瘤乏氧,降低肿瘤转移发生率。所述药物能够作为热疗的新辅助化疗药物。所述hifu在20分钟内可以将靶组织(如肿瘤)的温度从正常体温(~37℃)提升至目标温度(40
‑
80℃),并维持目标温度30分钟以上;调整所述hifu的功率可以改变目标温度。
[0052]
在本发明的一个实施方式中,所述肿瘤是乳腺癌,例如三阴性乳腺癌。
[0053]
在本发明的一个实施方式中,所述肿瘤治疗药物是阿霉素。
[0054]
本发明所述的硅质体在制备超声成像剂中的应用。
[0055]
本发明的有益效果:
[0056]
本发明的优点之一是硅质体作为氟化碳的包裹剂,可以提高药物递送系统的稳定性,减少药物在血液循环中的提前泄露,降低药物副作用。硅质体是一种无机
‑
有机杂化材料,它的结构与普通磷脂组成的脂质体类似,由两亲性的复合脂质自组装形成微米或纳米粒子。但是,与传统的磷脂脂质体相比,硅质体的表面是有机硅酸盐构成的单原子层网络结构,具有更高的结构稳定性和载药稳定性。而和硅纳米粒子相比,硅质体的磷脂层结构降低
了纳米粒子整体的硬度和密度,并减少了硅元素的注射剂量,提高了生物安全性。氟化碳类化合物疏水性强,采用硅质体包载含量极低。本发明通过研究改进了硅质体的微观结构,采用单分子层硅质体结构,巧妙地解决了强疏水性氟化碳类化合物的高含量包载问题,并保持了硅质体作为递送系统的稳定性。
[0057]
本发明的优点之二是本发明的硅质体包载的氟化碳类化合物的制剂可以响应高强度聚焦超声的刺激,通过高强度聚焦超声可以触发药物和氧气在靶区的快速释放。高强度聚焦超声(high
‑
intensity focused ultrasound,hifu)是临床上用于热消融治疗的一种非侵入性技术。hifu通过将超声能量转换为热能和机械能,可以精确地控制和升高目标组织的温度。hifu可以轻微升高靶组织的温度(<43℃),已被用于药物的热敏释放,而不会损伤组织。温度的轻微升高会增加肿瘤血流量,因此hifu本身也可以一定程度地缓解肿瘤乏氧。本发明的硅质体能响应hifu的刺激,从而释放其中包载的氧气或药物,能够实现精准地定点控制氧气或药物的释放,并结合hifu的升温作用更好地缓解肿瘤的乏氧。
[0058]
本发明的优点之三是本发明的硅质体包载的氟化碳类化合物的制剂可以同时将药物和氧气靶向递送到肿瘤部位。药物的靶向递送可以增加药物的药效,氧气的靶向递送可以改善肿瘤乏氧,缓解肿瘤的多药耐药和上皮
‑
间质转化,提高药效,减少肿瘤转移。
[0059]
本发明的优点之四是本发明的硅质体包载的氟化碳类化合物的制剂具有增强超声成像的效果,可以实现影像监测下的药物递送和药物释放,预估治疗效果。
附图说明
[0060]
图1:本发明所述的一种硅质体包载的氟化碳制剂的结构示意图。
[0061]
图2:实施例3制备的硅质体包载的氟化碳制剂的冷冻蚀刻扫描电镜图像,图像说明该纳米制剂为球形,平均粒径在100nm左右。
[0062]
图3:实施例3和实施例5分别制备的硅质体包载的氟化碳制剂、普通磷脂包载的氟化碳制剂的载药稳定性比较。
[0063]
图4:实施例3制备的硅质体包载的氟化碳制剂经过高强度聚焦超声处理后的药物控释效果评价。
[0064]
图5:实施例3制备的硅质体包载的氟化碳制剂的氧气控释效果评价。
[0065]
图6:实施例3制备的硅质体包载的氟化碳制剂的超声成像效果评价。
[0066]
图7:不同种类的氟化碳组成的制剂的载药稳定性。
[0067]
图8:m
‑
hifu(pwm=2%)聚焦超声处理1分钟之前(a
‑
d)和之后(i
‑
v)的不同的制剂的超声成像图:(a)和(i)d
‑
pfp
‑
vpcs,(b)和(ii)d
‑
pfh
‑
vpcs,(c)和(iii)d
‑
pfob
‑
vpcs,(d)和(iv)d
‑
pfce
‑
vpcs,(v)pbs。
[0068]
图9:制剂在细胞水平上的治疗效果。
[0069]
图10:制剂对药物的体内分布的改善情况。
[0070]
图11:制剂的体内化疗效果。
[0071]
图12:制剂降低肿瘤转移的效果。
[0072]
图13:制剂作为新辅助化疗手段的治疗效果。
具体实施方式
[0073]
以下结合实施例对本发明作进一步的详细说明。但本领域技术人员了解,本发明的保护范围不仅限于以下实施例。根据本发明公开的内容,本领域技术人员将认识到在不脱离本发明技术方案所给出的技术特征和范围的情况下,对以上所述实施例做出许多变化和修改都属于本发明的保护范围。
[0074]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备;所进行的操作都是本领域已知的,或者按照市售商品的用户手册进行。
[0075]
实施例1制备硅质体包载的氟化碳制剂
[0076]
(1)将9.5μmol复合脂质a,0.5μmol dspe
‑
mpeg2000,4μmol小分子药物阿霉素和20μl全氟己烷溶解在2ml乙醇中,注入20ml pbs中,注入的过程中和注入后的15分钟内,20ml pbs使用水浴超声仪(英国prima公司,型号pm3
‑
900tl)超声处理。之后,使用探头型超声仪(美国qsonica公司,型号q700),在冰浴条件下,以50%的输出功率,持续超声10分钟。通过高压挤出器(美国northern lipid公司)将液体用聚碳酸酯滤膜(孔径为100nm,美国whatman公司)反复挤出3次,获得平均粒径100nm左右的制剂。
[0077]
(2)为了除去没有包封到硅质体制剂中的阿霉素,首先将步骤(1)获得的制剂用0.45μm滤器过滤,再使用排阻色谱柱sephadex g
‑
50除去游离的阿霉素。
[0078]
(3)测定该制剂的氟化碳和药物的包载量:取0.1ml 100mg/ml的该制剂(所述制剂浓度是以该制剂冻干后的重量为溶质重量进行计算的,所述溶质相当于10mg的冻干后硅质体,冻干后硅质体因全氟己烷挥发而不再含有全氟己烷),用500μl的甲醇溶解,通过气相色谱仪检测全氟己烷的含量,测定结果为每10mg的冻干后硅质体对应含有6.24mg的全氟己烷。同样,取0.1ml 100mg/ml该制剂,用酸化异丙醇溶液(10%的1mol/l的盐酸,90%的异丙醇)溶解,通过荧光分光光度法检测阿霉素的含量,测定结果为每10mg冻干后硅质体对应含有1.75mg的阿霉素,据此计算阿霉素的载药量为17.5%,包封率为84.9%。
[0079]
(4)检测该制剂的硅质体外层是否为单层脂质分子层,方法一:通过冷冻电镜观察该制剂的外壳厚度,结果表明,其厚度约为2nm,由于磷脂双分子层的厚度约为4.5nm,因此,该制剂的外壳为脂质单分子层。方法二:将9.5μmol复合脂质a,0.5μmol dspe
‑
mpeg2000,2μmol小分子药物阿霉素和20μl全氟己烷,0.01μmol水溶性染料钙黄绿素钠和0.01μmol脂溶性染料尼罗红,溶解在2ml乙醇中,后续处理方法与步骤(1)和步骤(2)相同,检测制备出的制剂的紫外
‑
可见光吸收曲线。检测结果表明,制剂含有尼罗红的特征吸收峰,没有钙黄绿素钠的特征吸收峰,说明制剂没有水腔,因此外层为单层脂质分子层。
[0080]
实施例2制备硅质体包载的氟化碳制剂
[0081]
(1)将6μmol复合脂质b在酸性乙醇中40℃下水解30分钟。所述酸性乙醇溶液的制备是将稀盐酸加入乙醇溶液中,用ph计调节ph值为3.0。
[0082]
(2)将1μmol小分子药物紫杉醇,4μmol不含si的磷脂成分(包括3μmol的氢化大豆卵磷脂(hspc)和1μmol的dspe
‑
mpeg2000)溶解在2ml三氯甲烷中,加入步骤(1)的水解液,挥干液体形成磷脂薄膜,真空干燥过夜,加入5ml超纯水和20μl全氟冠醚,震荡水化60分钟后,使用探头型超声仪(美国qsonica公司,型号q700),冰浴条件下,以50%的输出功率,持续超声10分钟,获得平均粒径200nm左右的制剂。
[0083]
(3)为了除去没有包封到硅质体制剂中的紫杉醇,首先将步骤(2)获得的制剂用
0.45μm滤器过滤,再使用排阻色谱柱sephadex g
‑
50除去游离的紫杉醇。
[0084]
(4)采用实施例1相同的检测方法,测定结果为每10mg冻干后硅质体对应含有5.1mg的全氟冠醚和0.9mg的紫杉醇。
[0085]
实施例3制备带有肿瘤靶向分子的硅质体包载的氟化碳制剂
[0086]
(1)将9.5μmol复合脂质a,0.2μmol dspe
‑
mpeg2000,4μmol小分子药物阿霉素,20μl全氟己烷,0.3μmol dspe
‑
mpeg2000
‑
maleimide溶解在2ml乙醇中,注入20ml pbs中,注入的过程中和注入后的15分钟内,20ml pbs使用水浴超声仪(英国prima公司,型号pm3
‑
900tl)超声处理。之后,使用探头型超声仪(美国qsonica公司,型号q700),冰浴条件下,以50%的输出功率,持续超声10分钟。通过高压挤出器(美国northern lipid公司)将液体用聚碳酸酯滤膜(孔径为100nm,美国whatman公司)反复挤出3次,获得平均粒径100nm左右的制剂。
[0087]
(2)向制剂中加入0.6μmol肿瘤靶向分子var7多肽(var7可以借助肿瘤的酸性微环境,靶向到肿瘤部位,氨基酸序列为aceeqnpwarylewlfptetlllel,详细信息如文献所述:mol imaging biol(2016)18:686
‑
696),4℃搅拌24小时,使var7多肽的巯基与maleimide偶联。
[0088]
(3)为了除去没有包封到硅质体制剂中的阿霉素和没有连接到硅质体表面的var7,首先将步骤(2)获得的制剂用0.45μm滤器过滤,再使用排阻色谱柱sephadex g
‑
50除去游离的阿霉素和var7。
[0089]
(4)采用实施例1相同的检测方法,测定结果为每10mg的冻干硅质体对应含有5.9mg的全氟己烷和1.6mg的阿霉素。
[0090]
实施例4制备带有肿瘤靶向分子的硅质体包载的氟化碳制剂
[0091]
(1)将9.5μmol复合脂质b,0.2μmol dspe
‑
mpeg5000,2μmol小分子药物卟吩姆钠,20μl全氟溴辛烷,0.3μmol dspe
‑
maleimide(二硬脂酰磷脂酰乙醇胺改性马来酰亚胺)溶解在2ml乙醇中,注入20ml pbs中,注入的过程中和注入后的15分钟内,20ml pbs使用水浴超声仪(英国prima公司,型号pm3
‑
900tl)超声处理。通过高压挤出器(美国northernlipid公司)将液体用聚碳酸酯滤膜(孔径为10μm,美国whatman公司)反复挤出3次,获得粒径在10μm左右的制剂。
[0092]
(2)向制剂中加入0.6μmol肿瘤靶向分子sh
‑
peg5000
‑
folate(巯基
‑
聚乙二醇5000
‑
叶酸),4℃搅拌24小时,使sh
‑
peg5000
‑
folate的巯基与maleimide偶联。
[0093]
(3)为了除去没有包封到硅质体制剂中的卟吩姆钠和没有连接到硅质体表面的肿瘤靶向分子,将步骤(2)获得的制剂用50%的乙醇反复洗涤三次,保存在pbs中备用。
[0094]
(4)采用实施例1相同的检测方法,测定结果为每10mg的冻干硅质体对应含有7.3mg的全氟溴辛烷和0.7mg的卟吩姆钠。
[0095]
实施例5制备普通磷脂组成的脂质体包载的氟化碳制剂
[0096]
(1)将6.5μmol二硬脂酰基磷脂酰胆碱(dspc),0.2μmol dspe
‑
mpeg2000,3.0μmol胆固醇,2.0μmol小分子药物阿霉素,20μl全氟己烷,0.3μmol dspe
‑
mpeg2000
‑
maleimide溶解在2ml乙醇中,注入20ml pbs中,注入的过程中和注入后的15分钟内,20ml pbs使用水浴超声仪(英国prima公司,型号pm3
‑
900tl)超声处理。之后,使用探头型超声仪(美国qsonica公司,型号q700),冰浴条件下,以50%的输出功率,持续超声10分钟。通过高压挤出器(美国
northern lipid公司)将液体用聚碳酸酯滤膜(孔径为100nm,美国whatman公司)反复挤出3次,获得平均粒径100nm左右的制剂。
[0097]
(2)向制剂中加入0.6μmol肿瘤靶向分子var7多肽,4℃搅拌24小时,使var7多肽的巯基与maleimide偶联。
[0098]
(3)为了除去没有包封到制剂中的阿霉素和没有连接到脂质体表面的var7,首先将步骤(2)获得的制剂用0.45μm滤器过滤,再使用排阻色谱柱sephadex g
‑
50除去游离的阿霉素和var7。
[0099]
(4)采用实施例1相同的检测方法,测定结果为每10mg的冻干脂质体对应含有5.3mg的全氟己烷和1.4mg的阿霉素。
[0100]
实施例6评价硅质体包载的氟化碳制剂的载药稳定性
[0101]
(1)采用表面活性剂triton
‑
100增溶法来比较不同氟化碳制剂的载药稳定性。将实施例3和实施例5制备的氟化碳制剂分别分散在不同浓度的triton
‑
100溶液中。
[0102]
(2)室温震荡10分钟后,取样检测阿霉素的荧光强度,通过荧光去淬灭法,检测从制剂中释放出的阿霉素的药物量,计算释放出的阿霉素占制剂中总阿霉素的百分比,公式如下:
[0103][0104]
其中,f(t)是经过triton
‑
100处理后的样品的阿霉素的荧光强度,f0是没有经过triton
‑
100处理的对照样品的阿霉素的荧光强度,而fc是被1%triton
‑
100和1m盐酸彻底破坏后的五个样本的阿霉素的平均荧光强度。阿霉素的荧光强度通过荧光分光光度计检测,检测的激发波长为480nm,发射波长为580nm。
[0105]
检测结果如附图3所示,随着triton
‑
100浓度的增加,普通磷脂组成的脂质体包载的氟化碳制剂中的阿霉素会大量释放出来,但是,硅质体包载的氟化碳制剂中的阿霉素只有很少量被释放出来。说明硅质体包载的氟化碳比脂质体包载的氟化碳的稳定性高,更能抵抗表面活性剂的增溶作用。
[0106]
实施例7评价硅质体包载的氟化碳制剂在高强度聚焦超声控制下的药物释放能力
[0107]
(1)将实施例3制备的硅质体包载的氟化碳制剂分散在37℃的pbs中,使用高强度聚焦超声处理4分钟,高强度聚焦超声的脉宽调制(pulse width modification,pwm)为2%。在超声处理期间的不同时间,对溶液取样并立即在冰上冷却,以停止药物释放。
[0108]
(2)通过荧光去淬灭法检测药物的释放率,计算释放出的阿霉素占制剂中总阿霉素的百分比,公式如下:
[0109][0110]
其中,f(t)是经过高强度聚焦超声处理后、在冰上冷却并平衡回室温的样品的阿霉素的荧光强度,f0是没有经过高强度聚焦超声处理的对照样品的阿霉素的荧光强度,而fc是被1%triton
‑
100和1m盐酸彻底破坏后的五个样本的阿霉素的平均荧光强度。阿霉素的荧光强度通过荧光分光光度计检测,检测的激发波长为480nm,发射波长为580nm。
[0111]
检测结果如附图4所示,在高强度聚焦超声的处理下,硅质体包载的氟化碳制剂会将阿霉素快速释放出来。说明高强度聚焦超声可以触发该制剂释放药物,实现药物控释。
[0112]
实施例8评价硅质体包载的氟化碳制剂的氧气递送和控释能力
[0113]
(1)在无菌氧气室(o2流量=5l/min)中,将1ml实施例3制备的制剂充氧1分钟,然后迅速注入4ml pbs中。
[0114]
(2)在注入之前和注入之后,使用便携式溶氧仪(rex,型号jpbj
‑
608,中国)监测4mlpbs的溶解氧浓度。将不含有硅质体制剂的1ml充氧或不充氧的pbs注入到4ml pbs中,用作对照。
[0115]
(3)注入时定义为0分钟,分别在注入后的20、30、40和50分钟,将注入后的溶液用高强度聚焦超声(pwm=2%)照射处理2分钟,触发氧气从制剂的释放。使用溶氧仪总共记录氧气浓度约60分钟。
[0116]
检测结果如附图5所示,向4ml的pbs注入1ml充氧的制剂后,pbs的氧含量迅速增加,并且,每次用高强度聚焦超声处理时,制剂都会迅速释放氧气,在氧气浓度曲线中形成一个突起的“波动峰”,说明该制剂可以有效地携带氧气,并且高强度聚焦超声可以触发氧气的释放。
[0117]
实施例9评价硅质体包载的氟化碳制剂的超声成像效果和氟化碳控释效果
[0118]
(1)小鼠皮下瘤模型的建立:将4t1细胞以每只小鼠5
×
106个细胞的量接种于balb/c小鼠的大腿外侧皮下,小鼠于spf级环境下饲养;
[0119]
(2)肿瘤大小长到约100mm3时,经尾静脉注射200μl实施例3制备的制剂,注射剂量为5mg/kg的阿霉素;
[0120]
(3)使用高强度聚焦超声(pwm=2%)照射处理肿瘤部位,在高强度聚焦超声处理之前和持续照射不同时间之后分别进行超声成像。使用配备有l12
‑
3e超声探头(3
‑
12mhz)的resona 7超声系统(mindray,中国)采集肿瘤部位的超声图像,机械指数(mi)设定为0.044。
[0121]
检测结果如附图6所示,注射制剂后,肿瘤区域的超声信号没有明显变化。但是,注射制剂并且使用高强度聚焦超声(hifu)处理肿瘤后,随着hifu处理时间的增加,肿瘤的超声信号越来越强,最终在整个肿瘤区域都出现了明显增强的超声信号,说明hifu的处理可以将氟化碳制剂转化为气泡以产生超声信号。这也说明hifu的处理可以促进该制剂释放氟化碳。
[0122]
实施例10评价由不同种类的氟化碳组成的制剂的载药稳定性
[0123]
按照实施例3描述的方法,分别制备由全氟戊烷、全氟己烷、全氟溴辛烷和全氟冠醚组成的制剂,分别命名为d
‑
pfp
‑
vpcs,d
‑
pfh
‑
vpcs,d
‑
pfob
‑
vpcs,d
‑
pfce
‑
vpcs;按照实施例5描述的方法,制备由普通磷脂包载的全氟己烷制剂,命名为d
‑
pfh
‑
vpls。取1ml的该制剂分散在10ml的37℃胎牛血清中,并持续搅拌(500转/分钟),模拟不同制剂在体内血液循环系统所处的环境,在此期间,在不同的时间对溶液进行采样,并使用实施例6描述的荧光去淬灭法测定制剂释放阿霉素的药物量。
[0124]
结果如附图7所示,在48小时内,d
‑
pfp
‑
vpcs泄漏了约50%的阿霉素,而其他三种d
‑
vpcs则只泄漏了<10%的阿霉素,说明由全氟戊烷组成的制剂的载药稳定性较差。d
‑
pfh
‑
vpls的阿霉素泄漏量远高于d
‑
pfh
‑
vpcs,两者分别为78.5%和8.4%,说明该制剂相比于普通磷脂包载的全氟己烷制剂具有更好的载药稳定性。
[0125]
实施例11评价由不同种类的氟化碳组成的制剂的体外超声造影效果
[0126]
将前述实施例10中制备的不同种类的氟化碳组成的制剂装在乳胶管中,浸入37℃的水中,使用resona 7超声成像系统(mindray,中国)检测hifu处理之前和处理之后的超声信号,hifu使用2%pwm(pulse
‑
width modulation)的功率,超声成像系统使用l12
‑
3e超声检测探头(3
‑
12mhz),机械指数(mi)为0.044。
[0127]
如附图8所示,没有经过hifu预处理时,四种氟化碳组成的制剂都没有明显的超声信号。经过hifu(pwm=2%)预处理3分钟后,d
‑
pfp
‑
vpcs和d
‑
pfh
‑
vpcs出现明显的超声信号。但是,d
‑
pfob
‑
pcs和d
‑
pfce
‑
vpcs的超声信号不明显。因此,在hifu预处理后,只有全氟戊烷和全氟己烷组成的制剂具有超声造影能力,由全氟溴辛烷和全氟冠醚组成的制剂不具有超声造影能力。
[0128]
实施例12评价制剂在细胞水平上的治疗效果
[0129]
将4t1细胞接种到24孔板中(每孔104个细胞),在充满1%o2的低氧培养箱中培养24小时,诱导细胞进入乏氧状态。将按照实施例3方法制备的d
‑
pfh
‑
vpcs制剂分散在2mlrpmi
‑
1640培养基中(阿霉素的终浓度为5μg/ml),分别用氮气或氧气饱和(氮气或氧气经过0.22μm气体滤膜的过滤),加入到细胞中,立即用hifu(pwm=2%)处理3分钟,孵育4小时后,使用培养基洗涤细胞三次,继续培养细胞24小时,使用cck
‑
8试剂盒(dojindo,日本)检测细胞的存活率。
[0130]
结果如附图9所示,实验组“氮气饱和的该制剂+hifu处理”相比于实验组“氮气饱和的该制剂”,肿瘤细胞的存活率更低,说明hifu可以促进该制剂释放阿霉素。实验组“氧气饱和的该制剂+hifu处理”相比于实验组“氮气饱和的该制剂+hifu处理”,肿瘤细胞的存活率更低,说明该制剂携带的氧气可以改善肿瘤细胞的乏氧状态,降低肿瘤细胞的存活率。并且实验组“氧气饱和的该制剂+hifu处理”的抗肿瘤效果远高于实验组“游离阿霉素”。
[0131]
本发明人采用未包载阿霉素的pfh
‑
vpcs,在氮气或氧气饱和情况下,以pbs为对照,进一步研究了相关处理方法对肿瘤细胞多重耐药的相关基因和蛋白(hif
‑
1α,mdr1和p
‑
gp)表达水平的影响(mdr1基因的相对表达水平通过rt
‑
qpcr定量,hif
‑
1α和p
‑
gp蛋白的相对表达水平通过western blotting定量)。结果发现:细胞经过vpcs(o2)+hifu处理后,这三种标记物的表达水平都有显著降低,而vpcs(n2)+hifu或pbs(o2)+hifu对三种标志物的表达水平没有明显影响。所述结果表明,pfh
‑
vpcs能够携带氧气,且pfh
‑
vpcs(o2)+hifu在缓解肿瘤乏氧后,可以有效地下调肿瘤细胞多重耐药相关的基因和蛋白的表达水平。
[0132]
实施例13评价制剂对药物的体内分布的改善情况
[0133]
在balb/c小鼠的双侧后腿皮下种植4t1肿瘤,待两侧肿瘤体积增长到100mm3时,经静脉分别注射实施例3制备的制剂(d
‑
vpcs)、实施例5制备的由普通磷脂包载的全氟己烷制剂(d
‑
vpls)、游离阿霉素(free dox)、阿霉素脂质体给药剂量为每千克小鼠体重给予5mg阿霉素。注射后立即用hifu(pwm=2%)对其中一侧的肿瘤进行超声处理15分钟。24小时后,处死小鼠,收集肿瘤和主要器官,称重,向每50mg组织加入0.5ml的pbs,剪碎后,使用电动组织匀浆器彻底匀浆。向100μl匀浆液添加900μl沉淀缓冲液(其中包含800μl异丙醇,100μl 10vol%triton
‑
100和100μl水)中。在4℃条件下静置24小时,将混合物12000g离心,通过多功能酶标仪检测上清液的阿霉素的荧光强度。将已知浓度的阿霉素分散在未经给药处理的小鼠组织匀浆液中,经过上述相同的处理,检测阿霉素的荧光强度,制作每种组织对应的标准校正曲线。从相应的标准校准曲线计算阿霉素的组织含量,最终结果表示为
每克组织中阿霉素含量占阿霉素注射总剂量的百分比(id%/g)。
[0134]
结果如附图10所示,注射d
‑
vpcs后,使用hifu处理的一侧肿瘤比没有使用hifu处理的另一侧肿瘤有更高的阿霉素摄取量(34.2vs 1.2%id/g),这说明d
‑
vpcs有很好的hifu响应性。实验组“d
‑
vpcs+hifu”具有最高的肿瘤阿霉素摄取量,是实验组“free dox”的16.3倍(34.2vs 2.1%id/g),并且是实验组的8.8倍(34.2vs 3.9%id/g)。
[0135]
心肌损伤是阿霉素最严重的副作用之一。尽管与free dox相比,一定程度地减少了心肌对阿霉素的摄取量,但是,实验组“d
‑
vpcs+hifu”具有更低的心肌摄取量,与相比,降低了75.0%(0.3vs 1.2%id/g);与d
‑
vpls相比,降低了81.3%(0.3vs 1.6%id/g)。与free dox,和d
‑
vpls相比,经过“d
‑
vpcs+hifu”处理后的小鼠肾脏和肺中的阿霉素摄取量也最低。
[0136]
实施例14评价制剂的体内化疗效果
[0137]
准备荷4t1肿瘤balb/c小鼠,待肿瘤体积生长至约100mm3左右时,将小鼠随机分为九组,每组5只小鼠:(1)pbs;(2)vpcs(o2)+hifu;(3)free dox;(4)(5)d
‑
vpcs;(6)d
‑
vpls+hifu;(7)d
‑
vpcs+hifu;(8)d
‑
pcs+hifu;(9)d
‑
vpcs(o2)+hifu。d
‑
vpcs和vpcs分别为按照实施例3方法制备的包载阿霉素和不包载阿霉素的修饰了肿瘤靶向分子var7的制剂,d
‑
pcs为按照实施例3方法制备的包载阿霉素、没有修饰肿瘤靶向分子var7的制剂,d
‑
vpls为实施例5制备的普通磷脂包载的全氟化碳制剂(含阿霉素)。对于组(2)和(9),“(o2)”的含义是给予小鼠高氧呼吸处理:从注射前5分钟开始,给小鼠配带氧气呼吸面罩,持续呼吸高浓度氧气20分钟。所有组的给药试剂都是分散在pbs中,经尾静脉注射小鼠,每只小鼠注射体积为200μl。对于使用hifu超声处理的组,在注射后立即用hifu(pwm=2%)对肿瘤进行超声处理,以触发氧气和dox的释放。对于不含dox的组(1)和组(2),从第0天到第9天,每天注射两次药物,早晚各一次,共注射10天;对于其他含有阿霉素的组,分别在第0、2、4、6、8天,每天注射一次,每次阿霉素剂量为5mg/kg。对于第(9)组,在第0、2、4、6、8天,每天早上给予一次“d
‑
vpcs(o2)+hifu”,晚上给予一次“vpcs(o2)+hifu”,并且在第1、3、5、7、9天,每天给予两次“vpcs(o2)+hifu”,早晚各一次,如此一来,可以保证组(9)和组(2)的小鼠接收了相同次数的高氧呼吸。每两天测量一次肿瘤体积。肿瘤体积计算方法为v=(l
×
w2)/2,其中l和w分别是肿瘤的长度和宽度。在治疗结束时(第16天),计算肿瘤生长抑制率,计算公式为:(pbs组的肿瘤体积
‑
实验组的肿瘤体积)/pbs组的肿瘤体积。
[0138]
结果如附图11所示,首先,vpcs(o2)+hifu对肿瘤的生长没有明显影响,一定程度上说明了vpcs和hifu的良好的生物安全性。free dox和具有相似的肿瘤生长抑制率(20.6%vs 25.77%)。d
‑
vpcs+hifu的肿瘤治疗效果远好于d
‑
vpcs、free dox、肿瘤生长抑制率提高到83.2%。d
‑
vpcs+hifu还表现出比d
‑
pcs+hifu、d
‑
vpls+hifu更好的肿瘤生长抑制效果(p<0.05),这是由于d
‑
vpcs相比于d
‑
pcs,增加了肿瘤的靶向性,d
‑
vpcs相比d
‑
vpls,具有更好的载药稳定性。
[0139]
通过改善肿瘤乏氧,缓解乏氧介导的多药耐药,d
‑
vpcs(o2)+hifu的抗肿瘤效果比d
‑
vpcs+hifu更高。与free dox、d
‑
vpcs+hifu相比,d
‑
vpcs(o2)+hifu的肿瘤生长抑制率高达94.6%,相对于前三者,分别提高了3.59、2.67、0.14倍(p<0.0001)。
[0140]
实施例15评价制剂降低肿瘤转移的效果
[0141]
制备转染荧光素酶基因的4t1细胞(4t1
‑
luciferase)。准备荷4t1
‑
luciferase皮下瘤的balb/c小鼠,待肿瘤体积生长至约100mm3左右时,将小鼠随机分为7组,每组5只小鼠:(1)pbs;(2)vpcs;(3)vpcs(o2)+hifu;(4)free dox;(5)(6)d
‑
vpcs+hifu;(7)d
‑
vpcs(o2)+hifu。d
‑
vpcs和vpcs分别为按照实施例3方法制备的包载阿霉素和不包载阿霉素的修饰了肿瘤靶向分子var7的制剂;“(o2)”的含义是给予小鼠高氧呼吸处理。通过实施例14描述的给药方法处理小鼠。通过荧光素酶的生物发光成像可以动态监测4t1细胞在体内的转移和分布情况。在体内治疗实验结束时(第16天),使用ivis spectrum小动物活体成像系统(perkinelmer,美国)观测不同组的肿瘤的肺转移程度:小鼠经腹腔注射150mg/kg d
‑
荧光素钾盐,并在注射后15分钟成像。
[0142]
结果如附图12所示,体内治疗实验的第16天,pbs组(组1)的所有五只小鼠都发生了自发性肺转移。经过free dox(组4)或(组5)的五次给药,尽管原位瘤的生长受到了一定程度的抑制,但是,肺转移的程度明显加重。经过vpcs(o2)+hifu(组3)的多次处理,肺转移程度与pbs组(组1)相比,明显降低,说明vpcs(o2)+hifu可以通过减轻原位肿瘤的乏氧,抑制原位瘤的肺转移。d
‑
vpcs+hifu组(组6)的肺转移程度明显低于pbs组(组1)。d
‑
vpcs(o2)+hifu(组7)的肺转移程度低于d
‑
vpcs+hifu(组6),在所有五只小鼠中均未观察到明显的肺转移,说明该制剂可以改善原位瘤的乏氧程度,抑制肿瘤的自发肺转移。
[0143]
实施例16评价制剂作为新辅助化疗手段的治疗效果
[0144]
准备荷4t1皮下瘤的balb/c小鼠,待肿瘤体积生长至约100mm3左右时,将小鼠随机分为四组,每组6只小鼠,分别经过以下处理:(组1)pbs
→
ht;(组2)free dox
→
ht;(组3)
→
ht;(组4)d
‑
vpcs(o2)+hifu
→
ht。ht代表热疗:使用hifu(pwm=8%)照射肿瘤15分钟。在第0天给予ht,在第负4天和第负2天,分别给予一次化疗。给予化疗时,四组小鼠分别给予pbs,free dox,和d
‑
vpcs(o2)+hifu,药物经尾静脉注射,药物剂量为5mg/kg阿霉素,注射体积为200μl,其中,d
‑
vpcs(o2)+hifu的处理方法如实施例14所述:小鼠在吸入高氧的情况下,经尾静脉注射d
‑
vpcs(由实施例3制备),使用hifu(pwm=2%)照射肿瘤部位15分钟。
[0145]
结果如附图13所示,由于h
‑
hifu介导的热疗本身无法彻底根除所有肿瘤细胞,因此,通过pbs预处理(第
‑
4和
‑
2天)和热疗处理(第0天)的组1,所有小鼠的肿瘤均迅速复发。用free dox或进行化疗预处理(第
‑
4和
‑
2天)和热疗处理(第0天)的组2和组3,肿瘤的复发率分别降至66.7%和50.0%。通过d
‑
vpcs(o2)+hifu进行化疗预处理和热疗处理的组4,在其后的60天内未观察到肿瘤复发。因此,与free dox和相比,d
‑
vpcs(o2)+hifu更适合作为新辅助化疗(化疗预处理)手段,来抑制热疗后的肿瘤复发。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1