一种16S和宏基因组测序数据关联分析方法、系统及设备与流程
一种16s和宏基因组测序数据关联分析方法、系统及设备
技术领域
1.本发明涉及数据处理和分析领域,尤其涉及一种16s和宏基因组测序数据关联分析方法、系统及设备。
背景技术:
2.微生物与生物,环境,生产等方方面面息息相关,扮演着重要的作用,其中人体微生物更是被认为人类第二基因组学。随着科技技术的发展,高通量测序也在不断普及。16s测序技术和宏基因组测序技术是两种重要的高通量测序手段。16s测序技术一般仅侧重研究群落的多样性变化,宏基因组测序侧重挖掘微生物群落的功能结构。
3.现有技术存在如下缺陷:虽可通过软件根据数据进行16s测序数据进行通路功能分析,但分析准确性有限,且受引物特异性、扩增偏好性、分辨率等影响;虽已有分析可以挖掘宏基因组中多样性研究,但数据量不足和组装效果不理想等因素制约着数据分析。
技术实现要素:
4.为克服相关技术中存在的问题,本发明提供一种16s和宏基因组测序数据关联分析方法、系统及设备,从而解决单一16s测序数据和单一宏基因组测序数据分析中的缺陷。
5.根据本发明实施例的第一方面,提供一种16s和宏基因组测序数据关联分析方法。该方法包括:
6.对样本进行16s测序和宏基因测序,获取16s组学数据和宏基因组学数据;其中,所述16s组学数据包括16s物种丰度表和16s功能丰度表,所述宏基因组学数据包括宏基因物种丰度表和宏基因功能丰度表;
7.根据所述16s组学数据进行micropita分析,获得代表性样本;
8.根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有的物种/功能统计,并图示化展示;
9.根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种/功能丰度变化和鉴定到的主要物种/功能类型;
10.根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pea rson相关系数、mantel test分析、procrustes test分析;
11.根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析。
12.优选地,所述对样本进行16s测序和宏基因测序,获取16s组学数据和宏基因组学数据,具体包括:
13.对所述样本进行16s rdna测序,获得所述16s物种丰度表,利用picr ust2软件预测,获得所述16s功能丰度表;
14.对所述样本进行宏基因组测序,获得所述宏基因物种丰度表和所述宏基因功能丰度表;
15.其中,所述宏基因功能丰度表和所述16s功能丰度表都包括kegg功能的level b丰
度表和level c丰度表。
16.优选地,所述根据所述16s组学数据进行micropita分析,获得代表性样本,具体包括:
17.根据所述16s组学数据,利用筛选指标挑选代表性样本,其中,所述筛选指标包括diverse,extreme,representative,distinct,discriminant;
18.利用micropita软件在所述16s组学数据中获取所述代表性样本的样本信息;
19.利用r语言vegan包对16s otu丰度表进行bray
‑
curtis距离pcoa分析,获得所有样本在二维空间的分布特征,同时在图形中可标记micropita筛选出的代表性样本。
20.优选地,所述根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有的物种/功能统计,并图示化展示,具体包括:
21.基于各物种分类水平和kegg功能的level b丰度表和level c丰度表,使用r语言的venndiagram包和upsetr包,以0为阈值,进行韦恩分析;
22.使用韦恩图和upset图展示所述16s组学数据和所述宏基因组学数据的共有、特有元素的数量;
23.使用lefse软件在物种层级进化分支树上标记16s和宏基因组鉴定到的共有、特有物种。
24.优选地,所述根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种/功能丰度变化和鉴定到的主要物种/功能类型,具体包括:
25.挑选所述16s组学数据中各分类水平排名前10的物种/功能,并存储为第一16s物种/功能;
26.挑选所述宏基因组学数据中各分类水平排名前10的物种/功能,并存储为第一宏基因物种/功能;
27.对所述第一16s物种/功能与所述第一宏基因物种/功能取并集,并保存;
28.从所述宏基因组学数据和所述16s组学数据中除去所述第一16s物种/功能与所述第一宏基因物种/功能的并集,并将已知的物种/功能划标注为other,将未知物种/功能标注为unclassified;
29.使用r语言ggplot2包绘制两组学所有样本中物种/功能丰度变化和两组学鉴定到的主要物种/功能类型,其中,所述两组学包括所述16s组学数据和所述宏基因组学数据。
30.优选地,所述根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pearson相关系数分析、mantel test分析、procrustes test分析;
31.所述pearson相关系数分析具体操作为:将所述16s组学数据和所述宏基因组学数据中所有样本的丰度数据进行对数转化后,使用r语言psych包计算所述16s组学数据和所述宏基因组学数据的pearson相关系数,并使用fizh
‑
z 变换计算相关显著性,并图示化展示数据分析结果;
32.所述mantel test分析具体操作为:基于所述16s组学数据中的16s物种/ 功能数据,计算样本间的bray
‑
curtis距离矩阵;基于所述宏基因组学数据中的宏基因组物种/功能数据,计算样本间的bray
‑
curtis距离矩阵;使用r语言veg an包,对两组学的距离矩阵进行mantel test分析,进而计算各物种/功能分类水平下16s和宏基因组测序结果的相关性,
通过permutation置换检验999次,计算相关显著性,并图示化展示数据分析结果;
33.所述procrustes test分析具体操作为:使用r语言vegan包,根据各分类水平两组学物种/功能bray
‑
curtis距离的pcoa结果,开展procrustes分析,并使用permutation test进行相关显著性检验,并图示化展示数据分析结果;
34.其中,所述两组学包括所述16s组学数据和所述宏基因组学数据。
35.优选地,所述根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析,具体包括:
36.使用r语言psych包计算环境因子间的pearson相关系数;
37.使用r语言vegan包进行16s测序otu丰度表、16s测序的shannon多样性、宏基因组功能矩阵与每个环境因子的mantel test分析;
38.通过permutation置换检验999次,计算相关显著性;
39.利用r语言ggcor包生成热图和网络图结合的形式展示。
40.根据本发明实施例的第二方面,提供一种16s和宏基因组测序数据关联分析系统,该系统包括:
41.组学数据获取模块,对样本进行16s测序和宏基因测序,获取16s组学数据和宏基因组学数据;其中,所述16s组学数据包括16s物种丰度表和16 s功能丰度表,所述宏基因组学数据包括宏基因物种丰度表和宏基因功能丰度表;
42.micropita分析模块,根据所述16s组学数据进行micropita分析,获得代表性样本;
43.韦恩分析模块,根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有的物种/功能统计,并图示化展示;
44.物种/功能分布分析模块,根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种 /功能丰度变化和鉴定到的主要物种/功能类型;
45.相关性分析模块,根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pearson相关系数、mantel test分析、procrustes test分析;
46.环境因子关联分析模块,根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析。
47.优选地,一种16s和宏基因组测序数据关联分析系统,该系统还包括:
48.显示模块,用于显示所述组学数据获取模块、所述micropita分析模块、所述韦恩分析模块、所述物种/功能分布分析模块、所述相关性分析模块和所述环境因子关联分析模块中获得的关联分析图与关联分析数据;
49.存储模块,用于存储所述组学数据获取模块、所述micropita分析模块、所述韦恩分析模块、所述物种/功能分布分析模块、所述相关性分析模块和所述环境因子关联分析模块中获得的关联分析图与关联分析数据。
50.根据本发明实施例的第三方面,提供一种电子设备,包括存储器和处理器,所述存储器用于存储一条或多条计算机程序指令,其中,所述一条或多条计算机程序指令被所述处理器执行以实现所示的16s和宏基因组测序数据关联分析方法。
51.本发明的实施例提供的技术方案可以包括以下有益效果:
52.1、通过使用韦恩分析进行两组学共有、特有的物种和功能分析,并结合物种进化分支树对物种的共有性可视化展示明确组学之间的差异,实现两种组学数据的互相补充,支撑组学之间的互相完善。
53.2、通过判断两组学的一致性和相关性,使数据分析的更靠性,使用堆叠图直观展示两组学样本中物种/功能丰度变化规律,初步观察两组学的一致性,结合饼图展示两组学各自鉴定到的高丰度物种/功能,对比观察一致性,进而开展相关性分析,实现两种组学数据的互相验证,有效判断组学相关性。
54.3、在宏基因组与16s测序样本没有一一对应时,本方案通过micropita 分析,可根据16s测序的otu丰度表,评估出最具有代表性的样本,作为指导宏基因组分析样本选择的参考。
55.应当理解的是,以上的一般描述和后文的细节描述仅是示例性和解释性的,并不能限制本公开。
附图说明
56.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
57.图1是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法的流程图。
58.图2是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的组学数据获取流程图。
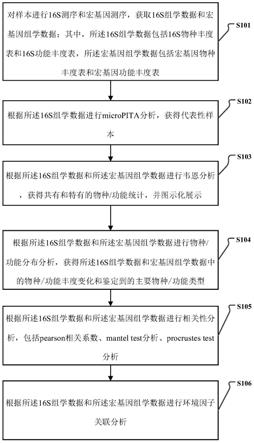
59.图3是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的micropita分析流程图。
60.图4是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的韦恩分析流程图。
61.图5是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的物种/功能分布分析流程图。
62.图6是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的散点图。
63.图7是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的mantel
‑
test检验图。
64.图8是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的procrustes分析图。
65.图9是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的环境因子关联分析流程图。
66.图10是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的环境因子mantel
‑
test分析图。
67.图11是本发明一个实施例的一种16s和宏基因组测序数据关联分析系统的结构图。
68.图12是本发明一个实施例的一种16s和宏基因组测序数据关联分析系统的模块处理关系示意图。
69.图13是本发明一个实施例的一种电子设备的结构图。
具体实施方式
70.为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。
71.在本发明的说明书和权利要求书及上述附图中的描述的一些流程中,包含了按照特定顺序出现的多个操作,但是应该清楚了解,这些操作可以不按照其在本文中出现的顺序来执行或并行执行,操作的序号如101、102等,仅仅是用于区分开各个不同的操作,序号本身不代表任何的执行顺序。另外,这些流程可以包括更多或更少的操作,并且这些操作可以按顺序执行或并行执行。需要说明的是,本文中的“第一”、“第二”等描述,是用于区分不同的消息、设备、模块等,不代表先后顺序,也不限定“第一”和“第二”是不同的类型。
72.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
73.微生物与生物,环境,生产等方方面面息息相关,扮演着重要的作用,其中人体微生物更是被认为人类第二基因组学。随着科技技术的发展,高通量测序也在不断普及。16s测序技术和宏基因组测序技术是两种重要的高通量测序手段之一。16s测序技术一般仅侧重研究群落的多样性变化,宏基因组测序侧重挖掘微生物群落的功能结构。
74.现有技术存在如下缺陷:虽可通过软件根据数据进行16s测序数据进行通路功能分析,但分析准确性有限,且受引物特异性/扩增偏好性/分辨率等影响;虽已有分析可以挖掘宏基因组中多样性研究,但数据量不足和组装效果不理想等因素制约着数据分析。
75.本发明技术方案提供16s与宏基因组测序数据的关联分析方法,使两组学数据相互补充,由宏基因组扩展数据研究的深度,并使两组学数据相互验证,由16s验证宏基因组数据分析的准确性。
76.图1是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法的流程图。优选地,16s和宏基因组测序数据关联分析方法,其特征在于,包括:
77.s101、对样本进行16s测序和宏基因测序,获取16s组学数据和宏基因组学数据;其中,所述16s组学数据包括16s物种丰度表和16s功能丰度表,所述宏基因组学数据包括宏基因物种丰度表和宏基因功能丰度表;
78.s102、根据所述16s组学数据进行micropita分析,获得代表性样本;
79.其中,micropita分析为基因筛选方法。
80.s103、根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有的物种/功能统计,并图示化展示;
81.s104、根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种/功能丰度变化和鉴定到的主要物种/功能类型;
82.s105、根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pearson相关系数、mantel test分析、procrustes test分析;
83.s106、根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析。
84.其中,pearson相关系数(pearson correlation coefficient)是用来衡量两个数据集合是否在一条线上面,它用来衡量定距变量间的线性关系;manteltest是对两个矩阵相关关系的检验;procrustes test为强制一致性检验。
85.在本发明实施例中,通过在进行16s测序和宏基因测序的基础上,利用 16s组学数据进行micropita分析,利用16s组学数据和宏基因组学数据进行韦恩图分析、物种/功能分布分析、相关性分析和环境因子关联分析。通过全方位的数据分析一方面能够获得两种组学数据的差别,另一方面可以发挥两种组学数据的各自优势,进而使两组学数据分析更加准确和全面。
86.图2是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的组学数据获取流程图。优选地,所述对样本进行16s测序和宏基因测序,获取16s组学数据和宏基因组学数据,具体包括:
87.s201、对所述样本进行16s rdna测序,获得所述16s物种丰度表,利用picrust2软件预测,获得所述16s功能丰度表;
88.其中,16srdna为编码该亚基的基因。
89.s202、对所述样本进行宏基因组测序,获得所述宏基因物种丰度表和所述宏基因功能丰度表;
90.其中,所述宏基因功能丰度表和所述16s功能丰度表都包括kegg功能的level b丰度表和level c丰度表。
91.其中,picrust2(phylogenetic investigation of communities byreconstruction of unobserved states)是一款基于标记基因序列来预测功能丰度的软件;kegg(kyoto encyclopedia of genes and genomes),是从大型分子数据集生成的基因组测序和其他高通量实验技术的实用程序数据库资源。
92.在本发明实施例中,首先通过16s的测序,获取了16s的物种丰度表,并通过picrust2软件预测到功能丰度表,其次通过宏基因组测序,获得物种丰度表和功能丰度表。最终,完成完整的16s和宏基因的测序,并保存了相应的物种和功能丰度,这部分是后续进行数据分析的数据基础。
93.图3是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的micropita分析流程图。优选地,所述根据所述16s组学数据进行 micropita分析,获得代表性样本,具体包括:
94.s301、根据所述16s组学数据,利用筛选指标挑选代表性样本,其中,所述筛选指标包括diverse,extreme,representative,distinct,discriminant;
95.s302、利用micropita软件在所述16s组学数据中获取所述代表性样本的样本信息;
96.s303、利用r语言vegan包对16s otu丰度表进行bray
‑
curtis距离pcoa分析,获得所有样本在二维空间的分布特征,同时在图形中可标记micropita 筛选出的代表性样本。
97.其中,r语言是用于统计分析、绘图的语言和操作环境。r语言是属于 gnu系统的一
个源代码开放的软件,用于统计计算和统计制图的工具。
98.在本发明实施例中,提供了组学数据的指标筛选方法的例子,包括 diverse(物种丰富度),extreme(极端(无分组)),representative(代表性),distinct(极端(有分组)),discriminant(区分度)等,进一步的, diverse为α多样性最高的样本,extreme为β多样性显示的最极端的样本, representative为β多样性显示的最能反映整体差异特征的样本,distinct为根据分组特征,分组之间β多样性距离最大的样本,discriminant为根据分组特征,分组内距离中心点最近的样本,在筛选的基础上,利用micropita分析即可获得代表性样本。
99.在获取组学数据的基础上,本实施例中将利用图4
‑
图12,分别进行基于 16s组学数据和宏基因组学数据的韦恩图分析、物种/功能分布分析、相关性分析和环境因子关联分析的详细介绍。
100.图4是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的韦恩分析流程图。优选地,所述根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有物种/功能的统计,图示化展示,即绘制物种进化分支图,具体包括:
101.s401、基于各物种分类水平和kegg功能的level b丰度表和level c丰度表,使用r语言的venndiagram包和upsetr包,以0为阈值,进行韦恩分析;
102.s402、使用韦恩图和upset图展示所述16s组学数据和所述宏基因组学数据的共有、特有元素的数量;
103.其中,upset图是指用利用upsetr绘制的图片。
104.s403、使用lefse软件在物种层级进化分支树上标记16s和宏基因组鉴定到的共有、特有物种。
105.本发明实施例中,通过韦恩分析可获得两组学间共有、特有物种和功能的统计,共有信息表明一致性,特有信息则可作为两组学互相补充的部分。使用r语言venndiagram包和upsetr包,基于16s和宏基因组分析各物种分类水平(门到种)和kegg功能(level b和levelc)丰度表,以0为阈值,其中所述0为阈值指物种和功能在该组学所有样本中丰度均值为0,则认为该组学无此物种/功能,进而进行韦恩分析,使用韦恩图和upset图展示共有、特有元素的数量。该方案可以直观展示共有、特有物种的进化关系特征,借助物种分类的进化分枝图,且标注两组学共有特有的物种,并使用lefse软件,基于物种分类层级信息构建物种的进化分枝树,为避免图形冗余,筛选在各组学中物种丰度均值大于1%的物种。
106.图5是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的物种/功能分布分析流程图。优选地,所述根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种/功能丰度变化和鉴定到的主要物种/功能类型,具体包括:
107.s501、挑选所述16s组学数据中各分类水平排名前10的物种/功能,并存储为第一16s物种/功能;
108.s502、挑选所述宏基因组学数据中各分类水平排名前10的物种/功能,并存储为第一宏基因物种/功能;
109.s503、对所述第一16s物种/功能与所述第一宏基因物种/功能取并集,并保存;
110.s504、从所述宏基因组学数据和所述16s组学数据中除去所述第一16s 物种/功能
与所述第一宏基因物种/功能的并集,并将已知的物种/功能划标注为other,将未知物种/功能标注为unclassified;
111.s505、使用r语言ggplot2包绘制两组学所有样本中物种/功能丰度变化和两组学鉴定到的主要物种/功能类型,其中,所述两组学包括所述16s组学数据和所述宏基因组学数据。
112.本发明实施例中,基于组学数据在所有样本中的丰度均值,挑选两组学各分类水平排名前10的物种/功能,取并集,其他已知物种/功能归为other,未知物种标注为unclassified,再使用r语言ggplot2包绘制堆叠图和饼图。优选的,其中以堆叠图展示两组学所有样本中物种/功能丰度变化,直观呈现两组学物种/功能在样本中的波动趋势;以饼图对比展示两组学鉴定到的主要物种/功能类型。
113.在一个或多个实施例中,优选的,所述根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pearson相关系数分析、mantel test分析、 procrustes test分析;
114.所述pearson相关系数分析具体操作为:将所述16s组学数据和所述宏基因组学数据中所有样本的丰度进行对数转化后,使用r语言psych包计算所述 16s组学数据和所述宏基因组学数据的pearson相关系数,并使用fizh
‑
z变换计算相关显著性,并图示化展示数据分析结果;
115.在一个或多个实施例中,基于物种/功能在所有样本中的丰度,将丰度数据进行对数转化后,使用r语言psych包计算两组学的pearson相关系数,并使用fizh
‑
z变换计算相关显著性。如图6所示的散点图直观展示所有样本两组学相关性数据的线性相关程度,在图6中一个点表示一个样本中的一个物种,点颜色表示不同物种/功能。横轴/纵轴分别表示物种/功能在宏基因组/16s两组学的丰度。r表示相关强度,越接近1,相关性越强。p值表示相关显著性,一般以0.05或0.01为显著性阈值标准。
116.所述mantel test分析具体操作为:计算样本间的bray
‑
curtis距离矩阵,使用r语言vegan包进行mantel test分析,进而计算物种/功能分类水平下16s 和宏基因组测序结果的相关性,通过permutation置换检验999次,计算相关显著性,并图示化展示数据分析结果;
117.在一个或多个实施例中,首先基于16s测序和宏基因组测序获得的物种 /功能丰度表,计算样本间的bray
‑
curtis距离矩阵,使用r语言vegan包进行 mantel test分析,然后计算各物种/功能分类水平下,16s和宏基因组测序结果的相关性。基于permutation置换检验999次,计算相关显著性。
118.在一个或多个实施例中,通过使用如图7所示的散点图直观展示距离矩阵的线性相关程度,相关程度高,表明两组学结果比较一致,可互相验证。图中一个点表示一对样本。横轴/纵轴分别表示一对样本在宏基因组测序和 16s测序数据中的距离。r表示相关强度,越接近1,相关性越强。p值表示相关显著性,一般以0.05或0.01为显著性阈值标准。
119.所述procrustes test分析具体操作为:使用r语言vegan包,根据各分类水平两组学物种/功能bray
‑
curtis距离的pcoa结果,开展procrustes分析,并使用permutation test进行相关显著性检验,并图示化展示数据分析结果,其中,所述两组学包括所述16s组学数据和所述宏基因组学数据。
120.在一个或多个实施例中,procrustes分析(普氏分析)是基于空间分布判断两组学的关联程度,应用于16s与宏基因组测序分析,即可评估两组学检测结果是否一致。使用r语言vegan包,基于各分类水平两组学物种/功能 bray
‑
curtis距离的pcoa结果,开展procrustes分析,并使用permutation test 进行相关显著性检验,进而绘制如图8所示的procrustes分析图,为叠加后的样本分布散点图展示分析结果。
121.图9是本发明一个实施例的一种16s和宏基因组测序数据关联分析方法中的环境因子关联分析流程图。
122.如图9所示,在一个或多个实施例中,优选地,根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析,具体包括:
123.s901、使用r语言psych包计算环境因子间的pearson相关系数;
124.s902、使用r语言vegan包进行16s测序otu丰度表、16s测序的shannon 多样性、宏基因组功能矩阵与每个环境因子的mantel test分析;
125.s903、通过permutation置换检验999次,计算相关显著性;
126.s904、利用r语言ggcor包生成热图和网络图结合的形式展示。
127.本发明实施例中,首先使用r语言psych包计算环境因子间的pearson 相关系数;然后使用r语言vegan包进行16s测序otu丰度表、16s测序的 shannon多样性、宏基因组功能矩阵与每个环境因子的mantel test分析,基于 permutation置换检验999次,计算相关显著性;最后使用r语言ggcor包以热图和网络图的形式组合呈现。具体的,环境因子mantel
‑
test分析图如图10 所示,清晰全面地展现对比16s和宏基因组数据与环境因子的关联。通过该方式从16s、宏基因组数据与环境因子关联分析中,既可以分析物种/功能与环境因子的关联特征,也可以对比两组学数据对环境因子的关联是否一致,从另一个角度分析两组学是否能够互相验证。
128.图11是本发明一个实施例的一种16s和宏基因组测序数据关联分析系统的结构图。优选地,所述16s和宏基因组测序数据关联分析系统的包括:
129.组学数据获取模块1101,对样本进行16s测序和宏基因测序,获取16s 组学数据和宏基因组学数据;其中,所述16s组学数据包括16s物种丰度表和16s功能丰度表,所述宏基因组学数据包括宏基因物种丰度表和宏基因功能丰度表;
130.micropita分析模块1102,根据所述16s组学数据进行micropita分析,获得代表性样本;
131.韦恩分析模块1103,根据所述16s组学数据和所述宏基因组学数据进行韦恩分析,获得共有和特有的物种/功能统计,并图示化展示;
132.物种/功能分布分析模块1104,根据所述16s组学数据和所述宏基因组学数据进行物种/功能分布分析,获得所述16s组学数据和宏基因组学数据中的物种/功能丰度变化和鉴定到的主要物种/功能类型;
133.相关性分析模块1105,根据所述16s组学数据和所述宏基因组学数据进行相关性分析,包括pearson相关系数、mantel test分析、procrustes test分析;
134.环境因子关联分析模块1106,根据所述16s组学数据和所述宏基因组学数据进行环境因子关联分析。
135.显示模块1107,用于显示所述组学数据获取模块1101、所述micropita 分析模块
1102、所述韦恩分析模块1103、所述物种/功能分布分析模块1104、所述相关性分析模块1105和所述环境因子关联分析模块1106中获得的关联分析图与关联分析数据;
136.存储模块1108,用于存储所述组学数据获取模块1101、所述micropita 分析模块1102、所述韦恩分析模块1103、所述物种/功能分布分析模块1104、所述相关性分析模块1105和所述环境因子关联分析模块1106中获得的关联分析图与关联分析数据。
137.图12是本发明一个实施例的一种16s和宏基因组测序数据关联分析系统的模块处理关系示意图。如图12所示,在一个或多个实施例中,通过在进行16s测序和宏基因测序的基础上,利用16s组学数据进行micropita分析,利用16s组学数据和宏基因组学数据进行韦恩图分析、物种/功能分布分析、相关性分析和环境因子关联分析。
138.在本发明实施例中,通过全方位的数据分析能够获得两种组学数据的差别,产生两种组学数据的互相补充和互相验证,判断两组学的相关性和一致性,使数据分析的更靠性。
139.图13是本发明一个实施例的一种电子设备的结构图。图13所示的电子设备为通用测序数据关联分析装置,其包括通用的计算机硬件结构,其至少包括处理器1301和存储器1302。处理器1301和存储器1302通过总线1303 连接。存储器1302适于存储处理器1301可执行的指令或程序。处理器1301 可以是独立的微处理器,也可以是一个或者多个微处理器集合。由此,处理器1301通过执行存储器1302所存储的指令,从而执行如上所述的本发明实施例的方法流程实现对于数据的处理和对于其它装置的控制。总线1303将上述多个组件连接在一起,同时将上述组件连接到显示控制器1304和显示装置以及输入/输出(i/o)装置1305。输入/输出(i/o)装置1305可以是鼠标、键盘、调制解调器、网络接口、触控输入装置、体感输入装置、打印机以及本领域公知的其他装置。典型地,输入/输出装置1305通过输入/输出(i/o) 控制器1306与系统相连。
140.本发明实施例中,提供16s与宏基因组测序数据的关联分析方法,使两组学数据相互补充,相互验证。可产生如下效果:
141.1、通过使用韦恩分析进行两组学共有、特有的物种和功能分析,并结合物种进化分支树对物种的共有性可视化展示明确组学之间的差异,实现两种组学数据的互相补充,支撑组学之间的互相完善。
142.2、通过判断两组学的一致性和相关性,使数据分析的更靠性,使用堆叠图直观展示两组学样本中物种/功能丰度变化规律,初步观察两组学的一致性,结合饼图展示两组学各自鉴定到的高丰度物种/功能,对比观察一致性,进而开展相关性分析,实现两种组学数据的互相验证,有效判断组学相关性。
143.3、在宏基因组与16s测序样本没有一一对应时,本方案通过micropita 分析,可根据16s测序的otu丰度表,评估出最具有代表性的样本,作为指导宏基因组分析样本选择的的参考。
144.本领域内的技术人员应明白,本发明的实施例可提供为方法、系统、或计算机程序产品。因此,本发明可采用完全硬件实施例、完全软件实施例、或结合软件和硬件方面的实施例的形式。而且,本发明可采用在一个或多个其中包含有计算机可用程序代码的计算机可用存储介质(包括但不限于磁盘存储器和光学存储器等)上实施的计算机程序产品的形式。
145.本发明是参照根据本发明实施例的方法、设备(系统)、和计算机程序产品的流程图和/或方框图来描述的。应理解可由计算机程序指令实现流程图和/或方框图中的每一流程和/或方框、以及流程图和/或方框图中的流程和/或方框的结合。可提供这些计算机程序指令到通用计算机、专用计算机、嵌入式处理机或其他可编程数据处理设备的处理器以产生一个机器,使得通过计算机或其他可编程数据处理设备的处理器执行的指令产生用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的装置。
146.这些计算机程序指令也可存储在能引导计算机或其他可编程数据处理设备以特定方式工作的计算机可读存储器中,使得存储在该计算机可读存储器中的指令产生包括指令装置的制造品,该指令装置实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能。
147.这些计算机程序指令也可装载到计算机或其他可编程数据处理设备上,使得在计算机或其他可编程设备上执行一系列操作步骤以产生计算机实现的处理,从而在计算机或其他可编程设备上执行的指令提供用于实现在流程图一个流程或多个流程和/或方框图一个方框或多个方框中指定的功能的步骤。
148.显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1