用于治疗神经退行性和代谢性疾患的化合物的制作方法
1.相关申请的交叉引用
2.本技术主张2019年5月14日提交的美国临时申请第62/847,600号的优先权的权益,该临时申请通过引用而整体并入本文并且用于所有目的。
3.政府资助的声明
4.本发明在政府支持下完成,由美国国立卫生研究院资助(项目编号:5r01ns085223和r21ns093488)。政府享有本发明的某些权利。
背景技术:
5.多种致命的神经退行性疾病(包括朊病毒病诸如克雅二氏症(cjd)、阿尔兹海默症(ad)、帕金森症(pd)、额颞叶痴呆(ftd)和肌萎缩性脊髓侧索硬化症(als))的特征在于引起蛋白质错误折叠的毒性并且成为蛋白质错误折叠神经退行性疾病(pmnd)。牵涉到这些疾病中的蛋白质错误折叠并形成各种尺寸的聚集体。这些聚集体中的一些对于神经元具有高毒性,这种现象也称为蛋白质毒性。从某种意义上来说,蛋白质聚集体也表现出“朊病毒样”性质,它们在细胞间传播并作为种子放大细胞内的错误折叠和聚集过程。此类有毒的错误折叠的蛋白质包括cjd中的朊病毒蛋白prp;ad中的aβ和tau;pd中的α-突触核蛋白和tau;ftd中的tau、tdp-43和c9orf72;als中的sod1、tdp43、fus和c9orf72。pd属于一类被称为共核蛋白病的更广泛的疾病,其特征在于错误折叠的α-突触蛋白聚集体的蓄积。路易体痴呆也是共核蛋白病。ftd属于另一类名为tau蛋白病的pmnd,这类疾病还包括慢性创伤性脑病变(cte)和进行性核上性麻痹(psp)。也存在前述蛋白质错误折叠的非神经系统疾病,诸如糖尿病,其中蛋白质iapp和胰岛素原形成对于胰腺β细胞有毒的蛋白质聚集体。
6.对神经毒性机制所知甚少阻碍了针对pmnd的有效疗法的开发。为研究此类机制,已经开发了使用错误折叠且有毒的朊病毒蛋白(tprp)的模型,并且特别地,tprp在细胞培养物中并且在进行脑内注射后可重现地诱导神经元死亡1。tprp以纳摩尔浓度诱导超过60%的所培养神经元的死亡,而朊病毒蛋白的天然折叠的对应物ntprp则不然。因此,该模型提供高效系统以研究在暴露于错误折叠蛋白之后的神经元死亡机制。某些针对朊病毒诱导的毒性的机制和如何阻止这些机制的知识被认为可以更广泛地应用于其他pmnd。因此,如本文所示,基于tprp的研究促进了用于治疗破坏性pmnd的新型神经保护方法的开发。
技术实现要素:
7.目前,没有疾病修正治疗可用于任何蛋白质错误折叠神经退行性疾病(pmnd)。目前的治疗措施,如果它们就任何意义而言存在,缓解某些疾病症状,但既不能减缓潜在致病机制的进展,也不能中止神经元损失。相比之下,本发明的化合物能够干扰与nad基质中的改变相关联的神经毒性的基本基质,从而使神经元免遭进一步损伤。因此,本文所述的方法可针对pmnd和其他与nad基质受损相关的疾病提供第一种的疾病修正治疗。
8.在各种实施方案中,提供了用于在患者体内抑制nad消耗和/或增加nad合成的方
法,包括向该患者给药有效量的化合物,该化合物包括本文中针对该目的公开并主张的化学结构的任何物质或任何族属成员。
9.在各种实施方案中,提供了用于在患者体内预防或抑制nad耗竭的方法或用于在患者体内改善与nad基质的改变相关联的病症的方法,包括向该患者给药有效量的化合物,该化合物包括本文中针对该目的公开并主张的化学结构的任何物质或任何族属成员。该病症可包括代谢性疾患、糖尿病、衰老、神经退行性疾病、与多发性硬化相关的神经退变、听力丧失或视网膜损伤、脑或心肌缺血、肾衰竭、创伤性脑损伤或轴突病变。
10.在各种实施方案中,提供了用于在患者体内提供保护以免于错误折叠的蛋白质的毒性的方法,包括向该患者给药有效量的化合物,该化合物包括本文中针对该目的公开并主张的化学结构的任何物质或任何族属成员。
11.在各种实施方案中,提供了用于预防或治疗患者的蛋白质错误折叠神经退行性疾病的方法,包括向该患者给药有效量的化合物,该化合物包括本文中针对该目的公开并主张的化学结构的任何物质或任何族属成员。该疾病可以是朊病毒病诸如克雅二氏症(cjd)、帕金森症(pd)或其他共核蛋白病、阿尔兹海默症(ad)、肌萎缩性脊髓侧索硬化症(als)、或tau蛋白病诸如额颞叶痴呆(ftd)、慢性创伤性脑病变(cte)和进行性核上性麻痹(psp)。
12.进一步提供了可能有用于本文所述方法的新颖化合物。
13.在各种实施方案中,提供了具有式(i)的化合物,
14.或其药学可接受的盐。
15.在式(i)中:
16.每个r1和r2独立地为h、(c
1-c4)烷基或(c
1-c4)烷氧基;并且
17.每个r3独立地选自h、任选地经oh取代的(c
1-c4)烷基、(c
1-c4)烷氧基或杂芳基,并且前提是两个r3不全为h;或者两个r3与其所键结的氮原子一起形成5至7元杂环基环,所述杂环基环包含至少一个选自o、s、s=o、s(=o)=o或nr的额外的杂原子,其中r为任选地经oh取代的(c
1-c4)烷基、或(c
1-c4)烷氧基。
18.在各种实施方案中,提供了具有式(ii)的化合物,
19.或其药学可接受的盐。
20.在式(ii)中:
21.每个r
a1
和r
a2
独立地为氢、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基、2至4元杂烷基、环烷基、杂环烷基、芳基或杂芳基;
22.每个r
b1
、r
b2
和r
b3
独立地为氢、卤代、(c
1-c4)烷基、-s(o)2rd、-s(o)2ord或(c
1-c4)卤代烷基;或者r
b2
和r
b3
接合在一起以形成芳基或杂芳基;
23.每个rc和rd独立地为氢或(c
1-c4)烷基;
24.ar为单环或双环的芳基或杂芳基,其任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基或杂芳基中的一者或多者取代;并且
25.n=2、3、4或5。
26.在各种实施方案中,提供了具有式(iii)的化合物,
27.或其药学可接受的盐。
28.在式(iii)中:
29.l1为键、c
1-c4亚烷基、或2至4元亚杂烷基;
30.r1为单环或双环的环烷基、杂环烷基、芳基、烷基芳基或杂芳基,其中所述环烷基、杂环烷基、芳基、烷基芳基或杂芳基任选地经选自卤代、(c
1-c4)烷基、羟基(c
1-c4)烷基、(c
1-c4)烷氧基、-c(=o)(c
1-c4)烷基、-c(=o)n(r)2或-c(=nr)(c
1-c4)烷基中的一者或多者取代,其中所述(c
1-c4)烷基未经取代或经杂环烷基取代;
31.每个r独立地为h、-oh、(c
1-c4)烷基或(c
1-c4)烷氧基,或者两个r与其所键结的氮原子一起形成杂环烷基,任选地进一步包含所述杂环基环中的o;
32.r2出现0、1或2次,并且为(c
1-c4)烷基、(c
1-c4)卤代烷基或so2n(r4)2;并且
33.每个r3和r4独立地为h或(c
1-c4)烷基。
34.在各种实施方案中,提供了具有式(iv)的化合物,
35.或其药学可接受的盐。
36.在式(iv)中:
37.r1为氢、(c
1-c4)烷基、-c(o)oh、
–
c(o)o-(c
1-c4)烷基、-c(o)nhnhr6、-c(o)nr
6-((c
1-c4)亚烷基)-nhr6、-c(o)nr6(c
1-c4)烷基或
–
c(o)nr
6-亚环烷基-nhr6;
38.r3为氢或(c
1-c4)烷基;
39.每个r2、r4、r5独立地为氢、卤代、(c
1-c4)烷基、
–
c(o)o-(c
1-c4)烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷基或cn;并且
40.每个r6为氢或(c
1-c4)烷基,
41.在各种实施方案中,提供了具有式(v)的化合物,
42.或其药学可接受的盐,
43.在式(v)中:
44.ar为单环或双环的芳基或杂芳基,任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、cn、-s(o)2nh2、氧代、-nh2、(c
1-c4)烷氧基或
–
nhc(o)(c
1-c4)烷基中的一者或多者取代;
45.每个r1和r2独立地为氢、(c
1-c4)烷基、芳基或杂芳基,其中所述芳基或杂芳基任选地经卤代或(c
1-c4)烷基取代,或者附接至氮的r1和r2接合在一起以形成5至6元杂环烷基;并且
46.r3为氢或羟基-(c
1-c4)烷基。
47.本发明的其他方法在下文公开。
附图说明
48.图1显示以384孔板格式进行的初步神经保护测定和验证性nad定量测定。如图所示,在存在或不存在所述剂量(剂量以μm计)的nad或筛选点击dmcm的情况下,将pk1成神经细胞瘤细胞(2000个细胞/孔)用2.5μg/ml tprp处理3天。如zhou,et.al.,proc natl acad sci u s a 109,3113-3118(2012)1中所述,制备tprp。dmso为0.3%。显示了一式五份的数据和sd。上图:使用(promega)进行的发光细胞存活率测定。下图:使用nad+/nadh-glotm(promega)进行的nad定量测定。
49.图2a、2b、2c、2d、2e、2f、2g、2h显示多个8个系列的先导化合物的结构和剂量应答曲线(在存在tprp的情况下的细胞存活率,红色;在存在tprp的情况下的nad定量,黑色;天然细胞中的细胞存活率的逆向筛选,绿色;天然细胞中的nad定量逆向筛选,蓝色):图2a:咔唑类;图2b:吡唑并嘧啶类;图2c:氨基噻唑类;图2d:三唑并酞嗪类;图2e:氨基酞嗪类;图2f:类黄酮类(川陈皮素);图2g:生物碱类(巴马汀);图2h:3-杂芳基喹啉类(dmpq)。
50.细胞存活率测定在1536孔板中进行。在5μl的总体积中,将pk1成神经细胞瘤细胞(80个细胞/孔)用4μg/ml tprp处理3天。如zhou,et.al.,proc natl acad sci u s a 109,3113-3118(2012)1中所述,制备tprp。将化合物添加到0.6%dmso中,总体积30nl,以所示剂量10点4对数滴定。使用(promega)测量细胞存活率。使用nad+/nadh-glotm(promega)对nad进行定量。
51.图3显示不存在通过结构上与dmcm无关的gaba
a r抑制剂进行的神经保护。将pk1细胞(1500个细胞/孔,96孔板)用氟马西尼(50nm)处理24小时,然后在具有或不具有dmcm(0.5或5μm)的tprp(3μg/ml)中暴露4天。如zhou,et.al.,proc natl acad sci u s a 109,
3113-3118(2012)1中所述,制备tprp。使用(promega)测量细胞存活率。在10点剂量应答实验(0.3-164nm,未显示)中,再一次显示不存在氟马西尼针对tprp毒性的保护。在该实验中,氟马西尼的用量为其针对gaba
a r抑制剂的ic
50
的100倍。
52.图4显示,没有gaba
a r活性的dmcm类似物(dmcm-10049)和氨基酰胺dmcm-8137具有神经保护活性。dmcm-8137和dmcm-10049在tprp神经保护测定中显示几乎相同的剂量应答曲线。将细胞(1500个细胞/孔,96孔板)在5μg/m的tprp和所示剂量的化合物中暴露4天。如zhou,et.al.,proc natl acad sci u s a 109,3113-3118(2012)1中所述,制备tprp。使用(promega)测量细胞存活率。
53.图5显示,dmcm在原代神经元中拯救tprp诱导的毒性。将原代小鼠皮层神经元(life technologies)在12μg/ml的tprp中暴露5天,并且用所示dmcm或nad进行处理。该测定以96孔板格式进行。如zhou,et.al.,proc natl acad sci u s a 109,3113-3118(2012)1中所述,制备tprp。显示了一式三份的数据
±
sd。使用(promega)测量细胞存活率。dmcm和nad治疗也抑制tprp暴露引起的神经断裂和神经元空泡化(未显示)。
54.图6显示,dmcm减轻由错误折叠的蛋白质引起的过度蛋白质adp核糖基化。在存在9nm fk866(以抑制从存在于培养基中的烟酰胺合成nad)和40μm生物素-nad(trevigen)的情况下,用5μg/ml tprp处理pk1细胞。在处理2或3天后收获细胞。通过sds-page分析细胞裂解液,使用链霉亲和素-hrp发现adp-核糖基化的、生物素化的蛋白质。在存在dmcm的情况下,对应于tprp特异性adp核糖基化的~80和130kd处的带减少(与每个时间点的泳道1&2相比)。在不存在tprp的情况下,dmcm无效(每个时间点的泳道3&4)。每条泳道加载10μg蛋白质。
55.图7显示,在细胞pd模型中,dmcm-10049保留了树突棘并且降低了pα-syn*水平。将人干细胞源性神经元(分化后30天)用50μg/ml预成形的α-突触核蛋白原纤维(pff)接种,用2μm dmcm-10049或媒介物处理,并固定17天,然后进行分析。对照细胞未接种pff。a:使用鬼笔环肽-ifluor 488(abcam),用标记物f-肌动蛋白标记树突棘。b:pα-syn*(红色,抗体gtx50222,genetex)+dapi(蓝色)染色。使用imagej(nih)进行定量,用单因素方差(prism7)进行统计学分析。显示了每种条件下8张图像(a)或6张图像(b)的平均值和sd。****p《0.0001;***p《0.001;ns=不显著。
56.图8显示dmcm-10049在帕金森症鼠模型中的治疗效果。从9月龄开始,在饮用水中用50mg/kg/天的dmcm-10049治疗tg(snca*a53t)小鼠。使用无糖草莓味明胶进行味觉掩蔽,并使用4%dmso进行化合物增溶。媒介物对照组接受不含化合物的相同混合物。对照组(n=48)的中值存活期为411天,治疗组(n=16)为488天。存活期延长显著(对数秩检验中,p=0.01,prism 7)。
57.图9显示dmcm-10049在als鼠模型中的治疗效果。
58.从70日龄开始,在饮用水中用50mg/kg/天的dmcm-10049治疗tg(sod1*g93a)小鼠。使用无糖草莓味明胶进行味觉掩蔽,并使用4%dmso进行化合物增溶。媒介物对照组接受不含化合物的相同混合物。对照组(n=7)的中值存活期为165天,治疗组(n=11)为177天。存活期延长显著(对数秩检验中,p=0.0007,prism 7)。
59.图10显示,口服瓦他拉尼(sr5-1457)治疗在als小鼠模型中延缓了运动功能损伤。
从47日龄开始,小鼠(全部为雌性)在其饮用水中每天接受50mg/kg瓦他拉尼。使用无糖草莓味明胶进行味觉掩蔽,并使用4%dmso进行化合物增溶。媒介物对照组接受不含化合物的相同混合物。转棒测试和悬挂测试按所述执行
2,3
。显示了平均值
±
sem。用双因素方差分析执行统计学分析,n=12;*p《0.05;**p《0.01;***p《0.001。
60.图11显示,神经保护/nad恢复效应的sar与瓦他拉尼类似物中的vegfr抑制的sar不相关。在我们的测定中,制备并测试了与瓦他拉尼(sr5-1457)有关的具有和不具有vegfr活性的化合物,包括甲基酮sr1-134005。显示了针对神经保护(红色)和nad水平(黑色)的剂量应答曲线,以及逆向筛选(绿色),该逆向筛选的目的为检测在不存在tprp的情况下非特异性地增加发光/细胞存活率的化合物。如图2中文字说明所述,以1536孔板格式执行这些测定。
61.图12显示gabaar活性小得多的瓦他拉尼3-甲基酮类似物(sr1-134005)在als小鼠模型中的治疗效果。从100日龄开始,在饮用水中用6mg/kg/天的sr1-134005治疗tg(sod1*g93a)小鼠。使用无糖草莓味明胶进行味觉掩蔽,并使用4%dmso进行化合物增溶。媒介物对照组接受不含化合物的相同混合物。执行悬挂测试以测量肌肉强度。显示了平均值
±
sem。治疗组(n=14)与对照组(n=19)之间的差异显著(对于除145天外的所有时间点,p《0.001,多个未配对t检验,prism 7)。
62.图13显示gabaar活性小得多的瓦他拉尼3-甲基酮类似物(sr1-134005)在帕金森症小鼠模型中的治疗效果。从8月龄开始,在饮用水中用25mg/kg/天的sr1-134005治疗tg(snca*a53t)小鼠。使用无糖草莓味明胶进行味觉掩蔽,并使用4%dmso进行化合物增溶。媒介物对照组接受不含化合物的相同混合物。对照组(n=58)的中值存活期为411天,治疗组(n=16)为462天。存活期延长显著(对数秩检验中,p=0.03,prism 7)。
63.图14显示在细胞pd/共核蛋白病模型中的神经保护。将小鼠干细胞源性神经元(分化后8天)用pff(a、b:4μg/ml;c、d:3μg/ml3μg/ml)接种,并且在分化的最后2天期间用所指示剂量的测试化合物处理或用媒介物dmso处理。对照细胞未接种pff。用相差显微镜对细胞照相。使用imagej(nih)的neuronj插件对神经突长度进行定量,通过比较经化合物处理的细胞与dmso对照,成对地进行统计学分析(学生t检验,prism8)。显示了每种条件下4个视野的平均值和sem。每个视野含有大约50至100个神经元。所显示的图像对应于一个区域的代表性区域。****p《0.0001;***p《0.001;**p《0.01;*p《0.05;ns=不显著。
64.图15显示通过咔唑(dmcm-10049)、氨基酞嗪(sr1-134005,又名sr-005)、吡唑并嘧啶(sr1-293229,又名sr-229)、三唑并酞嗪(sr5-22843,又名sr-843)和类黄酮(川陈皮素)治疗时,不存在parp-1抑制。abt-888是一种已知的parp1抑制剂,用作该测定的药物对照。“对照”指示该测定在不存在化合物的情况下执行。使用bps bioscience parp1化学发光测定试剂盒,根据制造商的说明执行测定。
65.图16显示氨基酞嗪类瓦他拉尼和sr1-134005(又名sr-005)为nampt活化剂。在比色nampt活性测定(abcam,ab221819)中测试化合物。每个数据点表示两个重复样品。a、b:nampt被sr-005活化,但不被dmcm、sr-229、sr-259、sr-186或川陈皮素活化。化合物以5μm(a)或20μm(b)测试。c、d:sr-005对nampt的剂量依赖性活化(c,0.5和2.5μm;d,5和20μm)。e:瓦他拉尼对nampt的剂量依赖性活化。帕唑帕尼(一种vegfr-1、vegfr-2和vegfr-3的潜在抑
制剂)在10μm下不活化nampt。该数据补充了先前的论证,即瓦他拉尼和sr-005的nad恢复效应与其vegfr活性无关。
66.主要组件符号说明
67.无。
具体实施方式
68.错误折叠的毒性朊病毒蛋白tprp导致负责细胞死亡的神经元nad的严重耗竭,因为nad的补充导致在体外和体内暴露于tprp损伤的细胞的完全恢复,尽管继续暴露于tprp2仍如此。鼻内nad治疗改善了小鼠朊病毒病中的受损运动功能和活性。进一步发现,暴露于tprp的神经元中的nad耗竭至少部分地由于细胞nad在被称为单adp核糖基化的代谢反应期间的过度消耗2。多-adp-核糖基化的抑制剂称为parp抑制剂,先前已经作为抗癌药物开发。可用的选择性parp抑制剂不缓解由tprp造成的nad耗竭和神经元死亡,表明面需要鉴定能够干扰错误折叠的蛋白质借以引起毒性的机制的新化合物。此类机制也可能在如本文所述的其他与nad代谢失衡相关的疾患中发挥作用。
69.使用tprp作为表现出高神经毒性的原型淀粉样变性错误折叠的蛋白质,已经开发了高通量筛选(hts)测定以鉴定具有以下效果的化合物:a)预防神经元死亡;和b)预防由tprp引起的nad耗竭(图1)。
70.使用scripps药物发现文库(sddl),在佛罗里达州的scripps执行hts活动。鉴定了几种有效的新颖且易化学处理的小分子,当以从低纳摩尔到低毫摩尔范围内的水平使用时,它们可提供完全的神经保护并保持nad水平。高活性化合物为dmcm,一种别构gabaa受体(gaba
a r)调节剂;和瓦他拉尼,一种vegf受体(vegfr)酪氨酸激酶抑制剂,其已经在用于治疗癌症的临床试验中进行研究。在这次尝试中,还鉴定了其他六类神经保护分子。
71.示例性的活性化合物提供于下:
72.1)咔唑类(示例:sr1-75869,又名dmcm);
[0073][0074]
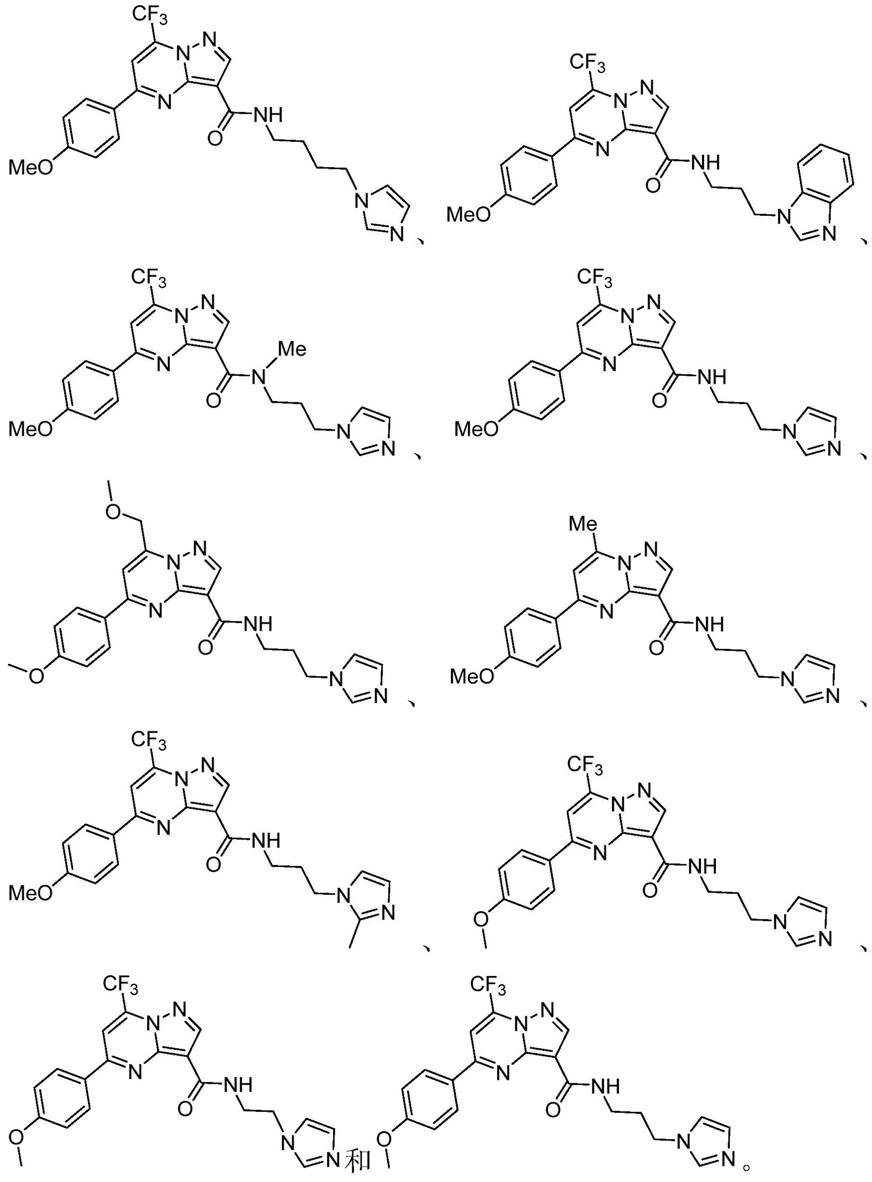
2)吡唑并嘧啶类(示例:sr1-293229);
[0075][0076]
3)氨基噻唑类(示例:sr1-477186);
[0077][0078]
4)三唑并酞嗪类(示例:sr1-115259);
[0079][0080]
5)氨基酞嗪类(示例:sr5-1457,又名瓦他拉尼,又名cgp79787,也称sr1-134005,一种gabaar活性低得多的瓦他拉尼类似物);
[0081][0082]
6)类黄酮类(示例:川陈皮素,又名sr1-712262);
[0083][0084]
7)生物碱类,包括异喹啉、阿朴啡和麦角生物碱(所显示的示例为异喹啉生物碱,名为盐酸巴马汀,又名sr1-841226);
[0085][0086]
8)3-杂芳基喹啉类(示例:dmpq,又名sr1-597975)
[0087][0088]
上述化合物系列的活性在图2a至2h中详细表征。每个系列的成员在旨在反映用于成功治疗如本文所述的几种神经退行性疾病中的潜力的神经保护测定中高度有效。再者,这些化合物多数对于先导开发具有有利的性质(例如,它们是无痛的4并且符合lipinski和veber药物相似性
5,6
规则)。
[0089]
定义
[0090]
本文所用的缩写须有它们在化学和生物学领域中的传统意义。本文中详述的化学结构和化学式根据化学领域中已知的化学价态的标准规则构建。
[0091]
若取代基通过其从左至右写出的常规化学式指定,则它们等同于涵盖将会导致从右至左写出该结构的化学式相同的取代基,例如,-ch2o-等同于-och
2-。
[0092]
除非另外指明,否则术语“烷基”自身或作为另一取代基的一部分意为直链(即不分支的)或支链碳链(或碳)或其组合,它可以是完全饱和的、单或多不饱和的,并且可以包括单价、二价和三价基团。烷基可以包括制定数目的碳(例如,c
1-c
10
意为1至10个碳)。烷基为未环化的链。饱和烃基的实例包括但不限于下述基团,例如,甲基(“me”)、乙基(“et”)、正丙基(“pr”)、异丙基(“ipr”)、正丁基(“bu”)、叔丁基(“tbu”)、异丁基、仲丁基、甲基、同系物和异构体,例如,正戊基、正己基、正庚基、正辛基等。不饱和的烷基是具有一个或多个双键或三键的烷基。不饱和烷基的实例包括但不限于,乙烯基、2-丙烯基、巴豆基、2-异戊烯基、2-丁二烯基、2,4-戊二烯基、3-(1,4-戊二烯基)、乙炔基、1-丙炔基、3-丙炔基、3-丁炔基、及高级同系物和异构体。烷氧基是经由氧链接基(-o-)附接至该分子剩余部分的烷基。烷基部分可以是烯基部分。烷基部分可以是炔基部分。烷基部分可以是完全饱和的。烯基可包括一个以上双键和/或除了包括一个或多个双键外还包括一个或多个三键。炔基可包括一个以上三键和/或除了包括一个或多个三键外还包括一个或多个双键。
[0093]
除非明确排除,否则术语“亚烷基”单独使用或作为另一取代基的一部分时意为源自烷基的二价基,例如但不限于,-ch2ch2ch2ch
2-。典型地,烷基(或亚烷基)将具有1至24个碳原子,其中本文中优选那些具有10个或更少碳原子的基团。“低级烷基”或“低级亚烷基”是较短链的烷基或亚烷基,通常具有8个或更少的碳原子。除非明确排除,否则术语“亚烯基”单独使用或作为另一取代基的一部分时意为源自烯烃的二价基。
[0094]
除非明确排除,否则术语“杂烷基”单独使用或与另一术语组合时意为稳定的直链或支链或其组合,包括至少一个碳原子和至少一个杂原子(如,选自o、n、p、si和s),并且其中该氮和硫原子可任选地被氧化,且该氮杂原子可任选地季铵化。该杂原子(例如,o、n、s、si或p)可置于该杂烷基的任何内部位置,或置于烷基借以附接至该分子剩余部分的位置。杂烷基为未环化的链。实例包括但不限于:-ch
2-ch
2-o-ch3、-ch
2-ch
2-nh-ch3、-ch
2-ch
2-n(ch3)-ch3、-ch
2-s-ch
2-ch3、-ch
2-s-ch2、-s(o)-ch3、-ch
2-ch
2-s(o)
2-ch3、-ch=ch-o-ch3、-si(ch3)3、-ch
2-ch=n-och3、-ch=ch-n(ch3)-ch3、-o-ch3、-o-ch
2-ch3和-cn.最多可连续出现两个或三个杂原子,例如,-ch
2-nh-och3和-ch
2-o-si(ch3)3。杂烷基部分可包括一个杂原子(例如,o、n、s、si或p)。杂烷基部分可包括两个任选地不同的杂原子(例如,o、n、s、si或p)。杂烷基部分可包括三个任选地不同的杂原子(例如,o、n、s、si或p)。杂烷基部分可包括四个任选地不同的杂原子(例如,o、n、s、si或p)。杂烷基部分可包括五个任选地不同的杂原子(例如,o、n、s、si或p)。杂烷基部分可包括至多8个任选地不同的杂原子(例如,o、n、s、si或p)。术语“杂烯基”本身或与另一术语组合时,除非另外指明,否则意为包括至少一个双键的杂烷基。杂烯基可任选地包括一个以上双键和/或除了包括一个或多个双键外还包括一个或多个三键。术语“杂炔基”本身或与另一术语组合时,除非另外指明,否则意为包括至少一个三键的杂烷基。杂炔基可任选地包括一个以上三键和/或除了包括一个或多个三键外还包括一个或多个双键。
[0095]
同样,除非明确排除,否则术语“亚杂烷基”单独使用或作为另一取代基的一部分时意为源自杂烷基的二价基,例如但不限于,-ch
2-ch
2-s-ch
2-ch
2-和-ch
2-s-ch
2-ch
2-nh-ch
2-。对于亚杂烷基,杂原子也可占据链的一端或两端(例如,亚烷基氧、亚烷基二氧、亚烷基胺基、亚烷基二胺基等)。更进一步,对于亚烷基和亚杂烷基链接基团,不通过该链接基团分子式的书写而暗示该链接基团的取向。例如,式-c(o)2r'-表示-c(o)2r'-和-r'c(o)
2-两者。如上所述,本文中使用的杂烷基包括那些通过杂原子附接至该分子剩余部分的基团,诸如-c(o)r'、-c(o)nr'、-nr'r”、-or'、-sr'、及/或-so2r'。若在具体杂烷基的描述后引用“杂烷基”诸如-nr'r”等,应理解为术语“杂烷基”和-nr'r”不是多余的或不相互排斥。尤其是当引用具体的杂烷基以更清晰地理解时。因此,术语“杂烷基”在本文中不应解释为排除具体的杂烷基,诸如-nr'r'’等。
[0096]
除非明确排除,否则术语“环烷基”和“杂环烷基”单独使用或与其它术语组合时分别意为环状的“烷基”和环状的“杂烷基”。环烷基和杂环烷基不是芳香族的。此外,对于杂环烷基,杂原子可占据该杂环借以附接至该分子剩余部分的位置。环烷基的实例包括但不限于,环丙基、环丁基、环戊基、环己基、1-环己烯基、3-环己烯基、环庚基等。杂环烷基的实例包括但不限于,1-(1,2,5,6-四氢吡啶基)、1-哌啶基、2-哌啶基、3-哌啶基、4-吗啉基、3-吗啉基、四氢呋喃-2-基、四氢呋喃-3-基、四氢噻吩-2-基、四氢噻吩-3-基、1-哌嗪基、2-哌嗪基等。“亚环烷基”和“亚杂环烷基”单独使用或作为另一取代基的一部分时分别意为源自环烷基和杂环烷基的二价基。
[0097]
在实施方案中,杂环烷基为杂环基。如本文所用,术语“杂环基”意为单环、双环或多环的杂环。杂环基单环杂环为3、4、5、6或7元环,其含有至少一个独立地选自由o、n和s组成的组的杂原子,其中该环是饱和的或不饱和的但不是芳香族的。3或4元环含有1个选自由o、n和s组成的组的杂原子。5元环可以含有零个或一个双键以及一个、两个或三个选自由o、
n和s组成的组的杂原子。6或7元环可以含有零个、一个或两个双键以及一个、两个或三个选自由o、n和s组成的组的杂原子。杂环基单环杂环通过包含在该杂环基单环杂环内的任何碳原子或任何氮原子连接至母体分子部分。杂环基单环杂环的代表性示例包括但不限于,氮杂环丁基、氮杂环庚基、氮杂环丙基、二氮杂环庚基、1,3-二氧杂环己基、3-二氧杂环戊基、1,3-二硫杂环戊基、1,3-二噻烷基、咪唑啉基、咪唑烷基、异噻唑啉基、异噻唑烷基、异噁唑啉基、异噁唑烷基、吗啉基、氧杂二唑啉基、氧杂二唑烷基、噁唑啉基、噁唑烷基、哌嗪基、哌啶基、吡喃基吡唑啉基、吡唑烷基、吡咯啉基、吡咯烷基、四氢呋喃基、四氢噻吩基、噻二唑啉基、噻二唑烷基、噻唑啉基、噻唑烷基、硫代吗啉基、1,1-二氧化硫代吗啉(硫代吗啉砜)、噻喃基和三噻烷基。杂环基双环杂环为单环杂环与苯基、单环的环烷基、单环的环烯基、单环杂环或单环杂芳基融合。杂环基双环杂环通过包含在该双环的环系中的单环杂环部分内的任何碳原子或任何氮原子连接至母体分子部分。双环杂环基的代表性示例包括但不限于,2,3-二氢苯并呋喃-2-基、2,3-二氢苯并呋喃-3-基、吲哚啉-1-基、吲哚啉-2-基、吲哚啉-3-基、2,3-二氢苯并噻吩-2-基、十氢喹啉基、十氢异喹啉基、八氢-1h-吲哚基和八氢苯并呋喃基。在实施方案中,杂环基任选地经一个或两个基团取代,该基团独立地为氧代或硫杂。在某些实施方案中,双环杂环基为5或6元单环杂环基环与苯基环、5或6元单环环烷基、5或6元单环环烯基、5或6元单环杂环基或5或6元单环杂芳基融合,其中该双环杂环基任选地经一个或两个基团取代,该基团独立地为氧代或硫杂。多环杂环基环系为单环杂环基环(基础环)与下列融合:(i)一个选自由双环芳基、双环杂芳基、双环环烷基、双环环烯基和双环杂环基所组成的组的环系;或(ii)两个独立地选自由苯基、双环芳基、单环或双环杂芳基、单环或双环的环烷基、单环或双环的环烯基、和单环或双环的杂环基所组成的组的其他环系。多环杂环基通过包含在该基础环内的任何碳原子或氮原子附接至母体分子部分。在实施方案中,多环杂环基环系为单环杂环基环(基础环)与下列融合:(i)一个选自由双环芳基、双环杂芳基、双环环烷基、双环环烯基和双环杂环基所组成的组的环系;或(ii)两个独立地选自由苯基、单环杂芳基、单环的环烷基、单环的环烯基和单环的杂环基所组成的组的其他环系。多环杂环基的示例包括但不限于,10h-吩噻嗪-10-基、9,10-二氢吖啶-9-基、9,10-二氢吖啶-10-基、10h-吩噁嗪-10-基、10,11-二氢-5h-苯并[b,f]吖庚因-5-基、1,2,3,4-四氢吡啶并[4,3-g]异喹啉-2-基、12h-苯并[b]吩噁嗪-12-基和十二氢-1h-咔唑-9-基。
[0098]
除非另外指出,否则术语“卤代”或“卤素”自身或作为另一取代基的一部分意为氟、氯、溴或碘原子。此外,术语诸如“卤代烷基”意为包括单卤代烷基或多卤代烷基。例如,术语“卤代(c
1-c4)烷基”包括但不限于,氟甲基、二氟甲基、三氟甲基、2,2,2-三氟乙基、4-氯丁基、3-溴丙基等。
[0099]
除非明确排除,否则术语“芳基”意为多不饱和的芳香族烃取代基,其可以是单环、或稠合(即,稠环芳基)或共价链接的多个环(优选1至3个环)。稠环芳基是指稠合在一起的多个环,其中该稠合的环中的至少一个为芳环。术语“杂芳基”是指含有至少一个杂原子(诸如n、o或s)的芳基,其中该氮和硫原子可任选地氧化,且该氮原子可任选地季铵化。因此,术语“杂芳基”包括稠环杂芳基(稠合在一起的多个环,其中该稠合的环中的至少一个为杂芳基环)。5,6-稠环亚杂芳基是指稠合在一起的两个环,其中一个环具有5个成员且另一个环具有6个成员,并且其中至少一个环为杂芳基环。同样,6,6-稠环亚杂芳基是指稠合在一起的两个环,其中一个环具有6个成员且另一个环具有6个成员,并且其中至少一个环为杂芳
基环。而6,5-稠环亚杂芳基是指稠合在一起的两个环,其中一个环具有6个成员且另一个环具有5个成员,并且其中至少一个环为杂芳基环。杂芳基可通过碳或杂原子附接至该分子的剩余部分。芳基和杂芳基的非限制性实例包括苯基、萘基、吡咯基、吡唑基、哒嗪基、三嗪基、嘧啶基、咪唑基、吡嗪基、嘌呤基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基、吡啶基、嘧啶基、苯并噻唑基、苯并噁唑基、苯并咪唑基、苯并呋喃基、异苯并呋喃基、吲哚基、异吲哚基、苯并噻吩基、异喹啉基、喹噁啉基、喹啉基、1-萘基、2-萘基、4-联苯基、1-吡咯基、2-吡咯基、3-吡咯基、3-吡唑基、2-咪唑基、4-咪唑基、哌嗪基、2-噁唑基、4-噁唑基、2-苯基-4-噁唑基、5-噁唑基、3-异噁唑基、4-异噁唑基、5-异噁唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡啶基、3-吡啶基、4-吡啶基、2-嘧啶基、4-嘧啶基、5-苯并噻唑基、嘌呤基、2-苯并咪唑基、5-吲哚基、1-异喹啉基、5-异喹啉基、2-喹噁啉基、5-喹噁啉基、3-喹啉基和6-喹啉基。上述每一芳基和杂芳基环系统的取代基选自下述可接受的取代基所组成的组。“亚芳基”和“亚杂芳基”单独使用或作为另一取代基的一部分时分别意为源自芳基和杂芳基的二价基。杂芳基取代基可以-o-键结至环杂原子氮。
[0100]
符号表示化学部分附接至分子或化学式剩余部分的附接点。
[0101]
术语“药学可接受的盐”意为包括活性化合物的盐,其使用相对无毒的酸或碱制备,取决于在本文所述化合物上所见的特定取代基。当本公开的化合物含有相对酸性的官能性时,可通过令中性形式的此类化合物与足量的所希望的碱接触而获得碱加成盐,该碱可以是纯净物形式或在适合的惰性溶剂中接触。药学可接受的碱加成盐的实例包括钠盐、钾盐、钙盐、铵盐、有机胺盐或镁盐,或类似的盐。当本公开的化合物含有相对碱性的官能性时,可通过令中性形式的此类化合物与足量的所希望的酸接触而获得酸加成盐,该酸可以是纯净物形式或在适合的惰性溶剂中接触。药学可接受的酸加成盐包括衍生自无机酸如盐酸、氢溴酸、硝酸、碳酸、一氢碳酸、磷酸、一氢磷酸、二氢磷酸、硫酸、一氢硫酸、氢碘酸或亚磷酸的那些,以及衍生自相对无毒的有机酸如醋酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、富马酸、乳酸、扁桃酸、棕榈酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸、草酸、甲磺酸等的盐。还包括氨基酸的盐如精氨酸盐等,以及有机酸如葡萄糖醛酸或半乳糖醛酸等的盐(参见,例如,berge et al.,“pharmaceutical salts”,journal of pharmaceutical science,1977,66,1-19)。本公开的某些具体化合物含有碱性和酸性两种官能性,使得该化合物可以转化为碱加成盐或酸加成盐。
[0102]
如本文中所用,术语“ec
50”或“半最大有效浓度”是指分子(例如,小分子、药物、抗体、嵌合抗原受体或双特异性抗体)的浓度,在该浓度下,该分子能够诱导处于基线应答与特定暴露时间后的最大应答之间的半数的应答。在实施方案中,ec
50
为分子(例如,小分子、药物、抗体、嵌合抗原受体或双特异性抗体)的浓度,在该浓度下产生该分子的最大可能效果的50%。
[0103]
如本文所用,术语“神经退行性疾患”是指其中受试者的神经系统的功能受损的疾病或病症。可以用本文所述的化合物、药物组合物或方法治疗的神经退行性疾病的示例包括亚历山大病、阿尔珀氏病、阿尔兹海默症、肌萎缩性脊髓侧索硬化症、共济失调毛细血管扩张、贝敦氏症(也称为spielmeyer-vogt-sjogren-batten氏症)、疯牛病(bse)、卡纳万病、慢性疲劳综合征、慢性创伤型脑病变、科克因综合症、皮质基底节变性、克雅二氏症、额颞叶痴呆、综合征、亨廷顿舞蹈症、hiv相关痴呆、肯尼
迪氏病、克腊伯氏病、库鲁病、路易体痴呆、马查多-约瑟夫病(脊髓小脑性共济失调3型)、多发性硬化、多系统萎缩症、肌痛性脑脊髓炎、发作性睡病、神经病变(neuroborreliosis)、帕金森症、佩梅二氏病、皮克氏病、原发性侧索硬化症、朊病毒病、雷弗素姆氏病、桑德霍夫病、谢耳德氏病、继发于恶性贫血的脊髓亚急性联合变性、精神分裂症、脊髓小脑性共济失调(多种类型,具有不同特征)、脊髓性肌萎缩症、steele-richardson-olszewski氏病、进行性核上性麻痹或脊髓痨。
[0104]
术语“治疗”或“处理”是指在治疗或改善损伤、疾病、病理或病症方面取得成功的任何标志,包括任何客观或主观参数,例如症候的减轻、消退、消除;或使得损伤、病理或病症更能够被患者耐受;减缓退变或恶化的速度;使得退变终点时不那么衰弱;改善患者的身心健康。症候的治疗或改善可以基于客观或主观参数,包括提个检查、神经精神病学检查和/或心理评估。术语“治疗”及其动词形式可包括预防伤害、病理、病症或疾病。在多个实施方案中,治疗为预防。在多个实施方案中,治疗不包括预防。
[0105]
如本文所用(并且如本领域所充分了解),“治疗”或“处理”也广泛地包括任何用于获得受试者情况的有益或所希望的结果(包括临床结果)的方法。有益或所希望的临床结果包括但不限于,缓解或改善一种或多种症候或症状;减轻疾病程度;稳定疾病状态(即,不恶化);阻止疾病传播或扩散;延迟或减缓疾病进展;改善或缓和疾病状态;减少疾病复发;以及疾病消退,无论是部分的或总体的并且无论是可检测的或不可检测的。换言之,如本文所用,“治疗”包括疾病的任何治愈、缓解和预防。治疗可以阻止疾病出现,抑制疾病扩散,减轻疾病症候,完全地或部分地去除疾病基本肇因,缩短疾病持续时间,或作出这些事件的组合。
[0106]
术语“预防”是指减少患者体内疾病症状的发生率。如上所示,预防可以是完全的(无可检测的症状)或部分的,使得观察到的症状少于不进行治疗时将会出现的。
[0107]“患者”、或“有此需要的受试者”是指正苦于或被证实患有可通过给药本文提供的药物组合物而治疗的疾病或病症的活体有机体。非限制性实例包括人、其它哺乳动物、牛、大鼠、小鼠、狗、猴、山羊、绵羊、牛、鹿和其他非哺乳动物类的动物。在一些实施方案中,患者为人。
[0108]“有效量”为化合物足以实现所指定的相对于不存在该化合物时的目的(例如,达成其所给药的效果、治疗疾病、降低酶活性、增加酶活性、降低信号传导路径、或减轻疾病或病症的一个或多个症状)的量。“有效量”的一个实例为足以对治疗、预防或减轻疾病的一种或多种症状有所贡献的量,其也可称为“治疗有效量”。一种或多种症状的“减轻”(和这一短语的语法等效物)意为症状的严重性下降或频率减少,或症状的消除。药物“预防有效量”为当将药物给药至受试者时将具有所期望的预防效果的量,该预防效果为例如预防或延迟伤害、疾病、病理或病症的发作(或复发)或减小伤害、疾病、病理或病症或其症状发作的可能性。通过一个剂量的给药,不必出现完全预防性效果,并且预防性效果可仅在给药一系列剂量之后出现。因此,预防有效量可在一次或多次给药中完成。如本文中所用,“降低活性的量“是指相对于不存在拮抗剂时降低酶活性所需的拮抗剂的量。如本文中所用,“破坏功能的量“是指相对于不存在拮抗剂时破坏酶或蛋白质功能所需的拮抗剂的量。确切的量将取决于治疗目的,并且可由该领域技术人员使用已知技术(参见,例如,《药物剂型》(lieberman,pharmaceutical dosage forms(vols.1-3,1992));《药物配伍科学技术》(lloyd,the art,
science and technology of pharmaceutical compounding(1999));《剂量计算》(pickar,dosage calculations(1999));和《雷明顿药物科学与实践(第二十版)》(remington:the science and practice of pharmacy,20th edition,2003,gennaro,ed.,lippincott,williams&wilkins))确定。
[0109]
对于本文所述的任何化合物,治疗有效量最初可以从细胞培养物测定中确定。目标浓度将为活性化合物能够实现本文所述方法的那些浓度,如使用本文所述方法或该领域已知方法测量的。
[0110]
如该领域中周知的,用于人类的治疗有效量也可从动物模型测得。例如,可配制用于人的剂量以实现已经发现在动物中有效的浓度。可通过监控化合物有效性并且上调或下调剂量来调整用于人类的剂量,如上所述。调整剂量以基于上述方法和其他方法实现在人体中的最大功效完全处于该领域技术人员的能力范围内。
[0111]
如本文所用,术语“治疗有效量”是指治疗剂足以缓解病变的量,如上所述。例如,对于给定参数,治疗有效量将显示至少5%、10%、15%、20%、25%、40%、50%、60%、75%、80%、90%、或至少100%的增加或减少。治疗功效也可表达为“倍”增或“倍”减。例如,治疗有效量可具有至少1.2倍、1.5倍、2倍、5倍于对照物或更大的效果。
[0112]
剂量可依据患者需求和所采用的化合物而变。在本公开的语境中,给药至患者的剂量应足以随着时间推移在患者体内产生有益的治疗性应答。剂量大小也将通过任何副反应的存在、属性和程度而确定。用于具体情况的适宜剂量的确定处于从业者的技术能力范围内。通常,以低于该化合物优化剂量的较小剂量开始治疗。之后,剂量以小的增量增加,直到达到这些境遇下的最优效果。可独立地调节剂量和间隔以提供对于被治疗的具体临床适应症有效的所给药化合物水平。这将提供与个体的疾病状态严重性相称的治疗方案。
[0113]
如本文中所用,术语“给药”意为口服给药、作为栓剂给药、外用接触给药、静脉内给药、肠胃外给药、腹腔内给药、肌肉内给药、病灶内给药、鞘内给药、鼻内给药或皮下给药至受试者,或者将缓释装置例如微量渗透泵移植到受试者体内。给药可通过任意途径进行,包括肠胃外和跨粘膜(例如,颊腔、舌下、腭部、牙龈、鼻腔、阴道、直肠或透皮)途径。肠胃外给药包括,例如,静脉内给药、肌肉内给药、动脉内给药、皮内给药、皮下给药、腹腔内给药、心室内给药和颅内给药。其他递送模式包括但不限于,使用脂质体制剂、静脉输注、透皮贴剂等。在实施方案中,给药不包括给药除所指定活性剂之外的任何活性剂。
[0114]
如本文中所用,“细胞”是指执行足以保留或复制其基因组dna的代谢或其他功能的细胞。可通过本领域周知的方法鉴定细胞,包括例如完整细胞膜的存在、通过特定染料染色、产生子代的能力,在配子的情况下,与第二种配子合并以产生可存活的子代的能力。细胞可包括原核细胞和真核细胞。原核细胞包括但不限于细菌。真核细胞包括但不限于酵母细胞和源自植物和动物的细胞,例如哺乳动物细胞、昆虫(例如,灰翅夜蛾属)细胞和人细胞。当细胞是天然地非粘附的或已经过处理(例如,通过胰蛋白酶化)而不粘附于表面时,细胞是可用的。
[0115]
化合物
[0116]
一方面,本文提供化合物,该化合物可提供完全的神经保护和对除神经元之外的细胞类型的保护以及nad水平的保持。当该化合物以从低纳摩尔到低微摩尔水平的剂量范围内使用时,可能在a)预防神经元和/或细胞死亡和b)预防由tprp诱导的nad耗竭中高度有
效,如通过神经保护测定所鉴定。
[0117]
一方面,化合物具有式(i),
[0118][0119]
在式(i)中,
[0120]
每个r1和r2独立地为氢、(c
1-c4)烷基或(c
1-c4)烷氧基;并且
[0121]
每个r3独立地选自氢、任选地经oh取代的(c
1-c4)烷基、(c
1-c4)烷氧基或杂芳基,并且前提是两个r3不全为h;或者两个r3与其所键结的氮原子一起形成5至7元杂环基环,所述杂环基环包含至少一个选自o、s、s=o、s(=o)=o或nr的额外的杂原子,其中r为任选地经oh取代的(c
1-c4)烷基、或(c
1-c4)烷氧基。
[0122]
式(i)化合物包括所有药学可接受的盐形式。
[0123]
在实施方案中,r1为甲基。
[0124]
在实施方案中,r2为甲基。
[0125]
在实施方案中,r3中的一个为氢且另一个r3为为甲基、
[0126]
在式(i)的一些化合物中,当r1和r2为甲基,一个r3为氢时,另一个r3不是或甲基。
[0127]
在式(i)的一些化合物中,当r1和r2为甲基时,两个附接至氮原子的r3不形成
[0128]
一方面,化合物具有式(ii),
[0129][0130]
在式(ii)中:
[0131]
每个r
a1
和r
a2
独立地为氢、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基、2至4元杂烷基、环烷基、杂环烷基、芳基或杂芳基;
[0132]
每个r
b1
、r
b2
和r
b3
独立地为氢、卤代、(c
1-c4)烷基、-s(o)2rd、-s(o)2ord或(c
1-c4)卤代烷基;或者r
b2
和r
b3
接合在一起以形成芳基或杂芳基;
[0133]
每个rc和rd独立地为氢或(c
1-c4)烷基;
[0134]
ar为单环或双环的芳基或杂芳基,其任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基或杂芳基中的一者或多者取代;并且
[0135]
n=2、3、4或5。
[0136]
式(ii)化合物包括所有药学可接受的盐形式。
[0137]
在实施方案中,r
b1
为氢。在实施方案中,r
b1
为c
1-c4烷基。在实施方案中,r
b1
为甲基。在实施方案中,r
b1
为乙基。在实施方案中,r
b1
为卤代。在实施方案中,r
b1
为-f。在实施方案中,r
b1
为c
1-c4卤代烷基。在实施方案中,r
b1
为-cf3。在实施方案中,r
b1
为-s(o)2rc。在实施方案中,r
b1
为-s(o)2ch3。
[0138]
在实施方案中,r
b2
为氢。在实施方案中,r
b2
为甲基。在实施方案中,r
b3
为氢。在实施方案中,r
b3
为甲基。在实施方案中,rc为氢。在实施方案中,rc为甲基。在实施方案中,rd为氢。在实施方案中,rd为甲基。
[0139]
在实施方案中,r
b2
和r
b3
接合在一起以形成苯基。
[0140]
在实施方案中,r
a2
为氢。在实施方案中,r
a2
为(c
1-c4)烷基。在实施方案中,r
a2
为甲基。
[0141]
在实施方案中,化合物具有下式:
[0142][0143]ra1
和r
b1
如本文所述。re独立地为卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基或杂芳基,并且z为0、1、2、3、4或5。
[0144]
在实施方案中,z为0、1、2或3。
[0145]
在实施方案中,r
b1
为氢、甲基、乙基、-f、-cf3或-s(o)2me。
[0146]
在实施方案中,r
a2
为氢或甲基。
[0147]
在实施方案中,n为2。在实施方案中,n为3。在实施方案中,n为4。
[0148]
在实施方案中,r
a1
为(c
1-c4)烷基。在实施方案中,r
a1
为甲基。在实施方案中,r
a1
为
乙基。在实施方案中,r
a1
为异丙基。在实施方案中,r
a1
为叔丁基。在实施方案中,r
a1
为(c
1-c4)卤代烷基。在实施方案中,r
a1
为-cf3。在实施方案中,r
a1
为杂环烷基。在实施方案中,r
a1
为在实施方案中,r
a1
为-ch
2-o-ch3。在实施方案中,r
a1
为苯基。
[0149]
在实施方案中,ar为苯基,其任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷氧基或杂芳基中的一者或多者取代。
[0150]
在实施方案中,ar为在实施方案中,ar为在实施方案中,ar为
[0151]
在实施方案中,ar为
[0152]
在式(ii)的一些化合物中,当r
a2
、r
b1
、r
b2
、r
b3
和rc为氢,n为3,ar为未取代或经-ch3或-ome取代的苯基时,r
a1
不是二氟甲基或三氟甲基。
[0153]
在式(ii)的一些化合物中,当r
a2
、r
b1
、r
b2
、r
b3
和rc为氢,n为3,ar为经-f、br、取代的苯基时,r
a1
不是三氟甲基。
[0154]
在实施方案中,式(ii)化合物包括:
[0155]
[0156]
[0157]
[0158][0159]
一方面,化合物具有式(iii),
[0160][0161]
在式(iii)中,
[0162]
l1为键、c
1-c4亚烷基、或2至4元亚杂烷基;
[0163]
r1为单环或双环的环烷基、杂环烷基、芳基、烷基芳基或杂芳基,其中所述环烷基、杂环烷基、芳基、烷基芳基或杂芳基任选地经选自卤代、(c
1-c4)烷基、羟基(c
1-c4)烷基、(c
1-c4)烷氧基、-c(=o)(c
1-c4)烷基、-c(=o)n(r)2或-c(=nr)(c
1-c4)烷基中的一者或多者取代,其中所述(c
1-c4)烷基未经取代或经杂环烷基取代;
[0164]
每个r独立地为氢、-oh、(c
1-c4)烷基或(c
1-c4)烷氧基,或者两个r与其所键结的氮原子一起形成杂环烷基,任选地进一步包含所述杂环基环中的o;
[0165]
r2出现0、1或2次,并且为(c
1-c4)烷基、(c
1-c4)卤代烷基或so2n(r4)2;
[0166]
每个r3和r4独立地为h或(c
1-c4)烷基。
[0167]
式(iii)化合物包括所有药学可接受的盐形式。
[0168]
在实施方案中,l1为键。在实施方案中,l1为亚甲基。在实施方案中,l1为-ch2ch
2-o。
[0169]
在实施方案中,r1为
[0170]
在实施方案中,r2不出现。在实施方案中,r2出现一次。
[0171]
在实施方案中,r3为氢。在实施方案中,r3为甲基。
[0172]
在实施方案中,化合物具有下式:
[0173][0174]
r1和r2如本文所述。
[0175]
在式(iii)的一些化合物中,当l1为键,r2不出现,且r3为氢时,r1不是并且
[0176]
在式(iii)的一些化合物中,当l1为亚甲基,r2不出现,且r3为氢时,r1不是
[0177]
在实施方案中,式(iii)化合物包括:
[0178]
[0179]
[0180][0181]
一方面,化合物具有式(iv),
[0182][0183]
在式(iv)中,
[0184]
r1为氢、(c
1-c4)烷基、-c(o)oh、
–
c(o)o-(c
1-c4)烷基、-c(o)nhnhr6、-c(o)nr
6-((c
1-c4)亚烷基)-nhr6、-c(o)nr6(c
1-c4)烷基或
–
c(o)nr
6-亚环烷基-nhr6;
[0185]
r3为氢或(c
1-c4)烷基;并且
[0186]
每个r2、r4、r5独立地为氢、卤代、(c
1-c4)烷基、
–
c(o)o-(c
1-c4)烷基、(c
1-c4)烷氧基、(c
1-c4)卤代烷基或cn;并且
[0187]
每个r6为氢或(c
1-c4)烷基,
[0188]
式(iv)化合物包括所有药学可接受的盐形式。
[0189]
在实施方案中,每个r4和r5独立地为氢或(c
1-c4)烷氧基。在实施方案中,每个r4和r5为-ome。在实施方案中,r4为氢且r5为-ome。在实施方案中,r5为氢且r4为-ome。
[0190]
在实施方案中,r1为氢、(c
1-c4)烷基。在实施方案中,r1为甲基。在实施方案中,r1为乙基。在实施方案中,r1为-c(o)oh或
–
c(o)och3。在实施方案中,r1为-c(o)nh(c
1-c4)烷基。在实施方案中,r1为-c(o)nhch3。
[0191]
在实施方案中,r1为-c(o)nh-((c
1-c4)亚烷基)-nh2或-c(o)nh-亚环烷基-nh2。在实施方案中,r1为-c(o)nh-ch2ch
2-nh2、-c(o)nch
3-ch2ch
2-nhch3、或-c(o)nhnh2。
[0192]
在实施方案中,r3为氢或甲基。
[0193]
在实施方案中,r2为氢、(c
1-c4)烷基或-c(o)o-(c
1-c4)烷基。在实施方案中,r2为氢。在实施方案中,r2为乙基。在实施方案中,r2为甲基。在实施方案中,r2为-c(o)och3。在实施方案中,r2为-c(o)och2ch3。
[0194]
在实施方案中,r6独立地为氢。在实施方案中,r6独立地为甲基。
[0195]
在式(iv)的一些化合物中,当r4和r5为-ome,r3为氢,且r2为乙基时,r1不是-coome。
[0196]
在式(iv)的一些化合物中,当r4为-ome,r5为氢,且r1和r2为氢时,r3不是甲基。
[0197]
在式(iv)的一些化合物中,当r2、r3、r4和r5为氢时,r1不是-c(o)nhch3。
[0198]
在实施方案中,式(iv)化合物包括:
[0199][0200]
一方面,化合物具有式(v),
[0201][0202]
在式(v)中:
[0203]
ar为单环或双环的芳基或杂芳基,任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、cn、-s(o)2nh2、氧代、-nh2、(c
1-c4)烷氧基或
–
nhc(o)(c
1-c4)烷基中的一者或多者取代;
[0204]
每个r1和r2独立地为氢、(c
1-c4)烷基、芳基或杂芳基,其中所述芳基或杂芳基任选地经卤代或(c
1-c4)烷基取代,或者附接至氮的r1和r2接合在一起以形成5至6元杂环烷基;并且
[0205]
r3为氢或羟基-(c
1-c4)烷基。
[0206]
式(v)化合物包括所有药学可接受的盐形式。
[0207]
在实施方案中,r1和r2为氢。在实施方案中,r1和r2中的一个为氢且另一个为(c
1-c4)烷基诸如甲基、乙基、丙基、异丙基、正丁基或叔丁基。在实施方案中,r1和r2独立地为(c
1-c4)烷基。例如,r1和r2独立地选自甲基、乙基、丙基、异丙基、正丁基或叔丁基。在实施方案中,r1和r2为甲基。
[0208]
在实施方案中,r1和r2中的一个为氢且另一个为可任选地经f、cl、br或(c
1-c4)烷
基取代的苯基。在实施方案中,r1和r2中的一个为氢且另一个为苯基、
[0209]
在实施方案中,附接至氮的r1和r2接合在一起以形成5至6元杂环烷基诸如
[0210]
在实施方案中,r3为氢。在实施方案中,r3为羟基-(c
1-c4)烷基。在实施方案中,r3为-ch
2-oh。
[0211]
在实施方案中,ar为吡啶基、苯基、萘基或噻唑基,其任选地经卤代、(c
1-c4)烷基、(c
1-c4)卤代烷基、cn、-s(o)2nh2、-nh2、(c
1-c4)烷氧基或-nhc(o)烷基中的一者或多者取代。在实施方案中,ar为在实施方案中,ar为
[0212]
在实施方案中,化合物具有下式:
[0213]
r1、r2和ar如本文所述。
[0214]
在式(v)的一些化合物中,当r1、r2和r3为氢时,ar不是
[0215]
在实施方案中,式(v)化合物包括:
[0216]
[0217]
[0218]
[0219][0220]
方法
[0221]
一方面,提供了一种用于在患者体内抑制nad消耗和/或增加nad合成的方法,该方法包括向所述患者给药有效剂量的本文所述化合物。
[0222]
该化合物可抑制蛋白质adp-核糖基化反应。该化合物可抑制通过蛋白质脱乙酰酶或糖水解酶进行的nad裂解。该化合物可增加nad合成。该患者罹患蛋白质错误折叠神经退行性疾病或另一种蛋白质错误折叠疾病或处于其风险下。
[0223]
蛋白质错误折叠神经退行性疾病包括朊病毒病、帕金森症或其他共核蛋白病、阿尔兹海默症、肌萎缩性脊髓侧索硬化症或tau蛋白病,并且蛋白质错误折叠疾病包括糖尿病。
[0224]
一方面,提供了一种用于在患者体内预防或抑制nad耗竭的方法。另一方面,提供了一种用于在患者体内改善与nad的改变相关联的病症的方法。该方法包括向患者给药有效量的本文所述化合物。
[0225]
该病症包括代谢性疾患、衰老、退行性疾病、神经退行性疾病、与多发性硬化相关的神经退变、听力丧失、视网膜损伤或多发性硬化、脑或心肌缺血、肾衰竭、肾病、创伤性脑损伤或轴突病变。
[0226]
一方面,提供了一种用于在患者体内提供保护以免受错误折叠的蛋白质的毒性的方法。该方法包括向患者给药有效量的本文所述化合物。该患者罹患朊病毒病、帕金森症或其他共核蛋白病、阿尔兹海默症、肌萎缩性脊髓侧索硬化症、tau蛋白病或糖尿病。
[0227]
一方面,提供了一种用于在患者体内预防或治疗蛋白质错误折叠神经退行性疾病的方法。该方法包括向患者给药有效量的本文所述化合物。该蛋白质错误折叠神经退行性疾病包括朊病毒病、帕金森症或其他共核蛋白病、阿尔兹海默症、肌萎缩性脊髓侧索硬化症或tau蛋白病。
[0228]
神经保护和gaba
a r抑制是dmcm的两种不同活性。
[0229]
dmcm是咔唑系列的一个成员(结构在前文和实施例中显示),具有已知的作用模式:它是所有亚型的gaba
a r7的反向激动剂,结合至其苯二氮(bz)位点。因此,它是体内惊厥药。因为致惊厥活性在神经保护剂中是不能容忍的,它不能直接改变用途8。已经测试了另一种结合至bz位点的药理学调节剂氟马西尼(ro 15-1788)
9,10
。它既不能拯救tprp诱导的毒性,也不能与dmcm的效果竞争(图3),表明神经保护似乎与在gaba
a r处的内在活性无关。
[0230]
已经进行了一项初步sar研究来进一步测试以下结论:dmcm的神经保护活性与gaba
a r结合活性是不同的。图4显示肼酰胺1,它是dmcm的一种高度类似物(dmcm-10049),据报导它缺乏gaba
a r活性(对于大多数gaba
a r亚型,效力降低了超过100倍)
11
。然而,它显示了非常接近于dmcm的神经保护活性(图4)。用二胺类(包括1,4-顺-二氨基环己烷)进行的
dmcm处理给出了氨基酰胺诸如化合物2(dmcm-8137)。该化合物也具有神经保护活性(图4)。此外,它被设计为具有允许缀合靶标标志的柄(图4)。
[0231]
综上所述,这些研究显示,dmcm作用于除gaba
a r之外的靶标以提供神经保护,这一发现使得将dmcm类似物作为神经保护剂进行优化成为可能,使得它们缺乏母体化合物已经使用的活性。dmcm在原代神经元中具有神经保护活性(参见图5)。
[0232]
dmcm阻止由tprp诱导的过度单-adp核糖基化。
[0233]
已经证明,tprp至少部分地通过过度的蛋白质单-adp核糖基化2(一种消耗nad的反应)而诱导nad耗竭。图6显示,dmcm阻止这一过度的蛋白质adp核糖基化。
[0234]
dmcm在帕金森症(pd)的细胞模型中具有神经保护活性。
[0235]
帕金森症,类似于朊病毒病,是由蛋白质的错误折叠和聚集导致的,在这种情况下,该蛋白质是α-突触核蛋白。因此,在pd诱导的神经退变的细胞模型中研究来自hts的化合物的神经保护性质(图7和14)。在这些模型中,暴露于预成形的α-突触核蛋白原纤维(pff)的神经元细胞经历突触和树突棘的丢失以及神经突的缩短和丢失(图7,参见暴露于pff的神经元与对照神经元)。接种pff的神经元蓄积α-突触核蛋白原纤维和对细胞有毒的特定类型的α-突触核蛋白聚集体(称为pα-syn*
12
)。图7显示,dmcm的肼酰胺(名为dmcm-10049)保留了暴露于pff的神经元中的树突棘并且减少了有毒的pα-syn*的量。
[0236]
dmcm-10049在帕金森症鼠模型中的治疗效果。
[0237]
此外,dmcm已经作为具有有利的pk性质的工具化合物用于体内
5,7-9,13,14
。因此,在pd的鼠模型((tg(snca*a53t)小鼠,荷有负责人类家族性pd的α-突触核蛋白突变)中测试dmcm-10049。用dmcm-10049治疗显著延长了这些小鼠的存活期(图8)。
[0238]
dmcm-10049在als小鼠模型中的治疗效果。
[0239]
在als的鼠模型((tg(sod1*g93a)小鼠,荷有sod1突变)中测试dmcm-10049
15
。突变体sod1占家族性als病例的15-20%和明显散发的als病例的1-2%,并且在不携带突变的als患者体内发现了错误折叠的sod1
16
。用dmcm-10049治疗显著延长了这些小鼠的存活期(图9)。
[0240]
瓦他拉尼系列(氨基酞嗪系列)的详细研究。
[0241]
瓦他拉尼,又名sr5-1457,最近已经作为口服给药的抗肿瘤药用用于后期临床开发中
17-21
。它是一种受体酪氨酸激酶抑制剂,具体地是一种有效的vegfr抑制剂,具有高细胞渗透性并且在人类和啮齿动物中具有整体上优异的pk性质
22-24
。这一化合物具有高的神经保护活性,ec
50
=39.9nm(tprp毒性拯救)且ec
50
=195nm(nad测定)。因为其高效力和优异的pk性质(包括高的脑渗透和高的口服生物利用性),我们选择快速推进这一化合物的体内神经保护研究。
[0242]
如图10中所示,als小鼠的瓦他拉尼治疗显著改善了它们的运动功能和肌肉强度,使用转棒测试和悬挂测试评估。但是,没有观察到存活时间的延长(经治疗动物的160
±
2天存活与对照组的157
±
2天)。
[0243]
尽管使用瓦他拉尼仅这一早期概念验证研究,但由于这一具体化合物具有强烈抑制vegfr的能力,这种能力提供抗血管生成效果,而这种效果被认为对该化合物的抗肿瘤性质负责,因此该化合物不适合直接改变用途,这可能是因为该化合物的神经保护作用模式与其已知的对于vegfr的活性无关,因为已知vegf/vegfr-2信号传导是神经保护性的(因
此,阻断vegfr预期将会提供轻度的神经毒性而不是神经保护)
25
。已知vegfr-2过表达在tg(sod1*g93a)小鼠中延迟脊髓运动神经元的神经退变,并且vegf给药已经显示在als模型中延迟肌肉衰弱
26-28
并且在pd模型中具有神经保护活性。
29,30
。因此,瓦他拉尼的vegfr已知活性应与神经保护的目标相反,因此,对于改变用途来治疗als或更通常而言任何需要长期治疗的神经退行性疾病而言,瓦他拉尼将会是糟糕的选择。本文所鉴定的nad保留活性可能与内在vegfr效果相反(并强于后者)。缺乏vegfr活性的类似物对于神经保护类药物先导物而言应该是更好的候选物。
[0244]
已知对于vegfr无活性或至少具有非常低的与vegfr的亲和性的瓦他拉尼类似物显示在图11中。注意:flt-1和kdr是vegfr的亚型,也分别称为vegfr-1和vegfr-2。如图所示,这些化合物的神经保护/nad恢复效应的活性与它们的vegfr抑制活性不相关
24
。例如,含有甲基酮的瓦他拉尼结构类似物sr1-134005(图11中的第二种结构)作为神经保护剂的效力超过瓦他拉尼的6倍(6.3nm对39.9nm),但同一化合物作为vegfr-1抑制剂的效力比瓦他拉尼低13倍(1μm对77nm)。类似地,类似物sr1-151915(第三种结构)的神经保护活性为瓦他拉尼的一半(ec
50
=71.4nm),但同一化合物也是不良的vegfr抑制剂(ic
50
》1μm)。相反,类似物sr1-151911(第四种结构)的神经保护活性极小(在6.4μm时仅44%神经保护),但它具有中等程度的vegfr活性(对于vegfr-1的ic
50
=793nm,仅比瓦他拉尼低约20倍,且对于vegfr,它比sr1-134005和sr1-151915更有效)。
[0245]
再者,在tprp测定中,其它可商购的结构上与瓦他拉尼无关的有效vegfr抑制剂(乐伐替尼、帕唑帕尼、替沃扎尼和索拉非尼)在高达10μm的浓度下完全没有神经保护活性(数据未显示)。
[0246]
显然,两种活性(神经保护和通过vegfr抑制产生的抗肿瘤效果)可以区分,并且这一明确的结果促使我们测试在als小鼠模型中具有低vegf-r活性的瓦他拉尼类似物sr1-134005。
[0247]
sr1-134005在als小鼠模型中的治疗效果。
[0248]
瓦他拉尼的甲基酮类似物sr1-134005(表现出比瓦他拉尼降低约13倍的vegfr-1活性,图11)提供与瓦他拉尼相同的总体内获益,此外,它在8倍较低剂量时是有效的(图12)。这一体内结果支持我们的体外发现,即sr1-134005是比瓦他拉尼更有效力的神经保护剂,并且进一步地,该神经保护与vegfr抑制无关,而vegfr抑制被认为是这一类化合物的抗肿瘤效应的原因。
[0249]
sr1-134005在帕金森症鼠模型中的治疗效果。
[0250]
用瓦他拉尼的甲基酮类似物sr1-134005进行治疗显著延长了tg(snca*a53t)小鼠的存活期(图13)。
[0251]
吡唑并嘧啶类(sr1-293229)、氨基噻唑类(sr1-477186)、三唑并酞嗪类(sr1-115259)、氨基酞嗪类(瓦他拉尼)和类黄酮类(川陈皮素、芹菜素)在pd(一种共核蛋白病)的细胞模型中的神经保护效果。
[0252]
类似于我们使用咔唑系列观察到的结果,其他先导系列的化合物在培养的神经元中针对由α-突触核蛋白pff诱导的神经退变进行保护。这些化合包括吡唑并嘧啶类(sr1-293229)、氨基噻唑类(sr1-477186)、三唑并酞嗪类(sr1-115259)、氨基酞嗪类(瓦他拉尼)和类黄酮类(川陈皮素、芹菜素)。图14示例性说明保护效果并且显示先导化合物阻止了由
pff暴露导致的神经突丢失。
[0253]
通过咔唑、氨基酞嗪、吡唑并嘧啶、三唑并酞嗪和类黄酮系列进行的nad拯救并非由于parp-1抑制。
[0254]
图15显示,本文所述先导系列中的至少五种不是parp-1抑制剂。
[0255]
氨基酞嗪类(瓦拉他尼和sr1-134005)是nampt活化剂。
[0256]
已经证明,tprp诱导过度的adp-核糖基化,并且设计该化合物筛选策略以捕获能恢复生理nad水平的化合物。图6显示,这可以通过阻止过度的adp-核糖基化来实现。图16显示,它也可以通过增强nad合成来实现,因为四个先导序列中的一个(氨基酞嗪类的瓦他拉尼和“sr-005”)通过活化nampt(nad合成中的限速酶)而发挥作用。
[0257]
第一次证明了nad代谢的失败是错误折叠的淀粉样蛋白(tprp)诱导神经毒性的基本机制,并且证明nad的补充具有神经保护作用2。因此,针对从蛋白质毒性中拯救出来而筛选了nad恢复性化合物,假设其他病症可以使用能通过任何基质护肤健康nad水平的化合物成功地治疗。事实上,nad失调现在也被认为牵涉到ad
31,32
、衰老
33-36
、与多发性硬化相关的神经元退变
37
、听力丧失
38
、肾损伤
39
、创伤性脑损伤
40
和轴突病变
41
中。在退行性肾病中发现了nad水平的实质性降低
42
。已经证明,nad增加(例如nad给药或通过酶过表达来增加nad合成)可减轻脑缺血
43
、心肌缺血/再灌注损伤
44,45
和急性肾损伤
42
。
[0258]
也已经证明,2型糖尿病(t2d)的小鼠模型中的nad代谢改变
46,47
。糖尿病中nad代谢的改变可以通过我们的发现解释,即错误折叠的蛋白质诱导nad失调。事实上,已经证明糖尿病是蛋白质错误折叠疾病,其特征在于胰腺β细胞功能失调和死亡,伴有聚集的胰岛淀粉样多肽(iapp)的沉积,而iapp是一种与胰岛素一起被胰腺β细胞共表达和分泌的蛋白质
48
。淀粉样iapp沉积是不同病因的糖尿病的共有特征
49
。类似于牵涉到其他蛋白质错误折叠疾病中的蛋白质,iapp形成有毒的寡聚物
48
。此外,前胰岛素,即胰岛素的前体,也被证实在β细胞中错误折叠。前胰岛素的错误折叠已经与2型、1型和一些单基因形式的糖尿病进展相关联
48,50,51
。最后,胰腺β细胞带有一些与神经元共有的生理性质
52
。因此,本文所述的化合物将能够用来在糖尿病的细胞模型中减轻胰腺细胞的功能失调和死亡,并且用来在糖尿病的动物模型中实现治疗受益,作为其用于治疗人类糖尿病的潜力的证明。为此,我们将使用啮齿动物来源的胰岛素分泌细胞系诸如min-6和ins-1细胞
52,53
;我们预计,当暴露于错误折叠和/或聚集形式的iapp和/或前胰岛素时,它们的β细胞功能、nad水平和活力将发生改变。再者,我们预计,这些改变将通过使用本文所述化合物进行治疗而得以校正。化合物的治疗受益将在啮齿动物模型中评估,例如,高脂肪饲养小鼠、ob/ob小鼠和db/db小鼠(分别为瘦素缺失和瘦素抗性)用于t2d
54
,以及经链霉菌毒素治疗的小鼠、非肥胖性糖尿病(nod)小鼠、易患糖尿病的生物繁殖(bb)大鼠用于1型糖尿病
55,56
。
[0259]
如本文所用,nad表示该辅因子的氧化形式(nad+)和还原形式(nadh)。尤其是,作为一种用于调节能量代谢路径诸如糖酵解、tca循环和导致atp产生的氧化磷酸化的辅酶,nad至关重要。此外,nad作为底物用于信号转导和翻译后蛋白质修饰(称为adp-核糖基化)。
[0260]
nad合成酶和nad消耗酶的活性平衡所致的生理细胞nad水平,可能是因为错误折叠的蛋白质诱导的nad失衡(并且在我们的表型测定中评估),因此将会是nad生物合成受损或nad消耗增加的结果。
[0261]
在哺乳动物细胞中,nad主要通过使用前体烟酰胺(nam)的补救路径合成。用于补
救路径中的nad合成的限速酶是烟酰胺磷酸核糖基转移酶(nampt)。其他nad合成路径是利用前体色氨酸的从头合成路径和利用前体烟酸(na)的preiss-handler路径。
[0262]
另一方面,nad在以下细胞反应期间被消耗:1)通过被称为nad水解酶或adp-核糖基环化酶(cd38和cd157)的酶从nad产生钙释放第二信使环状adp-核糖(cadpr)和adp-核糖(adpr);2)sirtuin介导的蛋白质脱乙酰作用;和3)蛋白质adp-核糖基化,在该反应中,nad的一个或几个adp-核糖部分通过单/寡-adp-核糖转移酶(marts)或多-adp核糖转移酶(称为parp)被转移到蛋白质中。
[0263]
已经证明,tprp诱导细胞蛋白质的过度adp核糖基化,并且进一步证明,毒性没有通过选择性parp1抑制剂减轻。因此,这些研究揭示了一种新的神经毒性机制,该机制与至少部分地由于过度的单或寡adp核糖基化反应所致的nad代谢失衡相关联。如上所述,对本文所呈现的8个化合物系列进行鉴定的hts活动和随访测定依赖于表型读出,并且因此就暴露于蛋白质毒性的细胞中的nad水平和活力基本维持基质而言,该研究有意地采取不确定性。该设计旨在鉴定通过任何作用机制(诸如增强nad合成或阻止过度nad降解/消耗)调节nad水平的化合物,并且旨在包括非parp1抑制剂。我们的数据显示:1)本文呈递的所有保护性化合物均具有神经保护活性并且保持细胞nad水平;2)这些化合物中的至少5种不是parp-1抑制剂(图15);3)如概念验证一样,至少一种测试化合物阻止由错误折叠的蛋白质引起的过度adp核糖基化(图6),并且至少一种测试化合物为nampt活化剂(图16)。
[0264]
实施例
[0265]
实施例1:细胞存活率测定和nad定量测定
[0266]
下表显示可用于实践本发明方法的化合物的具体实例的结构,与相应的数据诸如化合物标识符、分子量、化合物性质和生物学结果。
[0267]
在两种测定中将测试化合物的生物学活性定量:细胞存活率测定其评估化合物预防由错误折叠的蛋白质tprp诱导的神经元死亡的能力;和nad定量测定(nad+/nadh-glo
tm
),其评估化合物预防由错误折叠的蛋白质tprp诱导nad耗竭。显示了有效浓度(ec
50
值)。过程如图2(1536孔板格式)中针对那些存活率ec
50
和nad ec
50
两者均示出(表1至8)的那些化合物所述,如图4(96孔板格式)中针对仅示出存活率ec
50
(表9至12)的那些化合物所述。
[0268]
表1.三唑并噻嗪类:sr1-115259系列
[0269]
[0270]
[0271][0272]
表2.吡唑并嘧啶类:sr1-293229和相关化合物
[0273]
[0274]
[0275][0276]
表3.氨基酞嗪类:瓦他拉尼(vatalanib)和相关化合物
[0277]
[0278][0279]
表4.咔唑类:dmcm和相关化合物
[0280]
[0281][0282]
表5.氨基噻唑类:sr1-477186和相关化合物
[0283]
[0284]
[0285][0286]
表6.类黄酮:川陈皮素和相关化合物
[0287]
[0288]
[0289][0290]
表7.生物碱,包括异喹啉、阿朴啡和麦角生物碱家族的所选成员:
[0291]
[0292][0293]
表8.3-杂芳基喹啉类(dmpq)
[0294][0295]
表9.式(ii)化合物
[0296]
[0297]
[0298]
[0299]
[0300]
[0301]
[0302][0303]
表10.式(iii)化合物
[0304]
[0305]
[0306]
[0307]
[0308]
[0309][0310]
表11.式(iv)化合物
[0311]
[0312][0313]
表12.式(v)化合物
[0314]
[0315]
[0316]
[0317]
[0318]
[0319][0320]
参考文献
[0321]
1zhou,m.,ottenberg,g.,sferrazza,g.f.&lasmezas,c.i.highly neurotoxic monomeric alpha-helical prion protein.proc natl acad sci u s a 109,3113-3118,doi:10.1073/pnas.1118090109(2012).
[0322]
2zhou,m.et al.neuronal death induced by misfolded prion protein is due to nad+depletion and can be relieved in vitro and in vivo by nad+replenishment.brain 138,992-1008,doi:10.1093/brain/awv002(2015).
[0323]
3olivan,s.et al.comparative study of behavioural tests in the sod1g93a mouse model of amyotrophic lateral sclerosis.exp anim 64,147-153,doi:10.1538/expanim.14-0077(2015).
[0324]
4dahlin,j.l.et al.pains in the assay:chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging hts.j med chem 58,2091-2113,doi:10.1021/jm5019093(2015).
[0325]
5lipinski,c.a.,lombardo,f.,dominy,b.w.&feeney,p.j.experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.adv drug deliv rev 46,3-26(2001).
[0326]
6veber,d.f.et al.molecular properties that influence the oral bioavailability of drug candidates.j med chem 45,2615-2623(2002).
[0327]
7mele,l.,massotti,m.&gatta,f.neuropharmacology of several beta-carboline derivatives and their 9-acetylated esters.in vivo versus in vitro studies in the rabbit.pharmacol biochem behav 30,5-11(1988).
[0328]
8leppa,e.et al.agonistic effects of the beta-carboline dmcmrevealed in gaba(a)receptor gamma 2 subunit f77i point-mutated mice.neuropharmacology 48,469-478,doi:10.1016/j.neuropharm.2004.11.007(2005).
[0329]
9atack,j.r.,smith,a.j.,emms,f.&mckernan,r.m.regional differences in the inhibition of mouse in vivo[3h]ro 15-1788 binding reflect selectivity for alpha 1 versus alpha 2 and alpha 3 subunit-containing gabaa receptors.neurop
sychopharmacology 20,255-262,doi:10.1016/s0893-133x(98)00052-9(1999).
[0330]
10lista,a.,blier,p.&de montigny,c.the benzodiazepine receptor inverse agonist dmcm decreases serotonergic transmission in rat hippocampus:an in vivo electrophysiological study.synapse 6,175-178,doi:10.1002/syn.890060209(1990).
[0331]
11huang,q.et al.pharmacophore/receptor models for gaba(a)/bzr subtypes(alpha1beta3gamma2,alpha5beta3gamma2,and alpha6beta3gamma2)via a comprehensive ligand-mapping approach.j med chem 43,71-95(2000).
[0332]
12grassi,d.et al.identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in parkinson's disease.proc natl acad sci u s a 115,e2634-e2643,doi:10.1073/pnas.1713849115(2018).
[0333]
13walters,w.p.going further than lipinski's rule in drug design.expert opin drug discov 7,99-107,doi:10.1517/17460441.2012.648612(2012).
[0334]
14crestani,f.,assandri,r.,tauber,m.,martin,j.r.&rudolph,u.contribution of the alpha1-gaba(a)receptor subtype to the pharmacological actions of benzodiazepine site inverse agonists.neuropharmacology 43,679-684(2002).
[0335]
15gurney,m.e.et al.motor neuron degeneration in mice that express a human cu,zn superoxide dismutase mutation.science 264,1772-1775(1994).
[0336]
16pokrishevsky,e.et al.aberrant localization of fus and tdp43 is associated with misfolding of sod1 in amyotrophic lateral sclerosis.plos one 7,e35050,doi:10.1371/journal.pone.0035050(2012).
[0337]
17brander,d.et al.phase ii open label study of the oral vascular endothelial growth factor-receptor inhibitor ptk787/zk222584(vatalanib)in adult patients with refractory or relapsed diffuse large b-cell lymphoma.leuk lymphoma 54,2627-2630,doi:10.3109/10428194.2013.784969(2013).
[0338]
18raizer,j.j.et al.a phase ii trial of ptk787/zk 222584 in recurrent or progressive radiation and surgery refractory meningiomas.j neurooncol 117,93-101,doi:10.1007/s11060-014-1358-9(2014).
[0339]
19dragovich,t.et al.phase ii trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy(pcrt o4-001).cancer chemother pharmacol 74,379-387,doi:10.1007/s00280-014-2499-4(2014).
[0340]
20gupta,p.et al.a phase ii study of the oral vegf receptor tyrosine kinase inhibitor vatalanib(ptk787/zk222584)in myelodysplastic syndrome:cancer and leukemia group b study 10105(alliance).invest new drugs 31,1311-1320,doi:10.1007/s10637-013-9978-z(2013).
[0341]
21jain,r.k.,duda,d.g.,clark,j.w.&loeffler,j.s.lessons from phase iii clinical trials on anti-vegf therapy for cancer.nat clin pract oncol 3,24-40,doi:10.1038/ncponc0403(2006).
[0342]
22remko,m.,boh
á
c,a.&kov
á
cikov
á
,l.molecular structure,pka,lipophilicity,solubility,absorption,polar surface area,and blood brain barrier penetration of some antiangiogenic agents.struct chem 22,635-648(2011).
[0343]
23bold,g.et al.new anilinophthalazines as potent and orally well absorbed inhibitors of the vegf receptor tyrosine kinases useful as antagonists of tumor-driven angiogenesis.j med chem 43,3200(2000).
[0344]
24bold,g.et al.phthalazines with angiogenesis inhibiting activity.wo 98/35958.(1998).
[0345]
25pronto-laborinho,a.c.,pinto,s.&de carvalho,m.roles of vascular endothelial growth factor in amyotrophic lateral sclerosis.biomed res int 2014,947513,doi:10.1155/2014/947513(2014).
[0346]
26ruiz de almodovar,c.,lambrechts,d.,mazzone,m.&carmeliet,p.role and therapeutic potential of vegf in the nervous system.physiol rev 89,607-648,doi:10.1152/physrev.00031.2008(2009).
[0347]
27storkebaum,e.et al.treatment of motoneuron degeneration by intracerebroventricular delivery of vegf in a rat model of als.nat neurosci 8,85-92,doi:10.1038/nn1360(2005).
[0348]
28zheng,c.,nennesmo,i.,fadeel,b.&henter,j.i.vascular endothelial growth factor prolongs survival in a transgenic mouse model of als.ann neurol 56,564-567,doi:10.1002/ana.20223(2004).
[0349]
29yasuhara,t.et al.neuroprotective effects of vascular endothelial growth factor(vegf)upon dopaminergic neurons in a rat model of parkinson's disease.eur j neurosci 19,1494-1504,doi:10.1111/j.1460-9568.2004.03254.x(2004).
[0350]
30piltonen,m.et al.vascular endothelial growth factor c acts as a neurotrophic factor for dopamine neurons in vitro and in vivo.neuroscience 192,550-563,doi:10.1016/j.neuroscience.2011.06.084(2011).
[0351]
31sorrentino,v.et al.enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity.nature 552,187-193,doi:10.1038/nature25143(2017).
[0352]
32hou,y.et al.nad(+)supplementation normalizes key alzheimer's features and dna damage responses in a new ad mouse model with introduced dna repair deficiency.proc natl acad sci u s a 115,e1876-e1885,doi:10.1073/pnas.1718819115(2018).
[0353]
33massudi,h.et al.age-associated changes in oxidative stress and nad+metabolism in human tissue.plos one 7,e42357,doi:10.1371/journal.pone.0042357
(2012).
[0354]
34zhu,x.h.,lu,m.,lee,b.y.,ugurbil,k.&chen,w.in vivo nad assay reveals the intracellular nad contents and redox state in healthy human brain and their age dependences.proc natl acad sci u s a 112,2876-2881,doi:10.1073/pnas.1417921112(2015).
[0355]
35zhang,h.et al.nad(+)repletion improves mitochondrial and stem cell function and enhances life span in mice.science 352,1436-1443,doi:10.1126/science.aaf2693(2016).
[0356]
36mouchiroud,l.et al.the nad(+)/sirtuin pathway modulates longevity through activation of mitochondrial upr and foxo signaling.cell 154,430-441,doi:10.1016/j.cell.2013.06.016(2013).
[0357]
37penberthy,w.t.&tsunoda,i.the importance of nad in multiple sclerosis.curr pharm des 15,64-99(2009).
[0358]
38brown,k.d.et al.activation of sirt3 by the nad(+)precursor nicotinamide riboside protects from noise-induced hearing loss.cell metab 20,1059-1068,doi:10.1016/j.cmet.2014.11.003(2014).
[0359]
39lin,j.b.et al.nampt-mediated nad(+)biosynthesis is essential for vision in mice.cell rep17,69-85,doi:10.1016/j.celrep.2016.08.073(2016).
[0360]
40satchell,m.a.et al.a dual role for poly-adp-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving nad+depletion and ribosylation of 14-3-3gamma.j neurochem 85,697-708(2003).
[0361]
41vaur,p.et al.nicotinamide riboside,a form of vitamin b3,protects against excitotoxicity-induced axonal degeneration.faseb j 31,5440-5452,doi:10.1096/fj.201700221rr(2017).
[0362]
42ralto,k.m.,rhee,e.p.&parikh,s.m.nad(+)homeostasis in renal health and disease.nat rev nephrol 16,99-111,doi:10.1038/s41581-019-0216-6(2020).
[0363]
43ying,w.et al.intranasal administration with nad+profoundly decreases brain injury in a rat model of transient focal ischemia.front biosci 12,2728-2734,doi:2267[pii](2007).
[0364]
44hsu,c.p.,oka,s.,shao,d.,hariharan,n.&sadoshima,j.nicotinamide phosphoribosyltransferase regulates cell survival through nad+synthesis in cardiac myocytes.circ res 105,481-491,doi:10.1161/circresaha.109.203703(2009).
[0365]
45yamamoto,t.et al.nicotinamide mononucleotide,an intermediate of nad+synthesis,protects the heart from ischemia and reperfusion.plos one 9,e98972,doi:10.1371/journal.pone.0098972(2014).
[0366]
46yoshino,j.,mills,k.f.,yoon,m.j.&imai,s.nicotinamide mononucleotide,a key nad(+)intermediate,treats the pathophysiology of diet-and age-induced diabetes in mice.cell metab 14,528-536,doi:10.1016/j.cmet.2011.08.014(2011).
[0367]
47trammell,s.a.et al.nicotinamide riboside opposes type 2 diabetes and neuropathy in mice.sci rep6,26933,doi:10.1038/srep26933(2016).
[0368]
48costes,s.targeting protein misfolding to protect pancreatic beta-cells in type 2 diabetes.curr opin pharmacol 43,104-110,doi:10.1016/j.coph.2018.08.016(2018).
[0369]
49ueberberg,s.et al.islet amyloid in patients with diabetes due to exocrine pancreatic disorders,type 2 diabetes and non-diabetic patients.j clin endocrinol metab,doi:10.1210/clinem/dgaa176(2020).
[0370]
50liu,m.et al.proinsulin misfolding and diabetes:mutant ins gene-induced diabetes of youth.trends endocrinol metab 21,652-659,doi:10.1016/j.tem.2010.07.001(2010).
[0371]
51sun,j.et al.proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes.mol aspects med 42,105-118,doi:10.1016/j.mam.2015.01.001(2015).
[0372]
52atouf,f.,scharfmann,r.,lasm
é
zas,c.&czernichow,p.tight hormonal control of prp gene expression in endocrine pancreatic cells.biochem and biophys res commun 201,1220-1226(1994).
[0373]
53green,a.d.,vasu,s.&flatt,p.r.cellular models for beta-cell function and diabetes gene therapy.acta physiol(oxf)222,doi:10.1111/apha.13012(2018).
[0374]
54al-awar,a.et al.experimental diabetes mellitus in different animal models.j diabetes res 2016,9051426,doi:10.1155/2016/9051426(2016).
[0375]
55lenzen,s.animal models of human type 1 diabetes for evaluating combination therapies and successful translation to the patient with type 1 diabetes.diabetes metabres rev 33,doi:10.1002/dmrr.2915(2017).
[0376]
56cheta,d.animal models of type i(insulin-dependent)diabetes mellitus.j pediatr endocrinol metab 11,11-19,doi:10.1515/jpem.1998.11.1.11(1998).
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1