基于分治整合策略的成对蛋白质相互作用网络比对方法
1.本发明涉及蛋白质相互作用网络比对领域,具体涉及一种基于分治整合策略的成对蛋白质相互作用网络比对方法。
背景技术:
2.随着生物信息学的发展,人们研究集中在蛋白质和dna等生物大分子.蛋白质分子在生物体内执行着各项重要任务,而蛋白质相互作用是维持细胞结构和功能的基础,因此对蛋白质相互作用网络的研究具有重要意义.其中一类很重要的研究工作就是对蛋白质相互作用网络进行比对分析,通过网络比对可进行蛋白质功能预测以及保守功能模块的挖掘等。
3.传统技术存在以下技术问题:
[0004]“spinal:scalable protein interaction network aligment”(期刊出处:bioinformatics.2013,4(29):917
–
924)算法使用一种由粗粒度和细粒度步骤组成的双通道比对算法。粗粒度阶段通过考虑在前一次迭代中计算出的与相邻节点匹配的置信度,迭代地改进每对节点估计匹配置信度的矩阵p。p收敛后开始细粒度阶段,该阶段使用种子
‑
扩展算法来构造比对。此外,在种子
‑
扩展过程的每次迭代中,都执行局部搜索,以直接增加保守边的数量。这种算法存在的问题:过多的考虑拓扑信息,导致最终比对的生物功能质量不高。
[0005]“modulealign:module
‑
based global alignment ofprotein
‑
protein interaction networks”(期刊出处:bioinformatics,2016,32(17):658
‑
664)算法提出了一种同源得分函数,它依靠模块相似性来计算结点的同源得分,并采用了动态匈牙利算法进行求解。这种算法存在的问题:模块化方法选取不当此算法通过模块相似性计算方法比较繁琐且计算方式不当,导致产生错误的生物相似性得分,从而使其生物功能质量较差。
[0006]“alignet:alignment ofprotein
‑
protein interaction networks”(期刊出处:bmc bioinformatics,2020,21(suppl 6):1
‑
22)算法采用了模块化思想,首先对将网络划分为若干个模块,将模块进行枚举比对,最后将所有比对结果合并处理为最终比对。这种算法存在的问题:需要将所有模块进行枚举比对,大大增加了时间复杂度。
[0007]“hubalign:an accurate and efficient method for global alignment of protein
‑
protein interaction networks”(期刊出处:bioinformatics,2014,30(17):438
‑
444)算法认为在ppi网络中充当枢纽的蛋白质在功能和拓扑上更为重要,提出了importance中心性,并使用贪心的种子
‑
扩展算法,根据蛋白质的importance分数和序列相似度组合对蛋白质进行排序。这种算法存在的问题:算法随机选择结点作为边拆分的起点,不同的起点可能会得到不同质量的比对结果。
[0008]“magna++:maximizing accuracy in global network alignment via both node and edge conservation”(期刊出处:bioinformatics,2015,31(14):2409
‑
2411)算法采用了遗传算法的思想进行网络比对,通过不停迭代交叉变异产生新解。有效解决了算
法陷入局部最优解问题。这种算法存在的问题:需要进行上千次的迭代,花费时间较长。
[0009]“index:incremental depth extension approach forprotein
‑
protein interaction networks alignment”期刊出处:biosystems,2017,162(2017):24
‑
34)算法提出了一种新的比对策略,考虑了比对分数和对齐核的增长,使得得到的公共连通子图比以往的方法具有更大的边数。这种算法存在的问题:比对的生物质量较差,未能达到较好的生物与拓扑质量的平衡。
技术实现要素:
[0010]
本发明要解决的技术问题是提供一种基于分治整合策略的成对蛋白质相互作用网络比对方法,解决了对同一网络中结点间相似性文件的依赖问题;使用已有蛋白对的匹配关系来预测模块的匹配关系,从而计算模块相似性,解决了模块相似性计算问题;使用度中心性和特征向量中心性捕捉结点的拓扑特性,提高了算法的生物质量和拓扑质量。
[0011]
为了解决上述技术问题,本发明提供了一种基于分治整合策略的成对蛋白质相互作用网络比对方法,包括:
[0012]
步骤1:读取源网络和目标网络及blast相似性文件;
[0013]
步骤2:采用基于结点和路径相结合的方法分别计算两个网络中结点的相似性得分,结合所述相似性得分分别对两个网络进行模块划分;
[0014]
步骤3:获取同源蛋白对,根据同源蛋白对和blast相似性分别计算来自不同网络模块间的相似性;根据相似性将来自不同网络的模块进行一对一匹配;
[0015]
步骤4:根据特征向量中心性和blast相似性计算每对已匹配模块中结点间的相似性,并进行模块内比对,将得到的子比对结果合并为候选结果集;
[0016]
步骤5:对候选结果集使用超图匹配算法得到最终一对一比对结果。
[0017]
在其中一个实施例中,步骤2中,计算相似性得分具体如下:
[0018]
采用度和最短路径长度衡量两个结点间的相似性,结点相似性计算如公式(1)
[0019][0020]
其中g为网络,u,v为g中的结点,deg
u
指结点u的度,deg
g
指图g中的最大度,d(g)指图g的直径,d
g
(u,v)指结点u,v的最短路径长度。
[0021]
在其中一个实施例中,步骤2中,模块划分步骤如下:
[0022]
(1)对源网络g使用公式(1)进行相似性计算,得到相似性矩阵s;
[0023]
(2)对于矩阵s中的每一行,根据数值大小,将相似性在前75%的结点构成一个模块,模块中心为该行矩阵的行名。
[0024]
在其中一个实施例中,步骤3具体如下:
[0025]
生成同源蛋白对,并根据蛋白对在模块中的集体行为计算模块的同源相似性,使用公式(2)将同源蛋白对文件转换为同源矩阵π,其中i,j分别为来自两个网络的蛋白质;
[0026][0027]
则根据矩阵π得到的模块m1,m2的同源相似性得分为:
[0028][0029]
公式(3)受限于公式(4):
[0030][0031]
模块相似性计算公式为:
[0032]
s(m1,m2)=hs(m1,m2)+blast(c1,c2)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(5)
[0033]
其中c1,c2分别为模块m1,m2的簇中心;blast为序列相似性;
[0034]
根据公式(5)得到模块间相似性矩阵s,对s使用匈牙利算法求解,则可得到一对一的模块匹配关系。
[0035]
在其中一个实施例中,步骤4具体如下:对于模块比对阶段得到的每一对匹配上的模块,首先计算模块内结点的相似性矩阵,再对两个模块内的结点进行比对;更具体过程如下:
[0036]
根据特征向量中心性和序列相似性计算不同模块中两个结点间的相似性,见公式(6)
[0037][0038]
其中t(u,v)表示结点u,v的特征向量中心性相似得分,其计算方法见公式(7):
[0039][0040]
其中c
u
指结点u的特征向量中心性;
[0041]
将所有配对模块生成的子比对合并为候选集,此时的候选集中一个结点可能会和来自另一个网络中的多个结点形成比对关系,因此候选集为多对多匹配集合。
[0042]
在其中一个实施例中,根据公式(6)进行模块内比对步骤如下:(1)首先将模块m1,m2的模块中心c1,c2比对上;(2)分别获取c1,c2的邻居,deg(c1),deg(c2);(3)从f中提取行名和列名在deg(c1)和deg(c2)中的子矩阵,并使用匈牙利算法将deg(c1),deg(c2)结点进行比对;(4)将已扩展结点(c1,c2)移除,并对剩余已比对结点对依次重复步骤(2)(3)。
[0043]
在其中一个实施例中,步骤5具体如下:将候选集抽象为超图,其中源网络中的结点为超图的源结点,目标网络中的结点为超图的目标结点,每个子比对对应超图的一条超弧;使用加权二部超图匹配算法将超图提取为仅包含一对一比对关系的二部图,即得到最终的结点匹配关系。
[0044]
基于同样的发明构思,本申请还提供一种计算机设备,包括存储器、处理器及存储在存储器上并可在处理器上运行的计算机程序,所述处理器执行所述程序时实现任一项所述方法的步骤。
[0045]
基于同样的发明构思,本申请还提供一种计算机可读存储介质,其上存储有计算机程序,该程序被处理器执行时实现任一项所述方法的步骤。
[0046]
基于同样的发明构思,本申请还提供一种处理器,所述处理器用于运行程序,其中,所述程序运行时执行任一项所述的方法。
[0047]
本发明的有益效果:
[0048]
使用基于结点和基于路径的相似性计算方法,代替自相似性文件,解决了对自相似性文件的依赖问题;从primalign获取同源蛋白对,并使用已有蛋白对的匹配关系来预测模块的匹配关系,从而计算模块相似性,解决了模块相似性计算问题;对特征向量中心性进行进一步优化,同时捕获结点自身的中心性和两个结点间的中心性差异,从而更好捕捉结点的拓扑特性,提高了算法的质量。
附图说明
[0049]
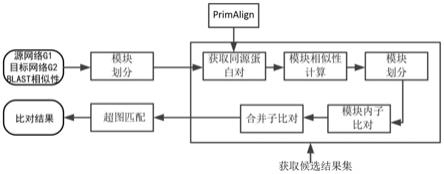
图1是一种基于分治整合策略的成对蛋白质相互作用网络比对方法的流程图。
[0050]
图2是不同算法在isobase数据集上的ec,ics,s3和fc得分示意图。
[0051]
图3是不同算法在合成数据集(dmc,dmr)上的ec,ics,s3和fc得分示意图。
[0052]
图4是本发明分别使用现有算法获取同源对时和其原有算法在ec,ics,s3和fc得分示意图。
[0053]
图5是本发明使用不同的现有算法获取同源对进行比对所需时间示意图。
具体实施方式
[0054]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0055]
参阅图1到图5,一种基于分治整合策略的成对蛋白质相互作用网络比对方法,包括:
[0056]
步骤1:读取源网络和目标网络及blast相似性文件。
[0057]
步骤2:采用基于结点和路径相结合的方法分别计算两个网络中结点的相似性得分,结合所述相似性得分分别对两个网络进行模块划分。
[0058]
为了更充分挖掘同一网络中结点的相似性信息,采用了基于结点和基于路径的相结合方法计算结点间的相似性,即采用度和最短路径长度衡量两个结点间的相似性,结点相似性计算如公式(1)。
[0059][0060]
其中g为网络,u,v为g中的结点,deg
u
指结点u的度,deg
g
指图g中的最大度,d(g)指图g的直径,d
g
(u,v)指结点u,v的最短路径长度。
[0061]
模块划分步骤如下:
[0062]
(1)对源网络g使用公式(1)进行相似性计算,得到相似性矩阵s;
[0063]
(2)对于矩阵s中的每一行,根据数值大小,将相似性在前75%的结点构成一个模块,模块中心为该行矩阵的行名。
[0064]
步骤3:获取同源蛋白对,根据同源蛋白对和blast相似性分别计算来自不同网络模块间的相似性;根据相似性将来自不同网络的模块进行一对一匹配。
[0065]
使用primalign生成同源蛋白对,并根据蛋白对在模块中的集体行为计算模块的
同源相似性,使用公式(2)将同源蛋白对文件转换为同源矩阵π,其中i,j分别为来自两个网络的蛋白质。
[0066][0067]
则根据矩阵π得到的模块m1,m2的同源相似性得分为:
[0068][0069]
公式(3)受限于公式(4):
[0070][0071]
模块相似性计算公式为:
[0072]
s(m1,m2)=hs(m1,m2)+blast(c1,c2)
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(5)
[0073]
其中c1,c2分别为模块m1,m2的簇中心。blast为序列相似性。
[0074]
根据公式(5)得到模块间相似性矩阵s,对s使用匈牙利算法求解,则可得到一对一的模块匹配关系。
[0075]
步骤4:根据特征向量中心性和blast相似性计算每对已匹配模块中结点间的相似性,并进行模块内比对,将得到的子比对结果合并为候选结果集;
[0076]
对于模块比对阶段得到的每一对匹配上的模块,首先计算模块内结点的相似性矩阵,再对两个模块内的结点进行比对,具体过程如下:
[0077]
根据特征向量中心性和序列相似性计算不同模块中两个结点间的相似性,见公式(6)
[0078][0079]
其中t(u,v)表示结点u,v的特征向量中心性相似得分,其计算方法见公式(7):
[0080][0081]
其中c
u
指结点u的特征向量中心性。公式(6)不仅考虑结点u,v的特征向量中心性之差,同时考虑了结点自身的中心性值,该种设计有助于首先将在模块中具有较强的中心性地位且中心性相近的蛋白质先比对上。
[0082]
根据公式(6)进行模块内比对步骤如下:
[0083]
(1)首先将模块m1,m2的模块中心c1,c2比对上;
[0084]
(2)分别获取c1,c2的邻居,deg(c1),deg(c2);
[0085]
(3)从f中提取行名和列名在deg(c1)和deg(c2)中的子矩阵,并使用匈牙利算法将deg(c1),deg(c2)结点进行比对;
[0086]
(4)将已扩展结点(c1,c2)移除,并对剩余已比对结点对依次重复步骤(2)(3)。
[0087]
将所有配对模块生成的子比对合并为候选集,此时的候选集中一个结点可能会和来自另一个网络中的多个结点形成比对关系,因此候选集为多对多匹配集合。
[0088]
步骤5:对候选结果集使用超图匹配算法得到最终一对一比对结果。
[0089]
将候选集抽象为超图,其中源网络中的结点为超图的源结点,目标网络中的结点为超图的目标结点,每个子比对对应超图的一条超弧。使用加权二部超图匹配算法将超图提取为仅包含一对一比对关系的二部图,即得到最终的结点匹配关系。
[0090]
下面给出本发明的一个具体应用场景:
[0091]
以isobase数据库中的sce和hsa两个网络为例:
[0092]
1.读取sce网络和hsa网络及blast相似性文件;
[0093]
2.根据公式(1)分别计算sce和hsa网络中结点的相似性值;
[0094]
3.分别对sce和hsa网络进行模块划分;
[0095]
4.使用primalign获取同源蛋白对
[0096]
5.根据公式(5)计算来自不同网络模块间的相似性;
[0097]
6.根据相似性将来自不同网络的模块进行一对一匹配;
[0098]
7.根据公式(6)计算每对已匹配模块中结点间的相似性,并进行模块内比对;
[0099]
8.将7得到的子比对结果合并为候选结果集;
[0100]
9.对候选结果集使用超图匹配算法得到最终一对一比对结果。
[0101]
以上所述实施例仅是为充分说明本发明而所举的较佳的实施例,本发明的保护范围不限于此。本技术领域的技术人员在本发明基础上所作的等同替代或变换,均在本发明的保护范围之内。本发明的保护范围以权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1