一种基于X染色体的男胎cffDNA含量计算方法与流程
本发明主要涉及到基因测序及生物信息分析技术领域,特指一种基于x染色体的男胎cffdna含量计算方法。
背景技术:
孕妇血浆中游离胚胎dna分子的发现将产前检测带入了无创时代,循环的游离胚胎dna(简称cffdna)逐步被认为是以无创方式检测胎儿异常的重要载体。基于高通量测序的无创产前检测(nipt)的各种方法被开发出来,现如今已迅速转化为医疗实践。在这些方案中,cffdna是控制检测结果性能和对检测结果进行恰当临床解读时的至关重要的参数。
现有的基于生物信息方法估计和预测cffdna含量的方法主要有以下几种:
(1)基于y染色体的计算方法:这是最早的估测胚胎dna含量的方法,在早期,位于y染色体上的父系遗传的基因标记如sry基因、dys14基因和zfy基因,通过pcr芯片来验证cffdna的存在,进而可以利用y染色体与某个常染色体的序列含量的比值来估计cffdna含量;在基于大规模并行测序的无创产前检测时代,来自整个y染色体的基因测序序列的比例可以换算为胚胎dna含量,该方法直观准确,适用于检测男胎cffdna含量。
(2)基于孕妇血浆测序数据和父亲基因分型相结合的方法:简单来讲,在生物学父亲和生物学母亲都是纯和且基因分型不同的单核苷酸多态性(snp)位点上,胎儿在该位点的基因分型将表现为杂合,继而可以通过遗传自父亲的等位基因对cffdna含量进行定量。虽然该方法能直接准确评估cffdna含量,但在实际医学实践中,生物学父亲的基因分型结果通常是无法简单直接获取的,因此该方法在实际应用时受到限制。
(3)为克服上述方法(2)在实际应用中的缺陷,有从业者提出了基于靶向高深度测序的cffdna含量计算方法,该方法是通过对孕妇外周血dna进行超高深度靶向测序,采用混合二项分布模型对孕妇和胎儿的四种隐含的基因型组合{aaaa,aaab,abaa,abab}对应的等位基因数量进行建模,在模型中采用极大似然估计对cffdna含量进行计算。该方法产生的结果与上述方法(2)十分接近,该方法的缺点在于:需要对样本进行超高深度测序,测序深度通常需要达到120x以上才能检测出胎儿的等位基因,且靶向测序只能覆盖基因组的一部分,无法实现对胎儿全基因组水平的染色体异常检测,因此在实际应用中也受到限制。
(4)孕妇外周血低深度测序与孕妇基因分型相结合的方法,该方法基于的原理是孕妇自身纯和位点观测到的其他等位基因理论上都是胎儿特有的,这样首先采用基因芯片对孕妇白细胞进行基因分型,然后从孕妇外周血测序数据中鉴定出与母亲纯和位点不同即理论上来自父亲的等位基因;如果假定测序和其他技术原因引入的错误偏差在不同样本中保持恒定的话,则cffdna含量将与这些胎儿杂合位点的比例线性相关,因此可通过对已知cffdna的样本进行上述分析来构建线性回归模型,用来估计和预测其他未知样本的cffdna。通常当测序数据达到1m条序列以上时,通过该方法计算得到的cffdna与上述方法(2)的相关系数可达到0.995以上。然而,模型中的参数可能因测序平台和分型芯片的不同导致噪声的分布特性不同,训练所得的模型不具有普适性;另一方面,杂合位点的比例在不同种族的群体中是不同的,这些固有因素都会影响cffdna预测的准确性。
(5)仅基于孕妇外周血低深度测序数据的seqff方法,该方法试图直接从常规的无创产检数据中估计出cffdna,其基本方法为:首先对孕妇外周血进行单端随机测序,分析常染色体(除13,18,21号染色体之外)上每个50kb窗口中归一化后的read数目以拟合出一个高维的弹性网络和降秩回归模型。该方法与基于y的计算方法的皮尔逊相关系数可达到0.93,但该高维模型的训练需要大规模的训练集样本,且当cffdna含量低于5%时,该方法的准确性也无法保证。
(6)基于胎儿甲基化标记物的方法,该方法是通过胎盘特有的甲基化标志物来估计cffdna含量。举例来讲,rassf1a启动子区域的序列在孕妇和胎儿中的甲基化状态不同,通过对该区域用甲基化敏感的酶进行酶切,来自胎儿的高甲基化的序列不受影响,来自孕妇的低甲基化序列则被酶切破坏,以此实现从母体背景序列中分离出胎儿的序列,用以分析cffdna含量。然而,基于亚硫酸氢盐的甲基化测序方法成本昂贵,且亚硫酸氢盐可能降解dna片段,在常规的无创产检中难以大规模应用。
(7)基于游离dna片段分布的方法,该方法基于的原理是孕妇游离dna中的dna片段与胎儿的dna片段的片段大小呈现出不同的分布特征,来自胎儿的dna片段通常更短,进而通过双端测序,可以基于不同长度片段之间的比例来估计cffdna含量。通常以[100,150]与[163,169]区间的片段数目的比值作为预测因子,通过拟合线性模型估计cffdna含量,该方法与y计算的cffdna含量的相关性系数为0.83,准确性难以满足无创产检的要求。
(8)基于游离dna核小体定位方法,已有研究表明,孕妇外周血中游离dna的片段长度的主峰是166bp,还有一些小的类似刺突的小峰是以10bp为间隔周期的,而胎儿游离dna分子长度的主峰是143bp;科学家推测166bp包含了核小体主体和一个连接子,相反143bp主峰的dna分子则缺少了该连接子作为其组成部分,基于这个假设模型,科学家基于核小体定位方法开发出一种预测cffdna的方法,但该方法准确性不高,难以满足临床要求。
综上所述,基于孕妇外周血游离dna低深度大规模并行测序的方法仍然是无创产前检测的主流方法,基于y染色体的计算方法被认为是男胎cffdna计算的金标准方法,然而该方法需要男性的游离dna作为对照,才能准确估算特定测序平台和生物信息处理流程计算得到的y染色体的含量,进而准确推断cffdna含量;但在常规无创产检中检测男性对照样本将增加测序的成本和流程管理的复杂性,另外,如果胎儿存在y染色体的非整倍体异常(如缺少y染色体,或存在多条y染色体),则无法准确估测cffdna含量。
技术实现要素:
本发明要解决的技术问题就在于:针对现有技术存在的技术问题,本发明提供一种原理简单、操作简便、准确率高、检测效率高的基于x染色体的男胎cffdna含量计算方法。
为解决上述技术问题,本发明采用以下技术方案:
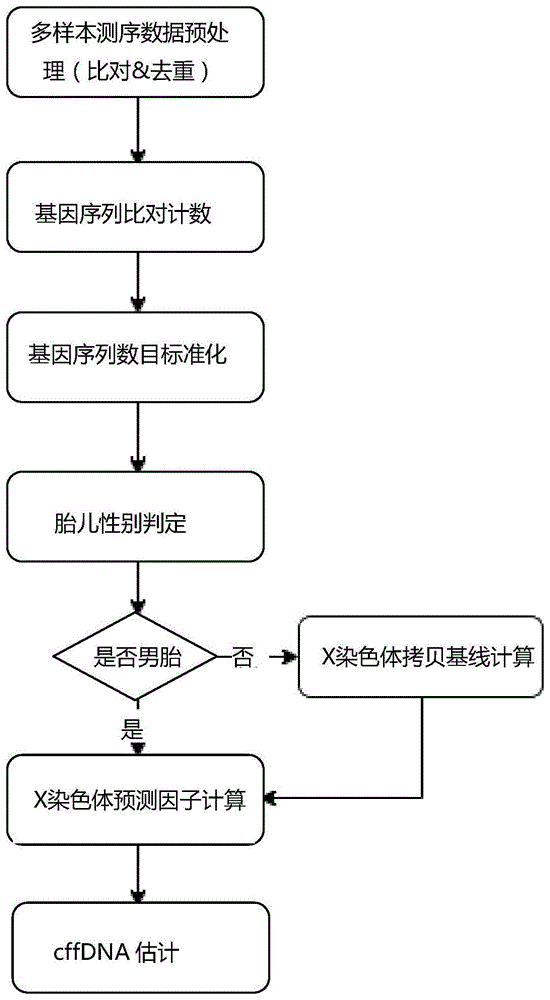
一种基于x染色体的男胎cffdna含量计算方法,其步骤包括:
步骤s1:获得原始测序基因序列;
步骤s2:对测序基因序列计数;
步骤s3:对测序基因序列数目进行标准化;
步骤s4:胎儿性别鉴定;
步骤s5:女胎x染色体窗口内拷贝基线计算;
步骤s6:男胎x染色体预测因子计算及异常点检测;
步骤s7:通过下式获得男胎cffdna含量:
其中,smale为男胎样本集合,i表示样本集中序号为i的样本,
作为本发明方法的进一步改进:所述步骤s1中,对无创产前检测孕妇外周血样本进行低深度测序,获得原始测序基因序列。
作为本发明方法的进一步改进:所述步骤s1中,对原始测序基因序列进行预处理;所述预处理包括将原始测序基因序列比对到人类参考基因组,并对比对结果去重复。
作为本发明方法的进一步改进:所述步骤s2中包括:
统计每个样本1-22号常染色体和性染色体上的唯一比对基因片段数目umi,j,其中1≤i≤n,j∈{1,2,…,22,x,y};
统计x染色体上大小为k的窗口内唯一比对的基因片段数目umi,x,k,其中1≤i≤n,
性染色体统计不计入假常染色体区域内的唯一比对基因序列片段。
作为本发明方法的进一步改进:所述步骤s3包括以下步骤:
步骤s301:计算1-22常染色体的唯一比对基因片段的总数
步骤s302:计算每个样本x染色体和y染色体的相对含量:xci=umi,x/umi,yci=umi,y/umi,1≤i≤n;
步骤s303:计算每个样本x染色体上m个大小为k的窗口内唯一比对的基因片段数目标准值:
作为本发明方法的进一步改进:所述步骤s4中,当y染色体含量yci大于给定阈值σ,则判定胎儿性别为男胎,否则判定为女胎。
作为本发明方法的进一步改进:所述阈值σ为0.0005。
作为本发明方法的进一步改进:所述步骤s5中包括以下步骤:
步骤s501:选取上步胎儿性别鉴定结果为女胎的样本集合sfemale;
步骤s502:计算所有女胎样本x染色体上m个大小为k的窗口内序列片段标准值的中值mnumx,k=median(numi,x,k),i∈sfemale,
作为本发明方法的进一步改进:所述步骤s6包括:
步骤s601:选取性别鉴定结果为男胎的样本集合smale;
步骤s602:计算smale集合中每个样本x染色体上m个大小为k的窗口内的预测因子pfi,x,k=log2(numi,x,k/mnumx,k),i∈smale,
步骤s603:去除预测因子中的异常点。
作为本发明方法的进一步改进:所述步骤s603包括:
步骤s6031:对样本i,计算所有预测因子的0.05和0.95分位数αi,0.05及αi,0.95;
步骤s6032:将小于αi,0.05和大于αi,0.95的预测因子去除,记去除异常后的预测因子集合为
与现有技术相比,本发明的优点在于:
1、本发明的一种基于x染色体的男胎cffdna含量计算方法,通过选取x染色体含量作为男胎cffdna含量的预测因子,解决了当胎儿y染色体存在非整倍体异常时,利用y染色体无法准确估测cffdna含量的问题。
2、本发明的一种基于x染色体的男胎cffdna含量计算方法,通过选取x染色体作为男胎cffdna含量的预测因子,由于x染色体为y染色体的3倍长,同等统计准确性下,需要的测序数据量是y染色体方法的1/3,进一步降低了检测成本.
3、本发明的一种基于x染色体的男胎cffdna含量计算方法,无需检测男性游离dna作为对照,而是以女胎孕妇外周血游离dna测序数据作为对照,在常规无创产前检测中数据可直接获取,降低了额外的测序成本及流程管理复杂性。
4、本发明的一种基于x染色体的男胎cffdna含量计算方法,采用滑动窗口方式对数据进行上采样,提高了统计功效,降低了对测序量的要求,进一步压缩检测成本。
附图说明
图1是本发明在具体应用实例中的流程示意图。
具体实施方式
以下将结合说明书附图和具体实施例对本发明做进一步详细说明。
人类有23对染色体,其中1-22号染色体为常染色体,x和y为性染色体,男性的性染色体组合为xy,女性的性染色体组合为xx;对于男胎孕妇来讲,其外周血中的游离dna是孕妇自身dna与胎儿dna的混合,孕妇与胎儿的性染色体组合为xxxy。假设cffdna含量为f,则x染色体含量的期望值为2*(1-f)+f=2-f,由于x染色体含量与cffdna含量相关,因此可基于x染色体间接预测得到cffdna含量;进一步,对于x染色体每个固定长度的子区域,也具有上述性质。
如图1所示,本发明基于以上原理提出一种基于x染色体的男胎cffdna含量计算方法,其步骤包括:
步骤s1:获得原始测序基因序列;
对n例(n>30)无创产前检测孕妇外周血样本进行低深度测序,获得原始测序基因序列,并对原始测序基因序列进行预处理;
步骤s2:对测序基因序列计数;
步骤s3:对测序基因序列数目进行标准化;
步骤s4:胎儿性别鉴定;
步骤s5:女胎x染色体窗口内拷贝基线计算;
步骤s6:男胎x染色体预测因子计算及异常点检测;
步骤s7:通过下式获得男胎cffdna含量:
其中,smale为男胎样本集合,i表示样本集中序号为i的样本,
在具体应用实例中,上述步骤s1中,进一步对原始测序基因序列进行预处理;所述预处理包括将原始测序基因序列比对到人类参考基因组(如在实施中可以选取基因组版本为hg19),并对比对结果去重复。
在具体应用实例中,上述步骤s2中,包括:
统计每个样本1-22号常染色体和性染色体上的唯一比对基因片段数目umi,j,其中1≤i≤n,j∈{1,2,…,22,x,y};
统计x染色体上大小为k(例如具体实施取k=5000)的窗口内唯一比对的基因片段数目umi,x,k,其中1≤i≤n,
性染色体统计不计入假常染色体区域(par区域)内的唯一比对基因序列片段。
在具体应用实例中,上述步骤s3可以包括以下步骤:
步骤s301:计算1-22常染色体的唯一比对基因片段的总数
步骤s302:计算每个样本x染色体和y染色体的相对含量:xci=umi,x/umi,yci=umi,y/umi,1≤i≤n;
步骤s303:计算每个样本x染色体上m(实施取m=12)个大小为k的窗口内唯一比对的基因片段数目标准值:
在具体应用实例中,上述步骤s4中,当y染色体含量yci大于给定阈值σ,则判定胎儿性别为男胎,否则判定为女胎。根据实际需要,通过专家数据库可以在具体实施中取σ=0.0005。
在具体应用实例中,上述步骤s5可以包括以下步骤:
步骤s501:选取上步胎儿性别鉴定结果为女胎的样本集合sfemale;
步骤s502:计算所有女胎样本x染色体上m个大小为k的窗口内序列片段标准值的中值mnumx,k=median(numi,x,k),i∈sfemale,
在具体应用实例中,上述步骤s6可以包括以下步骤:
步骤s601:选取步骤s4性别鉴定结果为男胎的样本集合smale;
步骤s602:计算smale集合中每个样本x染色体上m个大小为k的窗口内的预测因子pfi,x,k=log2(numi,x,k/mnumx,k),i∈smale,
步骤s603:去除预测因子中的异常点,这是因为孕妇的游离dna含量显著高于cffdna含量,如果孕妇在相应的统计窗口内存在拷贝数变异,则估计得到的cffdna含量将受到孕妇背景序列的影响,导致计算结果失真。
进一步,步骤s603可以包括以下流程:
步骤s6031:对样本i,计算所有预测因子的0.05和0.95分位数αi,0.05及αi,0.95;
步骤s6032:将小于αi,0.05和大于αi,0.95的预测因子去除,记去除异常后的预测因子集合为
以上仅是本发明的优选实施方式,本发明的保护范围并不仅局限于上述实施例,凡属于本发明思路下的技术方案均属于本发明的保护范围。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理前提下的若干改进和润饰,应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!