活性药物成分的纳米悬浮液在载体上的沉积的制作方法
活性药物成分的纳米悬浮液在载体上的沉积
1.本发明提供一种制备难溶性药物成分(api)的药物组合物的方法,该药物组合物被装载在载体上并通过其稳定化。特别地,本发明涉及在干燥状态下载体上的纳米微粒api的组合物,其被加工为具有改善的释放曲线和生物利用度的所述api的药物制剂。
2.要解决的发明问题的背景
3.药物溶解度差和生物利用度低仍然是制药行业的一个重要且普遍的问题。原则上,对于活性药物成分(api)的生物利用度,溶解度和溶出速率是基本参数。如果要改善难溶性药物的生物利用度,就必须影响这两个因素。因此,过去在这方面研究了许多不同的药物。
4.影响生物利用度的另一个因素在于活性物质吸收到新陈代谢中和因此活性成分转化为体液,借此活性成分可以到达作用部位。取决于活性成分的物理和化学性质,必须以在特别调整的制剂中提供。
5.了解活性成分的物理化学特性(溶解度、渗透性、粒度分布、多态性等)是该过程的先决条件,尤其有助于将分子置于生物制药分类系统(bcs)中。在此上下文中,基于脂质的制剂促进属于bcs ii类和iv类的许多难溶性候选药物的胃肠道吸收的能力已在已发表的文献中得到充分记录。然而,该技术的知识与其应用所需的专有技术之间仍然存在相当大的差距。[hauss,david;“oral lipid-based formulations“;advanced drug delivery reviews 59(2007)667
–
676]。与此同时,自此综述发表以来,情况并没有发生显著变化,尽管40%的已上市药物属于bcs ii和iv类药物,并且这是日益严重的问题,因为高达80%的所有药物开发物质属于这些bcs类别。
[0006]
此外,制药行业面临着许多挑战:
[0007]-需要更快地开发创新产品,
[0008]-需要将自己的产品与竞争对手的产品区分开来,
[0009]-增加研发成本,
[0010]-由于产生“差溶解度或/和低生物利用度”的物质结构的在开发过程中的高失败率。
[0011]
早在100多年前,noyes and whitney[a.a.noyes,w.r.whitney;“the rate of solution of solid substances in their own solutions”;j.am.chem.soc.,19(1897),pp.930-934]研究了固体在溶剂中的溶出度和溶解度之间的关系,并用数学方程表示了这种关系。后来,溶出速率和溶解度之间的关系通过改进的noyes-whitney方程表征[nernst,w.,1904.“theorie der reaktionsgeschwindigkeit in heterogenen systemen“;z.phys.chem.47,52
–
55.]:
[0012][0013]
dq/dt=溶出速率
[0014]
d=扩散系数
[0015]
h=扩散层厚度
[0016]cs
=溶解度
[0017]
cb=本体溶液浓度
[0018]
a=颗粒的表面积
[0019]
在这个方程中,除了扩散系数以外,要溶出的活性物质的表面以及溶解度是最重要的变量,通过它们可影响溶出速率。
[0020]
最近,已经开发了多种技术并努力改善难溶性bcs ii类和iv类药物的生物利用度,这些方法尽管有许多优点,但也有许多缺点。特别地,希望通过仅添加少量添加剂,但优选不添加添加剂来尽可能改善生物利用度。
[0021]
现在,回到修改后的noyes-whitney方程,用于改善难溶性药物的溶解度的一种方式应该增加难溶性固体活性成分的表面积(a)。这可以通过微粉化或纳米化方法来完成。减小粒度和增加表面积的一种直接可能性是使用研磨工艺。用于此的另一种已知技术是例如在超临界流体存在下的共研磨。
[0022]
改善这些难溶性活性物质的溶解度(cs)的另一种选择是改变活性物质应溶解在其中的液体环境的性质。这可以通过将溶剂与溶解度增强剂(如环糊精)或与由例如油、表面活性剂、助表面活性剂、助溶剂和溶解的药物物质组成的脂质制剂(其正在形成自乳化药物递送系统(sedds))混合或通过自微乳化药物递送系统(smedds)(这是一种改进的sedds,其在胃肠道的温和搅动下,无需溶出过程即可形成直径小于50nm的细小水包油液滴)来完成。
[0023]
另一方面,可以利用的是活性物质经常以各种多晶型形式存在,虽然它们的药效差别不是非常大,但溶解度不同。此外,向易溶盐的转化可以改善在水溶液中的溶解度或活性成分的共晶体的使用。
[0024]
另一种改善活性成分生物利用度的方法是通过适当的措施扩大表面积(a)以及改善溶解度(cs)。这可以例如通过使用热熔挤出或喷雾干燥技术或介孔二氧化硅制备固体分散体来实现,其中将难溶性活性物质施加到惰性载体上。特别适合作为支持物的是生物相容的多孔无机材料,其以具有合适粒度的相应粉末形式存在。在工业中开发制剂的制药科学家能够利用这三种技术来改变api的物理状态,将难溶性药物从其结晶形式转化为稳定的无定形结构,具有显著增强的溶解度和口服药物吸收。
[0025]
喷雾干燥和热熔挤出常用于制造固体分散体和固溶体。这种方法可以追溯到sekiguchi和obi,他们首先引入低共熔混合物作为增强溶解度的手段。[sekiguchi,k.and n.obi,man.chemical&pharmaceutical bulletin,1961.9(11):p.866-872.]。在固体分散体中,api通常以结晶或无定形状态或者在固体和玻璃状溶液的情况下在分子水平上分散或溶出在聚合物基质中,产生所谓的无定形固体分散体(asd))。[dhirendra,k.等人,pakistan journal of pharmaceutical sciences,2009.22(2):p.234-246]
[0026]
如上所述,理论上难溶性活性成分的溶解度可以通过扩大表面积(a)和最小化药物物质的粒径来改善。研磨药物化合物以实现更小粒径的微粉化方法已得到很好的建立。这些方法的详细概述在ep 1 401 401 b1(elan pharma int.ltd.[ie])中给出。在该文献中,公开了一种方法,该方法包括使用小型研磨机将难溶性化合物的粒度减小到约1μm或更小。在该过程中产生的产品是纳米颗粒模型物质的分散体,其包含一种或多种表面稳定剂,其被吸附到化合物的表面上。粒度的减小导致化合物的溶解度和/或分散性增加。因此,生
产相应纳米材料的最重要和最普遍的方法是所谓的纳米研磨法,该方法可以使用粘度增强剂进行,并且只有添加稳定剂才能实现。然而,因此,研磨过程并不总是成功,特别是如果研磨过程导致电荷的产生,这可能导致与未研磨的药物一样大甚至更大的小颗粒的聚集[lin s.-l.;menig j.;leon lachmann;“independence of physiological surfactant and drug particle size on the dissolution behavior of water-insoluble drugs”;j.pharm.sci.(1968);57(12);2143-8]。
[0027]
第二种可能性是通过较小的颗粒的聚集,例如通过超临界沉淀的自下而上的开发。在纳米颗粒药物载体的自下而上开发中,颗粒系统从分子分散型状态制备,并允许与固体颗粒的随后形成相关联。因此,自下而上的技术寻求将较小的组分排列成复杂结构的组件,例如通过超临界沉淀。
[0028]
然而,这里不考虑生产高表面积药物颗粒的这些特定方法,因为除其他外,它们的生产非常复杂且昂贵。
[0029]
用于研磨药物颗粒的一种经过充分研究的方法是湿磨。在此上下文中,使用珠磨机进行湿磨是降低api粒度的最有效方法之一。使用这种技术,大的药物晶体悬浮在研磨介质中。研磨介质由含有研磨珠和api的流体组成,api必须不溶于研磨介质。研磨珠需要表现出比待纳米研磨的药物更高的物理稳健性,并且通常必须针对高剪切力是稳定的。为了进行研磨,将由药物、水和稳定剂组成的粗浆料送入研磨室。在研磨室中,药物晶体受到研磨介质提供的高能量输入。该过程可以以分批模式或再循环方式运行。将api研磨至约200nm平均直径的典型停留时间在分批模式下在30至60分钟范围内[e.merisko-liversidge,g.g.liversidge,e.r.cooper,nanosizing:a formulation approach for poorly-water-soluble compounds,eur.j.pharm.sci.,18(2003)113-120]。然而,所需的时间框架是药物特有的,在其他情况下,可能需要数小时甚至数天才能达到所需的药物晶体尺寸[f.kesisoglou,s.panmai,y.wu,nanosizing-oral formulation development and biopharmaceutical evaluation,adv drug deliv rev,59(2007)631-644;j.u.junghanns;r.h.muller;“nanocrystal technology,drug delivery and clinical applications”;int j.nanomedicine,3(2008)295-309]。
[0030]
用于研磨的珠子的选择取决于它们在研磨过程中抗磨损的能力,磨损会导致不希望的产品污染。由玻璃或锆制成的珠子可能会经受住研磨过程,但即使使用这些珠子,也必须仔细考虑研磨珠碎片对产品的潜在污染。
[0031]
图1示意性地显示了用于执行湿珠纳米研磨工艺的可能程序和装置组件。用于此目的的相应设施是可商购的,并且甚至可以在单个装置中实现。
[0032]
纳米研磨的主要缺点是结晶api使用粘度增强剂以液体状态生产,这导致在储存过程中降低的稳定性和活性成分重结晶的趋势。因此,稳定是必须的。然而,后者只有在这种材料通过纳米研磨以悬浮液形式制备并且仅在添加稳定剂之后才有可能。
[0033]
通常,从湿磨悬浮液生产干燥材料的下一步是通过喷雾干燥或冷冻干燥、喷雾造粒或甚至通过在烘箱(有或没有真空)中的标准干燥来完成。[s.bose;d.schenck;i.ghosh;a.hollywood;e.maulit;c.ruegger“application of spray granulation for conversion of a nanosuspension into a dry powder form“;european journal of pharmaceutical sciences 47(2012)35
–
43]
[0034]
本发明的目的
[0035]
因此,纳米晶活性物质悬浮液的生产比活性化合物的微粉化和相应悬浮液的制备要求更高,因为当粒径低于微米范围时会出现一些挑战。传统的研磨方法如锤磨或喷射研磨由于其结构原理和物理限制,无法实现纳米尺寸的目标。
[0036]
因此,需要一种用于将相应的api研磨到纳米范围内并提供可重现的粒径的合适的制备方法。此外,还需要提供一种稳定所产生的纳米级药物颗粒的方法。此外,需要在纳米研磨步骤后不久将api悬浮液进一步加工成最终施用形式,并且此类制剂包含生产纳米级药物颗粒所需的所有添加剂。
[0037]
发明概述
[0038]
本发明提供了一种制备药物组合物的方法,该药物组合物具有药物活性成分(特别是但不限于属于bcs ii类和iv类并且通常是难溶性候选药物的api)的增强的生物利用度。所制备的本发明的药物组合物包含bcs ii类和iv类药物活性成分和药学上可接受的载体或赋形剂,并且为固体颗粒或粉末或颗粒剂形式。这些固体颗粒、粉末或颗粒剂可进一步填充到胶囊中或任选与添加剂一起压制成片剂。本发明进一步提供了制备本发明的药物组合物的方法,其特征在于如权利要求1-9中给出的特征。
[0039]
发明详述
[0040]
通常,已经发现,一旦颗粒尺寸减小到一微米以下,可能会发生由奥斯特瓦尔德熟化引起的团聚或甚至颗粒生长,但必须避免这种情况。在这里,在制备药物制剂的情况下,选择合适的稳定剂(即聚山梨醇酯或聚维酮)对于加工过的活性成分的稳定性要求至关重要。此外,必须根据经验评估稳定剂的类型及其与药物的比例。如文献所知,药物与稳定剂的重量比通常为20:1至2:1[merisko-liversidge e.;liversidge gg.;“nanosizing for oral and parenteral drug delivery:a perspective on formulating poorly-water soluble compounds using wet media milling technology”;adv drug deliv rev.(2011)63(6),427-4054]。太低的比率将导致颗粒的团聚,而当纳米分散体中的比率太高时,少量的所包含的药物将已经溶出。这将由于颗粒尺寸之间的不平衡导致增加的奥斯特瓦尔德熟化,由于其不同的颗粒表面曲率导致颗粒之间的质量重新分布。奥斯特瓦尔德熟化过程的基本方案是药物颗粒之间不均匀的尺寸分布导致较小颗粒的溶出和溶出的药物沉淀在较大的颗粒上。为了防止这种影响,生产工艺导致窄的粒度分布是重要的。
[0041]
原则上,生产难溶性api的稳定纳米悬浮液的主要方法有3种:喷射研磨法、使用珠磨机的湿磨法和高压均质化法。
[0042]
上面已经提到了湿磨法。在喷射研磨中,流体喷射研磨机使用流体(高压空气)的能量来实现药物粉末的超细研磨。在高压均质化法(hph)中,要粉碎的固体首先分散在合适的流体中,然后在压力下强制通过高压均质器的纳米尺寸孔径阀,这本质上是悬浮液以高流速通过然后突然经历突然的压力下降、湍流条件和气穴现象的瓶颈。
[0043]
如今,其中活性物质以微粉化形式或作为纳米颗粒存在的各种药物制剂可商购获得以供患者使用。下表列出了来自不同供应商的一些纳米级产品。
[0044]
商业化的纳米级药物在“advanced drug delivery reviews”中列出。这些药物是使用纳米颗粒技术制备的(来自[kesisoglou,f.;panmai,s.;wu,y.;advanced drug delivery reviews,volume 59,issue 7,30 july 2007,pages 631-644;“nanosizing
–
oral formulation development and biopharmaceutical evaluation]的修改列表)。此列表仅包括基于纳米颗粒的产品选择,但市场上还有更多产品。
[0045]
表1.商业化纳米级药物列表
[0046][0047]
[0048]
尽管此处讨论的尺寸减小技术方便且简单,但它们取决于药物物质的类型以及要微粉化或纳米化的颗粒有时是不合适且不利的。常规的尺寸减小方法常常已知具有某些典型的缺点,例如由于高能量需求而效率较低或由于药物的热和化学降解或最终产物的粒度分布不均匀而构成威胁。特别地,传统的研磨技术被认为是不受控制的过程,在控制尺寸、形状、形态、表面性质和静电荷方面存在局限性,并导致不均一的颗粒形状或甚至导致团聚的颗粒作为最终产品。为了克服这些限制并特别控制颗粒特性,已经开发了几种颗粒工程化技术并用于产生所需的颗粒尺寸和仔细控制颗粒特性。因此,尝试了不同的生产微粉化或纳米级药物颗粒的方法来减小水溶性差的药物的粒径,以增加它们的溶解度和溶出度,从而改善它们的生物利用度。
[0049]
将纳米研磨的api带入干燥阶段或最终制剂的一种解决方案是使纳米研磨的溶液和载体接触并结合。
[0050]
因此,已经发现在干燥阶段材料更容易处理,即使在用于直接压片方法中也是如此。
[0051]
因此,纳米研磨的api可以通过将不含稳定剂的悬浮液沉积在载体上来稳定。
[0052]
因此,在第一次尝试中,研究了通过冷冻干燥技术装载到二氧化硅颗粒上的纳米研磨的非诺贝特(纳米悬浮液)。在这项可行性研究中,测试了两种不同类型的二氧化硅材料。
[0053]
为此,将纳米研磨的活性药物成分(api)转移到载体上并通过首先制备包含api和颗粒载体的溶液的悬浮液然后通过冷冻干燥或标准干燥该悬浮液而进入干燥阶段。
[0054]
因此,通过在压片机上将接收的材料与已知的粘合剂一起(如果需要)压片,可以容易地将口服施用形式建立为最终制剂。
[0055]
因此,已经发现,在其干燥阶段,该材料更容易处理,并且甚至可以用于直接压片方法。然而,这也意味着纳米研磨的api可以很容易地通过支撑在载体上来稳定而不需要稳定剂,并且除了稳定之外,在不添加稳定剂的情况下对于应用的api还可以实现更高的保质期。
[0056]
下面给出的实施例显示了本发明的意想不到的有利特性和效果。实施例中使用的api在其化学性质(酸性或弱碱性)上不同:
[0057]
在这些实施例中
[0058]-测试api是非诺贝特和伊曲康唑
[0059]-纳米研磨的api悬浮液用稳定剂制备
[0060]-api的替代制剂是在没有任何稳定剂的情况下制备的
[0061]-将纳米研磨的api装载到载体(例如二氧化硅)上是通过
[0062]
浸渍法
[0063]
或通过
[0064]-纳米研磨的api二氧化硅悬浮液的冷冻干燥完成的
[0065]
储存样品用于比较,并检查纳米研磨后悬浮液的稳定性。
[0066]
如本文所用,术语“稳定的”是指物理稳定性,如稍后描述的测量粒度分布。
[0067]
通过使用机械方法在水性介质中湿磨悬浮液来实现所应用的药物(此处为非诺贝特和伊曲康唑,这些药物被例示为微溶模型物质)的尺寸减小。优选地,研磨在合适的球磨
机中进行。如上所述,研磨也可以在其他合适的研磨机中进行,条件是其中可以以期望的方式和在合适的条件下减小粒度。这种研磨机可以是例如喷射研磨机、介质研磨机,例如砂磨机、研磨机或珠磨机。这些研磨机中的研磨介质可以包括球形颗粒,例如不锈钢珠或氧化锆球。
[0068]
由于低可溶性活性成分的粒度减小优选在水分散体中进行处理,因此必须避免成分的漂浮以实现可靠的研磨结果。为了稳定分散体,可以根据要研磨的活性成分的性质添加各种物质。合适的稳定剂的例子包括但不限于明胶、酪蛋白、阿拉伯树胶(gum arabicum)、硬脂酸、硬脂酸钙、单硬脂酸甘油酯、脱水山梨糖醇酯、大粒凝胶醚如cetomacrogel 1000、聚氧乙烯蓖麻油衍生物、聚氧乙烯脱水山梨糖醇脂肪酸酯如聚氧乙烯硬脂酸酯、胶体二氧化硅、十二烷基硫酸钠、羧甲基纤维素钙、羧甲基纤维素钠、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素(hpmc)、聚乙烯吡咯烷酮(pvp)、泊洛沙姆如f 68和f 108、二辛基磺基琥珀酸钠(doss)、多库酯钠、十二烷基硫酸钠、20和80,以及聚乙二醇甘油酯,例如el。结合此处选择的模型物质,hpmc(羟丙甲纤维素)和doss已被证明是特别适合用于增强粘度和作为稳定剂的添加剂。
[0069]
下文公开的纳米研磨实例使用水性分散体进行。但取决于药物的性质,可能需要在另一种溶剂或溶剂混合物中进行纳米研磨。合适的液体的例子包括但不限于水、丙二醇、二丙二醇、聚丙二醇、乙二醇、聚乙二醇、甘油、丁二醇、己二醇、聚氧乙烯及其混合物。然而,优选地,研磨在水溶液中进行。
[0070]
在制备最终含活性成分的制剂的进一步过程中,可能需要在装载载体后添加其他添加剂,例如表面活性剂或抗氧化剂、防腐剂或片剂助剂,例如稀释剂、粘合剂、崩解剂润滑剂、助流剂。然而,在本发明的最优选实施方案中,不需要此类添加剂,特别是因为当使用此处实施例中使用的二氧化硅基载体时,装载活性成分后获得的自由流动粉末可以直接压制成片剂。如果需要添加合适的添加剂,本领域技术人员可以选择合适的添加剂。所制备的含有活性成分的制剂以固体颗粒的形式(如粉末或颗粒剂)获得,其可以填充到胶囊中或必要时与片剂助剂一起进一步加工,并压制成片剂。
[0071]
合适的表面活性剂的实例包括但不限于卵磷脂、脱水山梨糖醇单硬脂酸酯、由月桂酸、棕榈酸、硬脂酸和油酸制备的聚山梨醇酯、聚氧乙烯单酯如聚氧乙烯单硬脂酸酯(polyoxyethyl ethylene monostearate)、聚氧乙烯单月桂酸酯和聚氧乙烯单油酸酯、二辛基磺基琥珀酸钠、十二烷基钠硫酸盐和泊洛沙姆。
[0072]
合适的抗氧化剂的实例包括但不限于丁基羟基苯甲醚、丁基羟基甲苯、生育酚、棕榈酸抗坏血酸酯、抗坏血酸、焦亚硫酸钠、亚硫酸钠、硫代硫酸钠、没食子酸丙酯及其混合物。
[0073]
合适的防腐剂的实例包括但不限于对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯、苯甲酸钠、苯甲醇、山梨酸、山梨酸钾及其混合物。
[0074]
附图列表
[0075]
图1:示意性地显示了用于执行湿珠纳米研磨工艺的可能程序和设备组件。
[0076]
图2:显示ff_29062016_slc_500_001的dsc曲线,绘制在纯非诺贝特的dsc曲线旁
边。纯的结晶非诺贝特的吸热熔融峰在约80℃清晰可见
[0077]
图3:显示api释放(装载的二氧化硅载体+非诺贝特)的比较,50mg api,1000ml sgfsp+0.1%sds,75rpm,平均值[mg/l]+标准偏差[mg/l]
[0078]
图4:使用非诺贝特纳米悬浮液相对非诺贝特(结晶)获得的制剂的比较结果
[0079]
图5:在有机溶剂存在下装载无定形api(如前所述制备)相对通过纳米悬浮液装载(仍然是结晶api)获得的结果的比较
[0080]
图6:将装载的kieselgel si 5000批次(ff_29062016_si_5000_001)的纳米研磨颗粒的释放数据与第三批次的slc 500(ff_29062016_slc_500_003)的装载的纳米研磨悬浮液的数据进行比较。
[0081]
图7:施加到载体上的没有稳定剂的纳米研磨药物的溶出,其中slc 500和kieselgel si 5000在使用或不使用稳定剂时表现出非常相似的释放特性
[0082]
图8:显示结晶伊曲康唑的dsc曲线,以及slc 500和kieselgel si 5000的纳米悬浮液装载批次的曲线
[0083]
图9:显示批次icz_16092016_slc_2的结果,其以约3mg/l的最大浓度释放并显著快于比较的伊曲康唑结晶样品。
[0084]
下面通过不同的实验和实施例展示本发明。对这些实验的结果进行了详细解释、讨论和评估。这些附加的实施方案说明了本发明原理的一般适用性,因此,这些实施方案以及实施例包括在本发明的公开内容中。
实施例
[0085]
本说明书使本领域技术人员能够全面地应用本发明。即使没有进一步的评论,也假定本领域技术人员将能够在最广泛的范围内利用上述描述。
[0086]
实践者将能够通过常规实验室工作,使用本文的教导,在新工艺中制备包含如上文定义的制剂的活性成分。
[0087]
所描述的本发明可以通过以下实施例进一步说明,这些实施例仅用于说明目的并且无论如何不应被解释为限制本发明的范围。
[0088]
如果还有什么不清楚的地方,应理解应查询所引用的出版物和专利文献。因此,这些文件被视为本说明书的公开内容的一部分。
[0089]
为了更好地理解和说明本发明,下面给出一些实施例,这些实施例都在本发明的保护范围内。这些实施例还用于说明可能的变体。然而,由于所描述的本发明的原理的普遍有效性,这些实施例并不适合将本技术的保护范围仅缩小到这些实施例。
[0090]
此外,不言而喻,对于本领域技术人员来说,在给出的实施例和说明书的其余部分中,存在于组合物中的组分量基于整个组合物总和仅达到100%重量、体积或mol-%,并且不能超过这个值,即使在所示百分比范围内可能会出现更高的值。除非另有说明,否则%数据是重量%、体积%或mol-%,比率除外。
[0091]
实施例和说明书以及权利要求书中给出的温度始终以℃为单位。
[0092]
方法(见下文):
[0093][0094]
干燥失重(ir平衡)
[0095]
设备:mettler pm 400;mettler lp16(mettler toledo gmbh,gieβen,germany)
[0096]
重量:0.3g(最小)
[0097]
温度:105℃
[0098]
方法:0-100%
[0099]
constance:1位/10秒
[0100]
铝盘:me-13865
[0101]
测定次数:3
[0102]
理想情况下,干燥失重对于释放应低于1%。如果干燥失重较高,可能需要再次干燥。
[0103]
光学显微照片
[0104]
设备:light microscope zeiss stemi 2000-c(carl zeiss ag,oberkochen,germany),camera power shot a640(canon germany gmbh,krefeld,germany cold light lamp cl1500 eco(carl zeiss ag,oberkochen,germany)
[0105]
测量软件:axio vision rel.4.8(carl zeiss ag,oberkochen,germany)
[0106]
载玻片:76x26x1mm;iso 8037/1;边缘90
°
磨光,预清洁,无垫边(paul marienfeld gmbh&co.kg,lauda-germany)
[0107]
待测物质均匀分布在载玻片上,并调整照明条件和清晰度,直到达到所需的显示效果。
[0108]
差示扫描量热法
[0109]
设备:mettler toledo dsc 3+(mettler toledo,gieβen,germany),stare-excellence-software(mettler toledo,gieβen,germany)
[0110]
称重的量:对于40μl铝坩埚的2
–
4mg,对于100μl铝坩埚的30
–
40mg
[0111]
气氛:50.0ml/min n2[0112]
温度范围:25
–
350℃
[0113]
升温速率:见下文
[0114]
测定次数:至少2
[0115]
加热类型:
[0116]
程序1:(“40-100/5k”)
[0117]
以5k/min的加热速率将样品从30℃连续加热到120℃。
[0118]
程序2:(“40-100/30k”)
[0119]
以30k/min的加热速率将样品从30℃连续加热到120℃。
[0120]
程序3:(“40-60/10k_60iso_5min_60-90/2k_alu40_n2”)
[0121]
以2k/min的加热速率将样品从40℃连续加热至90℃,包括在60℃下5分钟的温度保持阶段。
[0122]
程序4:(“25-100/5k_(alu100_n-2)”)
[0123]
以5k/min的加热速率将样品从25℃连续加热到100℃。
[0124]
程序5:(“25-85/5k_85_5min_85-50/5k_(alu100_n-2)”)
[0125]
以5k/min的升温速率将样品从25℃连续加热到85℃,保温5min,以5k/min的冷却速率将样品从85℃冷却到50℃。
[0126]
活性成分的释放(sotax 1和2)
[0127]
设备:sotax 1和2,
[0128]
释放装置:sotax at 7smart(sotax ag,germany),光度计agilent 8453(agilent technologies,waldbronn,germany)
[0129]
容器数量:3或6
[0130]
方法:桨式(paddle)
[0131]
介质:sgfsp+0.1%十二烷基硫酸钠
[0132]
介质的量:1000毫升
[0133]
介质温度:37℃
[0134]
转速:75转
[0135]
持续时间:2小时
[0136]
采样时间:5、10、15、20、25、30、45、60、75、90、105、120分钟
[0137]
最后旋转:没有
[0138]
比色皿层厚:5mm
[0139]
波长:288nm
[0140]
活性成分剂量:50mg
[0141]
载药量:约16%
[0142]
过滤器去除站:gf/d 2.7μm
[0143]
样品体积:2.5ml
[0144]
每个样品都用自动取样器收集在试管中。然后通过hplc测定离线测量样品(参见通过hplc测定含量的方法)。
[0145]
通过hplc测定含量
[0146]
设备:hplc系统elite(hitachi europe gmbh,d
ü
sseldorf,germany)
[0147]
检测器:紫外检测器l-2400vwr hitachi(hitachi europe gmbh,d
ü
sseldorf,
germany)
[0148]
自动进样器:自动进样器l-2200vwr hitachi(hitachi europegmbh,d
ü
sseldorf,germany)
[0149]
柱子:125-4,100rp-18e(5μm)
[0150]
洗脱液:乙腈/milli-q水/三氟乙酸(700:300:1)
[0151]
洗涤液取样器:乙腈/milli-q-wasser(1:1)
[0152]
柱温箱-温度:50℃
[0153]
进样量:25μl
[0154]
波长-检测器:288nm
[0155]
流速:2.0ml/min(等度)
[0156]
持续时间-运行:5分钟
[0157]
序列:xxxxxxxx_01_pk_fenofibrate_disso_nanosuspensionen 1.seq
[0158]
方法:xxxxxxxx_01_pk_fenofibrate_method
[0159]
过滤器
–
样品制备:whatman
tm anotop
tm 10,0.02μm,cat.-no.:6809-1002
[0160]
在填充到小瓶中之前,首先使用带有luer-lock连接的注射器和用于样品制备的上述过滤器过滤来自释放的每个样品,以保留纳米悬浮液的任何颗粒并消除系统误差。这种系统误差可以在大多数科学论文和专利中发现,因为大多数评估没有通过使用适当的过滤器从样品中仔细去除仍然纳米研磨的颗粒。只应检测可溶性api含量。
[0161]
为了评估hplc结果,饱和浓度通过固定的实验室方法确定,并进行结晶非诺贝特从之前进行的实验室测试(在线测定)的释放。
[0162]
通过h
1-nmr测定含量
[0163]
方法:1h-nmr光谱
[0164]
条件:dmso-d6
[0165]
测量模式:含量[%]
[0166]
测量由外部分析命令进行。为此,将样品管装满一半并送到适当的地方。结果以%含量给出。
[0167]
粒度测定(zetasizer nano zs)
[0168]
设备:zetasizer nano zs(malvern instruments ltd,herrenberg,germany)
[0169]
量:几滴(稀释的,不混浊的溶液)
[0170]
分散介质:淡化水(粘度:0,8872cp)
[0171]
测量范围:0.3nm
–
10μm
[0172]
测量时间:几分钟
[0173]
温度:25℃
[0174]
平衡时间:60秒
[0175]
测量次数:6x12次测量
[0176]
测量方法:尺寸测量(数字)
[0177]
测量角度:173
°
反向散射(nibs默认)
[0178]
活性成分:非诺贝特(ri:1,547*;吸收:0.01)
[0179]
数据处理:通用
[0180]
比色皿类型:dts0012
–
一次性尺寸化比色皿
[0181]
比色皿:10x10x45mm聚苯乙烯/聚苯乙烯(polystyrol/polystyrene)(ref:67.754;sarstedt ag&co,numbrecht,germany)
[0182]
评估:形成至少3次测定的平均值
[0183]
(*来源:http://www.lookchem.com/fenofibrate/)
[0184]
将样品装入比色皿(最好是40μl比色皿)直至zetasizer的标记并进行测量。如果结果不是“好的”(见“专家建议”),用更稀的样品重复测量。为了利用zetasizer的最佳工作范围,样品应最多略微混浊。
[0185]
一般信息“纳米研磨”[0186]
湿磨:
[0187]
研磨将在-mill研究实验室(willy a.bachofen maschinenfabrik,muttenz,switzerland)进行
[0188]
填充体积:60
–
200毫升
[0189]
研磨球:sili zyp 0.2
–
0.3mm
[0190]
研磨球重量:200克
[0191]
搅拌速度:2000
–
4000rpm
[0192]
冷却液温度:-10℃
[0193]
装载两种不同孔径的载体slc 500/kieselgel si 5000
[0194]
设备:heidolph rzr 2102控制实验室搅拌器(heidolph instruments,schwalbach,germany),带搅拌叶片的头部搅拌器称重(二氧化硅):约10克
[0195]
称重(悬浮):约10克(和约20克)
[0196]
搅拌速度:70rpm
[0197]
套管类型:0.80x120mm bl/lb
[0198]
给药速度:约2g/min
[0199]
slc 500或kieselgel si 5000通过使用带有luer-lock盖和套管的10ml注射器均匀施加纳米悬浮液来装载。载体材料位于搅拌器直接装入其中的烧杯中。在施加期间,用搅拌器继续搅拌。如果载体材料的混合不完全,可以手动改变搅拌器的高度和浸入深度(例如,通过提升/降低烧杯)。
[0200]
其他设备:
[0201]
磁力搅拌器:-werke gmbh&co.kg,staufen,germany
[0202]
表1:材料:
[0203][0204]
slc 500是一种硅胶,具有500m2/g(bet测量)的比表面积和6nm的平均孔径。
[0205]
kieselgel si 5000来自merck kgaa的二氧化硅合成(通过使用nacl的添加和熔化来改变载体的孔径)。它具有3m2/g(bet测量)的比表面积和500nm的平均孔径。
[0206]
为了稳定纳米悬浮液,将hpmc和doss添加到悬浮介质中。在没有这些稳定剂的情况下,所生产的非诺贝特纳米悬浮液可能会由于大大增加的表面效应例如静电吸引力和溶出速率(奥斯特瓦尔德熟化)而易于快速形成聚集体和积聚较大颗粒。
[0207]
sgfsp:没有(sine)胃蛋白酶的模拟胃液
[0208]
sds:十二烷基硫酸钠
[0209]
实验的原因:
[0210]
以下实验的目的是确定当活性成分以纳米悬浮液的形式应用(其中api悬浮为纳米颗粒但仍处于结晶状态)时,通过浸渍装载法是否实现微溶活性成分的释放的改善。用于此目的的载体是slc 500和非诺贝特用作活性成分。
[0211]
此外,kieselgel si 5000用作支持材料并装载结晶非诺贝特纳米悬浮液。在此,研究了孔径对非诺贝特释放的影响。
[0212]
在hplc分析中最重要的是样品的适当过滤。在填充到小瓶中之前,首先使用带有luer-lock连接的注射器和用于样品制备的上述过滤器过滤来自释放的每个样品,以保留纳米悬浮液的任何颗粒并消除系统误差。这种系统误差可以在大多数科学论文和专利中发现,因为大多数评估没有通过使用适当的过滤器从样品中仔细去除仍然纳米研磨的颗粒。只应检测可溶性api含量。
[0213]
进行实验:
[0214]
在实验开始时,制备用hpmc和doss(二辛基磺基琥珀酸钠)稳定的非诺贝特悬浮液(参见实验1a的方法),然后进行纳米研磨(参见下面的纳米研磨方法)。获得的纳米悬浮液
在2至8℃的温度下储存在冰箱中。
[0215]
使用浸渍方法(如以下实施例中所述),载体slc 500和kieselgel si 5000以1:1或2:1(w/w)的比例的装载非诺贝特纳米悬浮液,找出最佳装载比,但在不影响装载量的情况下,看看是否也可以进行更高的装载。然后通过冷冻干燥进行干燥(参见下文中的“纳米研磨”方法)。由于slc 500具有吸湿性,所有生产的批次都储存在干燥器中,置于橙色凝胶上。
[0216]
表2:溶出度测量实验和比较实验的概述
[0217][0218]
方法
[0219]
实验1
[0220]
a)制备悬浮液
[0221]
将5g hpmc和0.2g doss放入烧杯中,通过用磁力搅拌器搅拌约40分钟使其溶解在154.88g去离子水中。当物质溶解时,将80.0g收到的溶液置于烧杯中,加入20g非诺贝特并通过搅拌(450rpm)10分钟悬浮在溶液中。
[0222]
理论非诺贝特含量:20%(w/w)
[0223]
a')饱和浓度的测定(用于分析测量)
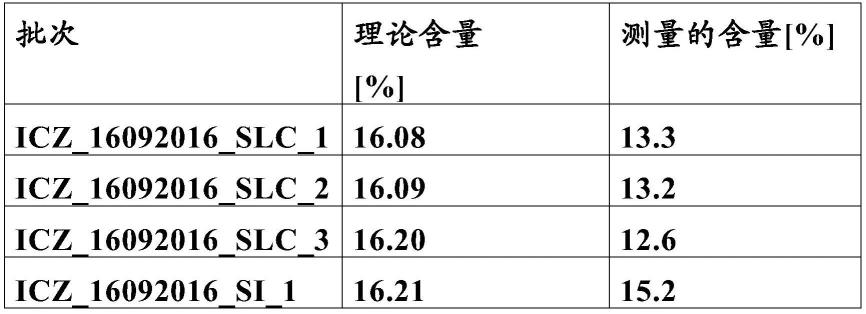
[0224]
为了制备饱和溶液,这里也使用了-werke gmbh&co.kg,staufen,germany的磁力搅拌器。
[0225]
为制备饱和非诺贝特溶液(测定饱和浓度所需),将1汤匙非诺贝特悬浮在200ml溶液(sgfsp+1%sds)中。将该悬浮液加热至40℃的温度并以300rpm搅拌至少24小时,此处为72小时。在此期间,烧杯被封口膜(parafilm)覆盖。随后,通过hplc测定测量饱和浓度。
[0226]
方法
[0227]
纳米研磨
[0228]
将悬浮液(实验1a)填充到研磨机的料斗中,并以2000rpm开始研磨过程。通过动态
[0243][0244]
将样品放入冷冻干燥机进行冷冻干燥,并关闭冷冻干燥机。
[0245]
水用于冷却(首先打开排水管,然后才打开入口!)。然后启动所需的程序。冷冻干燥后,产品的干燥失重应如所述地确定。如果干燥不充分,则进行进一步干燥
[0246]
d)分析所得产品
[0247]
a)dsc/xrd研究:检查api的物理状态
[0248]
b)hplc/nmr研究:测定二氧化硅上的药物含量
[0249]
c)释放-》离线,样品经过0.2μm ptfe过滤器(与加入二氧化硅前的纳米悬浮液比较)
[0250]
d)稳定性研究
[0251]
表6:生产具有以下量的批次:
[0252][0253]
实验2
[0254]
(无粘度增强剂的纳米研磨)
[0255]
a)制备悬浮液
12.98%,因此该批次在程序2“nanosus_pk”中再次干燥。
[0283]
干燥后的干燥失重为-1.01%。
[0284]
结果:光学评估
[0285]
ff_29062016_slc_500_001
[0286]
用非诺贝特纳米悬浮液装载slc 500后,支持材料略微结块。冷冻干燥后,这些团块仍然存在。但它们很容易用抹刀压碎。在此上下文中的一个影响因素可能是装载期间的计量速率。因为通过手动配料,所以配料率可能在此发生波动。其余为松散粉末,其具有良好的稠度(fine consistency)。
[0287]
ff_29062016_slc_500_002
[0288]
此外,在第二批次中,slc 500与非诺贝特纳米悬浮液的装载导致形成更小的团块。但是,相对于第一批次的那些,它们更小。这些团块也可以很容易地用抹刀压碎。
[0289]
ff_29062016_slc_500_003
[0290]
与其他批次的slc 500的装载一样,浸渍后会形成一些团块,其可以很容易地用抹刀压碎。
[0291]
在光学显微镜下,所有纳米研磨装载的slc 500颗粒只能识别为模糊结构。既不可识别所施加的纳米悬浮液也没有形成可见的非诺贝特晶体,证明分布在载体上的纳米研磨颗粒的成功装载。
[0292]
ff_29062016_si_5000_001
[0293]
与slc 500的装载期间类似,kieselgel si 5000的装载产生一些具有不同尺寸的团块。但与装载的slc 500相比,此处剩余的松散粉末呈粉状。
[0294]
ff_29062016_slc_500_001_a和进一步的实验(=20g装载)
[0295]
ff_29062016_slc_500_001_a和用于比较的装载20g纳米研磨悬浮液的其他样品的光学评估导致与10g装载量相似的粉末性质。材料部分结块在一起,但样品很容易转移到可流动的粉末上,这对于进一步加工如压片很重要。基于光学评估,我们对仅装载10g悬浮液的材料进行分析评估。
[0296]
纳米悬浮液的粒度分布
[0297]
如上所述,粒度分布是用zetasizer nano sz测量的。储存样品用于比较,并检查纳米研磨后悬浮液的稳定性(表7)。
[0298]
表7:
[0299][0300]
由于纳米悬浮液的颗粒的大大增加的比表面积,溶解过程在这种悬浮液中可能会加速,从而使非诺贝特溶出得更快。这可导致较大颗粒的生长,而较小的颗粒则完全溶出。这种效应称为奥斯特瓦尔德熟化。
[0301]
但在这个实验中,该效果是小的。这意味着悬浮晶体的生长缓慢,并且可以在储存10周后看到。在2周内,它们平均在可接受的范围内生长,以在较长时间内使用纳米悬浮液。因此,通过使用hpmc和doss,纳米悬浮液得到了足够好的稳定性。
[0302]
通过h
1-nmr测定含量
[0303]
表8:h
1-nmr-api含量评估显示以下值:
[0304][0305][0306]
测量的含量比理论含量多至低3.5%。这种偏差可能有很多原因:例如,在纳米研磨过程中,当悬浮液转移到纳米研磨机时,非诺贝特含量可能已经下降。转移完成时,运输容器中始终留有少量剩余悬浮液。有可能的是,尽管在振荡,但一定量的非诺贝特晶体已沉淀在那里,其在倾倒过程中仍留在容器中。由于所有批次具有减少的含量,因此可能是系统
误差。
[0307]
差示扫描量热法(api形态的评估)
[0308]
(dsc测量)
[0309]
表9:评估是否无定形/结晶形态:
[0310][0311]
参考结果在dsc测量的相应子项下找到。
[0312]
非诺贝特的熔融峰的鉴定
[0313]
图2显示了ff_29062016_slc_500_001的dsc曲线,绘制在纯非诺贝特的dsc曲线旁边。纯的结晶非诺贝特的吸热熔融峰在约80℃清晰可见。可以看出,非诺贝特纳米悬浮液在slc 500上的熔融峰显著地不太明显,并转移到较低的温度。虽然纯非诺贝特的非诺贝特峰急剧上升,但带电的slc 500更有可能在曲线上只有“凹痕”。在slc 500上它仍然是结晶的。
[0314]
ff_29062016_slc_500_001的dsc曲线也显示非诺贝特的熔点略有下降。由于样品中没有纯非诺贝特,使用的羟丙基甲基纤维素和doss都可以降低熔点。
[0315]
此外,可以发生通过slc 500的热传递的“屏蔽”,从而隐藏明确的、清晰的熔融峰。
[0316]
溶出度测量:装载的slc 500的批次
[0317]
图3显示了api释放的比较(装载二氧化硅载体+非诺贝特),50mg api,1000ml sgfsp+0.1%sds,75rpm,平均值[mg/l]+标准偏差[mg/l]。
[0318]
在释放研究中,slc 500批次1和3表明非诺贝特纳米悬浮液类似好地释放活性物质。两者都在仅5分钟后达到约15mg/l的饱和浓度,这明显更快,因为它通过溶出纯结晶非诺贝特进行。结晶非诺贝特在60分钟后达到饱和浓度。总体而言,slc 500的纳米悬浮液以及结晶活性成分仅略微超过了饱和浓度。正如预期的那样,(因为api的形态被测量为仍然是结晶的而不是无定形的)与未装载在载体上的非诺贝特相比,纳米微粒api装载的slc500的溶解度没有改善。在装载在载体上的纳米悬浮液和纯api之间的比较中非常有利的是装载的slc 500批次在开始时具有显著提高的溶出速率。
[0319]
图4显示了使用非诺贝特纳米悬浮液相对于非诺贝特(结晶)获得的结果的比较。
[0320]
通过在有机溶剂存在下装载无定形api(如前所述制备)相对通过纳米悬浮液(仍然是结晶api)装载获得的结果的比较(图5)
[0321]
与装载有纳米悬浮液(由于物理研磨api仍然是结晶的)的slc 500相
比,有机装载的slc 500(api以无定形形态装载)的释放显示出浓度的明显更高的初始增加(所谓的过饱和)。15分钟后达到约为47mg/l的最大值。随后是在90分钟后浓度渐近逼近25mg/l。与有机装载相比,两个样品纳米悬浮液装载的slc 500在释放后5分钟内达到其最大浓度。达到最大值后,浓度保持不变,与有机装载相反;该最大值略高于非诺贝特的饱和浓度。
[0322]
有机装载的slc 500的最大浓度为47mg/l;纳米颗粒装载的slc 500的最高释放浓度仅为25mg/l。然而,在此上下文中,积极的是没有再结晶,其中装载纳米研磨的非诺贝特的slc 500的浓度降低。
[0323]
在大多数科学论文和专利中都可以发现系统性误差,因为大多数评估在分析评估期间没有通过使用适当的过滤器从样品中仔细去除仍纳米研磨的颗粒。只应检测可溶性api含量,否则检测的api浓度高于api在结晶状态下的溶解度,因此通常会得出错误的结论。
[0324]
在图6中,将装载纳米研磨颗粒的kieselgel si 5000批次(ff_29062016_si_5000_001)的释放与装载相同量的纳米研磨悬浮液的第三批次slc 500(ff_29062016_slc_500_003)进行了比较。
[0325]
从这些比较测量得出的结论
[0326]
实验的目的是验证模型药物非诺贝特通过纳米研磨的释放,随后将悬浮液装载到slc 500上(测量孔径约为6nm),并将其溶出特性与相同程序装载的kieselgel si 5000载体(孔径在500nm范围内)进行比较。
[0327]
通过该实验发现,使用施加到kieselgel si 5000载体上的非诺贝特纳米悬浮液具有比结晶的微粉化非诺贝特显著更快的溶出速率。与装载的slc 500相比,没有观察到过饱和或更快的溶出,使用kieselgel si 5000装载的样品甚至可以测量稍微更小的总溶出。
[0328]
api的稳定和释放似乎是由slc 500和kieselgel si 5000的表面和孔隙性质造成的,而不是由孔径造成的。
[0329]
进一步的实验和测量必须证实这些结果,例如使用伊曲康唑。
[0330]
不添加稳定剂的进一步实验
[0331]
在以前的研究中,hpmc和doss已被用作纳米悬浮液生产的稳定剂和粘度增强剂。为了测试是否可以制备没有额外的稳定剂的纳米研磨的装载的载体的有利材料,以便能够在最终施用形式中使用这种不太复杂的材料,将纳米悬浮液装载到slc 500和kieselgel si 5000并研究释放曲线。
[0332]
如前所述,制备非诺贝特混悬液但不添加作为稳定剂的doss。然后对所得悬浮液进行纳米研磨。获得的纳米悬浮液在2至8℃的温度下储存在冰箱中。
[0333]
载体slc 500和kieselgel si 5000使用浸渍法各自以1:1(w/w)的比例装载非诺贝特纳米悬浮液。随后,通过冷冻干燥进行干燥。由于slc 500具有吸湿性,所有生产的批次都储存在干燥器中,置于橙色凝胶上。
[0334]
然后测定样品中活性成分的释放和样品的干燥失重。
[0335]
对于这些测量,制备的样品的理论非诺贝特含量为约18.0%(w/w)。
[0336]
载体的装载按照“slc 500/kieselgel si 5000的装载”中的描述进行。
[0337]
在这里,使用slc 500进行三个装载和一个kieselgel si5000装载,始终使用不添加稳定剂制备的纳米悬浮液(表10)。
[0338]
生产具有如下重量的批次:
[0339]
表10:
[0340][0341]
结果:
[0342]
无稳定剂的纳米悬浮液的制备
[0343]
制备不含稳定剂doss或粘度增强剂hpmc、仅含非诺贝特和milliq水的纳米悬浮液只可能在短研磨时间下进行,因此建立商业化工艺可能是不可行的。它涉及药物的漂浮和研磨机堵塞。添加hpmc(羟丙甲纤维素=pharmacoat 603)允许纳米研磨,甚至悬浮液与添加doss相比稍微多一些地起泡沫。
[0344]
无任何稳定剂的纳米研磨的称重:
[0345]
20.03g 非诺贝特
[0346]
80.0g
ꢀꢀ
milliq水
[0347]
使用hpmc的纳米研磨的称重:
[0348]
19.99g 非诺贝特
[0349]
2.52g
ꢀꢀ
hpmc/pharmacoat 603
[0350]
77.51g milliq水
[0351]
干燥失重
[0352]
表11:
[0353][0354]
样品的干燥失重刚好超过1%。样品的干燥失重不是在冷冻干燥后直接测定的,而是在几天后测定的。因此,可以假设,尽管在干燥器中储存,干燥失略有增加。由于这些值低于3%的自加标记,因此没有对样品进行后续干燥。
[0355]
没有装载在载体上的稳定剂的纳米研磨的非诺贝特的释放
[0356]
与结晶药物(未研磨)相比,没有施加至载体的稳定剂的纳米研磨的药物的溶出更好。非诺贝特纳米研磨(不含稳定剂)装载的作为载体的slc 500和kieselgel si 5000能够实现与纯结晶药物相比更快的释放。在片剂或胶囊剂的最终施用形式中使用不含稳定剂的制剂是有利的,因为在开发或临床阶段无需考虑稳定剂对api的额外影响或干扰。迄今为止报道的api纳米研磨制剂含有稳定剂,导致更复杂的施用形式,而不易预测添加剂的影响。
[0357]
没有施加至载体如slc 500和kieselgel si 5000的稳定剂的纳米研磨药物的溶出显示出在具有或不具有稳定剂的情况下非常相似的释放特性(图7)。在这两种情况下,纳米研磨的药物装载的载体都显示出比纯结晶药物更快的药物释放。
[0358]
总之,发现在不添加任何稳定剂的情况下进行纳米研磨是可能的,但是,活性物质漂浮在顶部并且没有获得进一步的可使用(workable)的纳米悬浮液。通过添加少量的hpmc,可以防止“漂浮”,并且可以生产纳米悬浮液。在所有情况下,与即使在测试的2小时后也达不到最大可能溶解度的纯结晶药物样品相比,装载在载体上的纳米研磨药物样品的释放更快(大约10分钟)。
[0359]
与具有伊曲康唑(弱碱的代表实例)作为活性成分的纳米悬浮液的比较
[0360]
与之前的实验一样,这里制备了伊曲康唑纳米悬浮液,将其应用于slc 500。研究是否可以通过应用纳米悬浮液来改善释放。
[0361]
将所得结果与非诺贝特纳米悬浮液的结果进行比较。
[0362]
相比之下,kieselgel si 5000装载有结晶伊曲康唑纳米悬浮液。在此,将研究孔径对伊曲康唑纳米悬浮液的释放的影响。此外,目标是验证装载伊曲康唑的载体与装载非诺贝特的载体所获得的分析结果,以确认不同api的api纳米研磨的装载载体的释放比没有研磨的结晶api更快且与孔径无关的结论。
[0363]
程序
[0364]
在实验开始时,制备伊曲康唑悬浮液,用羟丙基甲基纤维素(hpmc)和doss稳定,然后进行纳米研磨。获得的纳米悬浮液储存在2至8℃的冰箱中。
[0365]
使用浸渍法,载体slc 500和kieselgel si 5000以1:1(w/w)的比例装载伊曲康唑纳米悬浮液。干燥在冷冻干燥中进行(参见程序“nanosus_pk”)。由于程序“nanosus_pk”中的干燥不充分,再次干燥(程序:nanosus_pk_修改的)。由于slc 500的吸湿性,生产的所有批次都储存在干燥器中,置于橙色凝胶上。
[0366]
表12:批次概览
[0367][0368]
进行的测量
[0369]
所有批次干燥失重差示扫描量热法释放通过hplc测定含量通过h
1-nmr测定含量
[0370]
以与前述相同的方式和相同的设备和手段进行测量和确定。
[0371]
表13:
[0372]
使用的材料
[0373][0374]
为了稳定纳米悬浮液,将羟丙基甲基纤维素(hpmc)和doss添加到悬浮介质中。可以预期,在没有doss作为稳定剂的情况下,所生产的伊曲康唑纳米悬浮液可能会由于大大
增加的表面效应(例如静电吸引力)和溶出速率(奥斯特瓦尔德熟化)而迅速聚集并形成更大的颗粒。
[0375]
1.)
[0376]
伊曲康唑悬浮液的制备:
[0377]
伊曲康唑
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
19.870克
[0378]
hpmc 2.496克
[0379]
doss
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.500克
[0380]
milli-q水
ꢀꢀꢀꢀꢀꢀꢀꢀ
77.020克
[0381]
设备:磁力搅拌器(-werke gmbh&co.kg,staufen,germany)
[0382]
转速:600rpm
[0383]
温度:关
[0384]
其他:搅拌鱼(stirring fish)、烧杯、抹刀
[0385]
将hpmc和doss称重到vwr螺旋盖玻璃(250ml)中,并补充milli-q水至80.016g(称重,见上文)以制备用于纳米研磨的伊曲康唑悬浮液。将混合物用磁力搅拌器和搅拌鱼搅拌约2小时直至完全溶解。然后关闭螺旋罐。
[0386]
第二天,在研磨前不久,称重伊曲康唑。在搅拌下,加入伊曲康唑并悬浮约10分钟。随后,在纳米研磨机中研磨悬浮液。
[0387]
理论伊曲康唑含量:19.89%(w/w)
[0388]
2.)
[0389]
在sgfsp中制备饱和伊曲康唑悬浮液:
[0390]
伊曲康唑
ꢀꢀꢀꢀꢀ
约200mg
[0391]
sgfsp
ꢀꢀꢀꢀꢀꢀꢀꢀ
100ml
[0392]
设备:磁力搅拌器(-werke gmbh&co.kg,staufen,germany)
[0393]
转速:450rpm
[0394]
温度:40℃(恒温器)
[0395]
其他:搅拌鱼、烧杯、汤匙
[0396]
时间:72小时
[0397]
将约200mg伊曲康唑加入100ml sgfsp中,并在40℃下以450rpm悬浮72h。在此期间,烧杯用螺丝拧紧(screwed)。随后,通过hplc测量饱和浓度。
[0398]
纳米研磨
[0399]
设备:-mill research lab(willy a.bachofen ag-maschinenfabrik,muttenz,switzerland)
[0400]
称量:100克
[0401]
时间:30分钟
[0402]
研磨球:55.0ml氧化锆球(sili zyp 0.2
–
0.3mm,sigmund lindner gmbh,warmensteinach,germany)
[0403]
温度:-2℃(低温恒温器)
[0404]
转速:3000upm
[0420][0421]
将样品放入冷冻干燥机进行冷冻干燥,并关闭冷冻干燥机。
[0422]
水用于冷却(首先打开排水管,然后才打开入口!)。然后启动所需的程序。冷冻干燥后,产品的干燥失重应按描述确定。如果干燥不充分,则进行进一步干燥。
[0423]
由于程序1干燥后所有批次的干燥失重约为-10%,因此再次干燥。程序3:nanosus_pk_修改的用于此目的。在重复干燥后,再次测量干燥失重并且低于3%。此后,kieselgel si 5000的干燥失重仅为-2%。
[0424]
如上所述测定干燥失重。理想情况下,干燥失重应低于3%以用于释放。如果干燥失重较高,则可能需要进一步干燥。
[0425]
差示扫描量热法如前所述但根据程序1进行:
[0426]
方法:程序1:(“25-200/5k_(alu100_n-2)”)
[0427]
将样品以5k/min的加热速率从25℃连续加热到200℃
[0428]
使用与之前应用相同的设备确定活性成分的释放(sotax 1 and 2;freisetzungsapparatur sotax at 7smart(sotax ag,germany)
[0429]
sgfsp用作介质,释放测定在225nm波长下进行。每个样品都用自动取样器收集在试管中。随后,通过hplc离线测定样品的含量。有利地,纳米研磨装载的样品不会像结晶活性物质那样漂浮在释放介质上,而是更好地润湿。
[0430]
通过hplc测定含量
[0431]
设备:hplc系统elite(hitachi europe gmbh,d
ü
sseldorf,germany)
[0432]
检测器:紫外检测器l-2400 vwr hitachi(hitachi europe gmbh,d
ü
sseldorf,germany)
[0433]
自动进样器:自动进样器l-2200 vwr hitachi(hitachi europe gmbh,d
ü
sseldorf,germany)
[0434]
柱子:chromolith performance rp-18e 100
–
4.6mm(ob1108048)
[0435]
洗脱液:乙腈/tbahs-缓冲液(1.7g/l)/甲醇(450:450:100)
[0436]
洗涤液-进样器:乙腈/milli-q-wasser(1:1)
[0437]
柱温箱-温度:25℃
[0438]
进样量:15μl
[0439]
波长-检测器:225nm
[0440]
流速:2.0ml/min(等度)
[0441]
持续时间-运行:7分钟
[0442]
序列:xxxxxxxx_01_pk_____itaconazole_disso_nanosuspensionen 1.seq
[0443]
方法:xxxxxxxx_01_pk__itaconazole_method
[0444]
过滤器
–
样品制备:whatman
tm anotop
tm 10,0.02μm,cat.-no.:6809-1002
[0445]
在填充到小瓶中之前,首先使用带有luer-lock连接的注射器和用于样品制备的上述过滤器过滤来自释放的每个样品,以保留纳米悬浮液的任何颗粒并消除系统误差。
[0446]
为了评估hplc结果,测定饱和浓度并获取来自实验的结晶伊曲康唑的释放。
[0447]h1-nmr:
[0448]
方法:1h-nmr光谱
[0449]
条件:dmso-d6
[0450]
测量方式:含量[%]
[0451]
结果:
[0452]
纳米悬浮液的粒度分布
[0453]
粒度分布用zetasizer nano sz测量。
[0454]
在制备纳米悬浮液后立即测量粒度。
[0455]
颗粒尺寸在较长时间内几乎保持不变,悬浮液中的晶体生长非常缓慢。这意味着纳米悬浮液通过使用hpmc和doss得到了足够好的稳定性。
[0456]
生产的批次的光学评估
[0457]
icz_16092016_slc_1
[0458]
用伊曲康唑纳米悬浮液装载slc 500后,支持材料略微结块。冷冻干燥后,这些团块仍然存在。它们很容易用抹刀分开。其余的松散粉末具有良好的稠度。颜色为与起始物质相同的白色。
[0459]
icz_16092016_slc_2
[0460]
slc 500的装载还导致在第二批次中形成较小的团块。与第一批次一样,可以用抹刀轻松压碎团块。
[0461]
icz_16092016_slc_3
[0462]
与其他批次一样,slc 500也有一些团块,其在浸渍后可以很容易地用抹刀压碎。
[0463]
icz_16092016_si_1
[0464]
与slc 500的装载类似,kieselgel si 5000的装载产生一些不同大小的团块。与slc 500相比,松散的剩余粉末呈粉状。
[0465]
通过h
1-nmr测定含量
[0466]
表17:
[0467]
对不同批次的样品的外部h
1-nmr含量测定显示以下活性成分含量:
[0468][0469]
dsc测量
[0470]
表18:物理状态无定形/结晶
[0471][0472][0473]
图8显示了结晶伊曲康唑的dsc曲线,以及slc 500和kieselgel si 5000的纳米悬浮液装载的批次的曲线。清晰可见从约166℃开始的纯伊曲康唑的吸热熔融峰。slc 500和kieselgel si 5000上的纳米悬浮液批次的熔融峰更加不明显,并转移到较低的温度。代替伊曲康唑的尖锐熔融峰,较早开始的装载批次出现加宽的熔融峰,这意味着在约155℃。
[0474]
由于纳米悬浮液不仅仅是纯伊曲康唑,使用的羟丙基甲基纤维素和doss都可导致熔点降低。
[0475]
此外,通过使用的二氧化硅支持物可以发生热传递的“屏蔽”,从而覆盖明确的、清晰的熔融峰。活性成分在所有样品中均以结晶形式存在。
[0476]
装载在不同载体上的活性成分伊曲康唑的释放
[0477]
图9显示了批次icz_16092016_slc_2的结果,其以约3mg/l的最大浓度释放,并比伊曲康唑结晶样品显著更快。由于分析评估的挑战,饱和溶解度超过约0.5mg/l。
[0478]
当结晶伊曲康唑漂浮在释放介质上时,装载的slc 500批次的所有样品在添加后迅速落到容器底部。只有一小部分样品漂浮在介质表面上。
[0479]
装载在不同载体上的纳米研磨的伊曲康唑的分析结果支持了之前报道的非诺贝特纳米研磨装载载体的评估。在所有情况下,装载在不同载体上的api(api特性的代表)都导致与纯结晶api相比显著更快的释放。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1