抑制血管新生因子的化合物及其用途的制作方法
1.本发明涉及用于抑制血管新生因子的化合物及其用途。此外,本发明涉及使用所述化合物治疗血管新生性疾病的方法。
背景技术:
2.血管新生是一种生理现象,其中,在先前存在的血管周围形成新的血管,并在发生异常时可引起疾病或紊乱。本技术中提到的血管新生包括血管生成(angiogenesis),其中,从先前存在的血管生长出新的血管,这两个术语可互换使用。
3.例如,癌细胞通过过量产生血管新生因子在过度增长的癌组织中形成微血管,并且癌细胞的生存力通过经由所述微血管供给氧和养分得以提高。此外,已知眼部结构中的血管新生因子过量产生引起各种眼部疾病。
4.治疗这种血管新生性疾病的现有研究主要集中在遏制vegf上,vegf是主要的血管新生因子[1,2]。具体而言,抗vegf疗法基于以下三种途径:首先是使用vegf配体特异性抗体(例如,贝伐单抗、阿柏西普、雷珠单抗)以防止vegf配体与vegf受体结合的方法;使用vegf受体特异性抗体(例如雷莫芦单抗)的方法;或给予抑制vegf受体的酪氨酸激酶抑制剂(tki;索拉非尼、舒尼替尼、帕唑帕尼)的方法。
[0005]
其中,使用抗体的途径存在抗体的生理局限性的问题[3]。尽管随着其分子量的增加,抗体的药代动力学特性得到改善,但由于血管通透性降低,其存在组织渗透能力降低的问题。单克隆抗体具有改善的药代动力学特性,但组织渗透能力劣化。诸如单链可变片段(scfv)等抗体片段具有改善的组织渗透能力,但药代动力学特性劣化。因此,大多数抗体疗法依赖于注射给药的方法,使得药物直接作用于异常组织,但非侵入性方法(例如口服给药和透皮递送)是不可能的。
[0006]
此外,通过抗体直接遏制vegf可能会导致副作用[4]。由于vegf是具有多种功能的生长因子,当vegf抑制剂全身起效时,它可能会影响组织的正常生长。因此,当抗vegf疗法未直接给予异常组织或者引起的治疗没有作用于异常组织时,抗vegf疗法存在安全性严重恶化的问题。由于这些缺点,尽管抗vegf疗法目前是主流疗法,但其局限性很明显,因此,需要研究经由新机制而非vegf抑制的疗法。
[0007]
已知trap-1对hif-1α进行调节,hif-1α是vegf的亲代调节物(parent moderator)之一。trap-1是hsp90的旁系同源物之一,hsp90的抑制剂已在相关领域中得到了积极研究,但是这种抑制剂在hsp90及其旁系同源物上非选择性地起效,因此存在高毒性的问题(请参阅韩国专利申请公开号kr20150109540a)。本技术的发明人认识到trap-1可能是能够替换vegf的治疗靶标,并发明了能够选择性抑制trap-1的新方法,由此得到本技术。
技术实现要素:
[0008]
技术问题
[0009]
本技术公开的一项任务是提供用于抑制血管新生因子的化合物或其药学上可接
受的盐。
[0010]
本技术公开的另一项任务是提供包含所述化合物或其药学上可接受的盐或其前药的药物组合物。
[0011]
本技术公开的再一项任务是提供用于治疗血管新生性疾病的方法,所述方法包括:开具包含所述化合物或其药学上可接受的盐或其前药的药物组合物的处方。
[0012]
进一步地,本技术公开的另一项任务是提供用于治疗血管新生性疾病中血管新生性眼部疾病的方法。
[0013]
进一步地,本技术公开的另一项任务是提供用于治疗血管新生性眼部疾病的用于滴眼给药的药物组合物。
[0014]
进一步地,本技术公开的另一项任务是提供治疗血管新生性眼部疾病的方法,该方法的特征是口服或通过滴眼剂给予所述药物组合物。
[0015]
技术方案
[0016]
本技术提供具有由以下[化学式1]表示的结构的化合物、其药学上可接受的盐或其前药。
[0017]
[化学式1]
[0018][0019]
其中,
[0020]
r1、r2、r3、r4和r5独立地选自于h、卤素、羟基、羟基c
1~5
烷基、c
1~5
烷基、c
1~5
烯基、c
1~5
炔基和c
1~5
烷氧基。此处,如果r1和r4中的一个是羟基,则另一个不是羟基,或者,如果r2和r5中的一个是羟基,则另一个不是羟基;
[0021]
l包含-(ch2)
n-;以及
[0022]
n是5以上且12以下的整数。
[0023]
或者,化学式1中的r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基。
[0024]
或者,化学式1中的r3和r4各自为c
1~5
烷氧基。
[0025]
或者,化学式1中的r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基;r3和r4各自为c
1~5
烷氧基。
[0026]
或者,化学式1中的r2和r5各自为羟基、烷氧基或卤素,在这种情况下,r2和r5中的至少一个是卤素。
[0027]
或者,在化学式1中,r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基;r3和r4各自为c
1~5
烷氧基;r2和r5各自为羟基、烷氧基或卤素,在这种情况下,r2和r5中的至少一个是卤素。
[0028]
此外,在化学式1中的l包含-(ch2)
n-,其中n是10。
[0029]
或者,化学式1提供了由选自于[化学式2]至[化学式5]中任一个表示的化合物:
[0030]
[化学式2]
[0031][0032]
[化学式3]
[0033][0034]
[化学式4]
[0035][0036]
[化学式5]
[0037][0038]
此外,本技术提供了用于治疗血管新生性眼部疾病的药物组合物,所述药物组合物包含具有由以下[化学式1]表示的结构的化合物、其药学上可接受的盐或其前药:
[0039]
[化学式1]
[0040][0041]
其中,
[0042]
r1、r2、r3、r4和r5独立地选自于h、卤素、羟基、羟基c
1~5
烷基、c
1~5
烷基、c
1~5
烯基、c
1~5
炔基和c
1~5
烷氧基。此处,如果r1和r4中的一个是羟基,则另一个不是羟基,或者,如果r2和r5中的一个是羟基,则另一个不是羟基;
[0043]
l包含-(ch2)
n-;以及
[0044]
n是5以上且12以下的整数。
[0045]
或者,化学式1中的r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基。
[0046]
或者,化学式1中的r3和r4各自为c
1~5
烷氧基。
[0047]
或者,化学式1中的r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基;r3和r4各自为c
1~5
烷氧基。
[0048]
或者,化学式1中的r2和r5各自为羟基、烷氧基或卤素,在这种情况下,r2和r5中的至少一个是卤素。
[0049]
或者,在化学式1中,r1为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基;r3和r4各自为c
1~5
烷氧基;r2和r5各自为羟基、烷氧基或卤素,在这种情况下,r2和r5中的至少一个是卤素。
[0050]
此外,在化学式1中的l包含-(ch2)
n-,其中n是10。
[0051]
或者,化学式1提供了由选自于[化学式2]至[化学式5]中任一个表示的化合物:
[0052]
[化学式2]
[0053][0054]
[化学式3]
[0055][0056]
[化学式4]
[0057][0058]
[化学式5]
[0059][0060]
另外,本技术提供了用于治疗血管新生性眼部疾病的药物组合物,所述药物组合物包含具有由以下[化学式6]表示的结构的化合物、其药学上可接受的盐或其前药:
[0061]
[化学式6]
[0062][0063]
其中,
[0064]
r1是甲基;
[0065]
l包含-(ch2)
n-;以及
[0066]
n是5以上且12以下的整数。
[0067]
或者,化学式6中的l为-(ch2)
10-。
[0068]
此外,根据本技术的药物组合物用于滴眼剂。
[0069]
或者,提供的是一种用于治疗血管新生性眼部疾病的药物组合物,其中,所述血管新生性眼部疾病是脉络膜血管新生性疾病、视网膜血管新生性疾病、视网膜下血管新生性疾病、角膜血管新生性疾病、虹膜血管新生性疾病或血管新生性青光眼。此外,血管新生性眼部疾病是选自于糖尿病性视网膜病变、早产儿视网膜病变或视网膜静脉阻塞的视网膜血管新生性疾病。优选地,视网膜血管新生性疾病是糖尿病性视网膜病变。
[0070]
此外,提供的是用于治疗血管新生性眼部疾病的药物组合物,其中,所述脉络膜血管新生性疾病是湿性年龄相关性黄斑变性(湿性amd)。
[0071]
有益效果
[0072]
使用本技术的药物组合物可以治疗血管新生性疾病。其中,特别是可以治疗血管新生性眼部疾病。使用本技术的药物组合物可以获得以下效果。
[0073]
使用本技术的药物组合物,可以进行非侵入性治疗。smx和本技术的新型化合物分子作为小分子而非蛋白质,具有高的组织渗透能力。因此,无需对异常组织直接注射给药,具有可以使用诸如口服给药或滴眼给药等非侵入性治疗的优势。
[0074]
此外,与现有的治疗剂或治疗方法相比,本技术公开的药物组合物或使用所述药物组合物的治疗方法具有更高的稳定性。由于现有的治疗剂靶向于在异常细胞和正常细胞中均被表达的那些因子,而本技术公开的药物组合物smx和新型化合物分子靶向于在异常细胞中过表达的那些因子,这使得异常细胞能够被特异性治疗。因此,本技术所公开的药物组合物的优势在于,其对正常细胞不显示出副作用而使毒性得到更大改善。
附图说明
[0075]
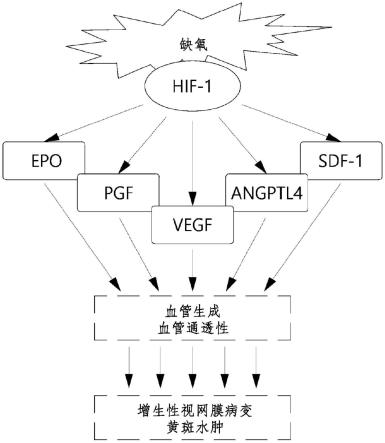
图1显示trap-1和相关血管新生因子的信号转导通路。
[0076]
图2显示trap-1和hif-1α之间的特定信号转导通路。
[0077]
图3显示trap-1的结构及其运作过程。
[0078]
图4是用trap-1+/+小鼠和trap-1+/
–
小鼠制备的氧诱导视网膜病变模型的一组视网膜血管造影。
[0079]
图5是对图4中的用trap-1+/+小鼠和trap-1+/
–
小鼠制备的氧诱导视网膜病模型的一组视网膜血管造影进行统计处理的一组结果。
[0080]
图6显示敲除trap-1的细胞的血管形成试验(tube formation assay)的结果。
[0081]
图7是对通过图6的血管形成试验进行统计处理的一组结果。
[0082]
图8显示蛋白质印迹的结果,证实了hif-1α的表达随smx的浓度而变化。
[0083]
图9显示蛋白质印迹的结果,证实了当使用smx处理时,包括hif-1α在内的血管新生因子表达的变化。
[0084]
图10显示在先前的研究中证实的hsp90与客户蛋白图10显示在先前的研究中证实的hsp90与客户蛋白的结合结构。
[0085]
图11是根据图10的hsp90和客户蛋白之间的结合位置以及根据图12的trap-1和smx之间的结合位置的重叠。
[0086]
图12显示trap-1和smx的结合结构的x射线晶体学分析的结果。
[0087]
图13显示在trap-1上参与smx结合的位置的种间保守分析的结果。
[0088]
图14显示用于验证smx竞争性结合至客户蛋白的pull-down试验的结果。
[0089]
图15是一组实验结果,证实将smx结合到trap-1上所涉及的位置与将客户蛋白结合到trap-1上所涉及的位置相同。左侧是在修饰相应位置后进行pull-down试验的结果,右侧是在修饰相应位置后进行sirt3活性实验的结果。
[0090]
图16显示用smx和pu-h71处理的野生型和突变体trap-1的atp酶活性试验的结果。
[0091]
图17显示氧诱导的视网膜病变小鼠模型(其中,眼内注射smx和eylea(阿柏西普))的视网膜血管分析的结果。
[0092]
图18是氧诱导的视网膜病变小鼠模型(其中,通过滴眼给予smx)的一组视网膜血管分析的照片。
[0093]
图19是对氧诱导的视网膜病变小鼠模型(其中,通过滴眼给予smx)的视网膜血管分析照片进行统计处理的一组结果。
[0094]
图20是显示制备(10-(2-溴-5-羟基-3,4-二甲氧基-6-甲基苯基)癸基)三苯基膦甲酸盐的步骤的示意图。
[0095]
图21是显示制备(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物的步骤的示意图。
[0096]
图22是显示制备(10-(2-溴-3,4,5-三甲氧基-6-甲基苯基)癸基)三苯基膦溴化物的步骤的示意图。
[0097]
图23是显示制备(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物的步骤的示意图。
[0098]
图24显示pull-down试验的结果,证实smx和新型化合物分子(sb-u005、sb-u009、sb-u011和sb-u012)与客户蛋白竞争性地结合至trap-1。
[0099]
图25显示当mio-m1 hre细胞系用smx和新型化合物分子(sb-u005、sb-u009、sb-u011和sb-u012)处理时的ic
50
值。
[0100]
图26显示蛋白质印迹的结果,该结果证实当arpe-19细胞系(视网膜色素上皮细胞系)被smx和新型化合物分子(sb-u005、sb-u009、sb-u011和sb-u012)处理时,血管新生因子(hif-1α)表达的变化。
具体实施方式
[0101]
现有技术文件
[0102]
非专利文件
[0103]
[1]kellen l.meadows and herbert i.hurwitz,anti-vegf therapies in the clinic,cold spring harb perspect med.2012oct 1;2(10).
[0104]
[2]katja zirlik and justus duyster,anti-angiogenics:current situation and future perspectives,oncol res treat.2018;41(4):166-171.
[0105]
[3]patrick chames,marc van regenmortel,etienne weiss and daniel baty,therapeutic antibodies:successes,limitations and hopes for the future,br j pharmacol.2009may;157(2):220-33.
[0106]
[4]t kamba and dm mcdonald,mechanisms of adverse effects of anti-vegf therapy for cancer,br j cancer.2007jun 18;96(12):1788-95.
[0107]
[5]peter a.campochiaro,ocular neovascularization,j mol med(berl).2013march;91(3):311-321.
[0108]
[6]barbara onnis,annamaria rapisarda and giovanni melillo,development of hif-1inhibitors for cancer therapy,j.cell.mol.med.vol13,no 9a,2009pp.2780-2786.
[0109]
[7]young chan chae,m.cecilia caino,sofia lisanti,and et al.control of tumor bioenergetics and survival stress signaling by mitochondrial hsp90,cancer cell.2012sep 11;22(3):331-44.
[0110]
[8]stuart k.calderwood and thomas l.prince,chaperones:methods and protocols,humana press,2018,pp.424-432.
[0111]
[9]daniel t.gewirth,paralog specific hsp90 inhibitors-a brief history and a bright future,curr top med chem.2016;16(25):2779-2791.
[0112]
[10]ionica masgras,carlos sanchez-martin,giorgio colombo and andrea rasola,the chaperone trap1 as a modulator of the mitochondrial adaptations in cancer cells,front oncol.2017mar 29;7:58.
[0113]
[11]verba ka,wang ry,arakawa a,liu y,shirouzu m,yokoyama s,and agard da,atomic structure of hsp90-cdc37-cdk4 reveals that hsp90traps and stabilizes an unfolded kinase,science.2016jun24;352(6293):1542-7.
[0114]
1.定义
[0115]
术语“烷氧基”是指结合有氧的烷基,优选结合有氧的低级烷基。代表性的烷氧基包括甲氧基、乙氧基、丙氧基、叔丁氧基等。
[0116]
术语“烷氧基烷基”是指用烷氧基取代的烷基,可以在化学通式中用烷基-o-烷基表示。
[0117]
本文所用的术语“烯基”是指包含至少一个双键的脂肪族官能团,也旨在包括“未取代的烯基”和“取代的烯基”,这意味着后者是具有取代基的烯基,所述取代基替代烯基基团的一个或多个碳上的氢。这些取代基可以存在于一个或多个碳上,这些碳被包含或不被包含在一个或多个双键中。此外,如下所述,除了稳定性成为问题的情况外,这些取代基包括所有可以考虑用于烷基基团的取代基。例如,可以考虑用一个或多个烷基、碳环基、芳基、杂环基或杂芳基等作为烯基的取代基。
[0118]“烷基”基团或“烷烃”是完全饱和的直链或支链的非芳香烃。通常,除非另有定义,否则直链或支链烷基具有1至20个碳原子。直链和支链烷基包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、戊基、己基、戊基和辛基。c
1-c6直链或支链烷基的另一个名称被称为“低级烷基”基团。
[0119]
此外,本说明书、实施例和权利要求中使用的术语“烷基”(或“低级烷基”)旨在包括“未取代烷基”和“取代烷基”,这意味着后者是具有取代基的烷基,所述取代基替代烃基的一个或多个碳上的氢。除非另有说明,所述取代基可包括例如:卤素、羟基、羰基(例如,羧基、烷氧羰基、甲酰基或酰基)、硫代羰基(例如,硫酯、硫代乙酸酯/盐或硫代甲酸酯/盐)、烷
氧基、磷酰基、磷酸酯/盐、膦酸酯/盐、次膦酸酯/盐、氨基、酰氨基、脒、亚胺、氰基、硝基、叠氮基、巯基、烷硫基、硫酸酯/盐、磺酸酯/盐、氨磺酰、亚磺酰氨基、磺酰基、杂环基、芳烷基,或芳基或杂芳基。本领域技术人员可以理解,当取代基合适时,烃链上的取代功能基团本身也可以被取代。例如,取代烷基的取代基可以包括氨基、叠氮基、亚氨基、酰氨基、磷酰基(包括磷酸酯/盐和次磷酸酯/盐)、磺酰基(包括硫酸酯/盐、亚磺酰氨基、氨磺酰基和磺酸酯/盐),以及取代形式和未取代形式的甲硅烷基,也可以包括乙醚、烷硫基、羰基(包括酮、醛、羧酸酯/盐和酯)、-cf3、-cn等。环烷基可以用烷基、烯基、烷氧基、烷硫基、氨基烷基、羰基取代的烷基、-cf3、-cn等另加取代。
[0120]
术语“羟基烷基”是指具有至少一个羟基取代基的烷基,例如,用一个或两个羟基取代的包含1至6个碳原子的线性单价烃自由基,或包含3至6个碳原子的支链单价烃自由基,条件是当存在两个羟基时,两个羟基不在同一碳原子上。具体来说,羟基烷基包括羟基甲基、2-羟基乙基、2-羟基丙基、3-羟基丙基、1-(羟基甲基)-2-甲基丙基、2-羟基丁基、3-羟基丁基、4-羟基丁基、2,3-二羟基丙基、1-(羟基甲基)2-羟基乙基、2,3-二羟基丁基、3,4-二羟基丁基和2-(羟基甲基)-3-羟基丙基等。
[0121]
本文所用的术语“炔基”是指包含至少一个三键的脂肪族官能团,也旨在包括“未取代的炔基”和“取代炔基”,这意味着后者是具有取代基的炔基,所述取代基替代炔基基团的一个或多个碳上的氢。这些取代基可以存在于一个或多个碳中,所述碳被包含或不包含在一个或多个三键中。此外,如下所述,除了稳定性成为问题的情况外,这些取代基包括所有可以考虑用于烷基基团的取代基。例如,可以考虑用一个或多个烷基、碳环基、芳基、杂环基或杂芳基等作为炔基的取代基。
[0122]
当与官能团(例如酰基、酰氧基、烷基、烯基、炔基或烷氧基)一起使用时,术语“c
x-y”旨在包括链中包含x至y个碳的官能团。例如,术语“c
x-y
烷基”是指取代或未取代的饱和烃基团,是指在链中包含x至y个碳的直链烷基和支链烷基以及包含例如卤代烷基基团如三氟甲基和2,2,2-三氟乙基。c0烷基在末端位置时指的是氢,在内部时指的是键。术语“c
2-y
烯基”和“c
2-y
炔基”分别是取代或未取代的不饱和脂肪族部分,对上述的烷基的长度和可能的取代基的定义适用于该部分,但旨在分别包含至少一个双键或三键。
[0123]
本文所用的术语“芳基”是指取代或未取代的单环芳香族基团,其中包括环的每个原子均为碳的基团。优选地,该环是5至7元环,更优选地是6元环。术语“芳基”还指包含两个或更多环的多环体系,其中两个相邻环共享两个或更多个碳,并且,至少一个环是芳香环,另外的环例如可以是环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。芳基基团包括苯、萘、菲、苯酚、苯胺等。
[0124]
本文中使用的术语“碳环、碳环的(carbocycle和carbocyclic)”是指饱和或不饱和环,其中环的每个原子均为碳。术语“碳环”既包括芳香碳环,也包括非芳香碳环。非芳香碳环包含其中所有碳原子均饱和的环烷烃环,以及包含至少一个双键的环烯烃环。
[0125]
术语“碳环”包括5-7元单环和8-12元双环。双环碳环的每个环可以选自于饱和环、不饱和环或芳香环。碳环包括其中两个环之间共享两个或三个以上原子的双环分子。术语“稠合碳环”是指双环碳环,其中各个环与其他环共享两个相邻的原子。稠合碳环的每个环可以选自于饱和环、不饱和环或芳香环。在示例性的实施例中,芳香环(例如,苯基)可以与饱和或不饱和环(例如环己烷、环戊烷或环己烯)稠合。只要在化合价允许的情况下,饱和双
环、不饱和双环以及芳香双环的所有组合均被包含在碳环的定义中。示例性的“碳环”包括环戊烷、环己烷、双环[2.2.1]庚烷、1,5-环辛二烯、1,2,3,4-四氢萘、双环[4.2.0]辛-3-烯、萘和金刚烷。示例性的稠合碳环包括萘烷、萘、1,2,3,4-四氢萘、双环[4.2.0]辛烷、4,5,6,7-四氢-1h-茚和双环[4.1.0]庚-3-烯。“碳环”可以在可以具有氢原子的任何一个或多个位置被取代。
[0126]“环烷基”基团是完全饱和的环状烃。“环烷基”包括单环和双环。除非另有定义,否则单环环烷基通常具有3至约10个碳原子,更通常而言,具有3至8个碳原子。双环环烷基的第二个环可以选自于饱和环、不饱和环以及芳香环。环烷基包括其中两个环之间共享一个、两个或三个以上原子的双环分子。术语“稠合环烷基”是指各个环与其他环共享两个原子的双环环烷基。稠合双环环烷基的第二个环可以选自于饱和环、不饱和环以及芳香环。“环烯基”基团是包含一个或多个双键的环状烃。
[0127]
术语“杂芳基(heteroaryl和hetaryl)”是指取代的或未取代的单个环结构,优选5至7元环,更优选5至6元环,并且包括其中包含至少一个杂原子的环结构,优选1至4个杂原子,更优选1或2个杂原子。
[0128]
术语“杂芳基(heteroaryl和hetaryl)”还指包含两个或更多个环的多环体系,其中两个相邻环共享两个或更多个碳,并且,至少一个环是杂芳环,另外的环可以为例如环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂芳基包括例如吡咯、呋喃、噻吩、咪唑、噁唑、噻唑、吡啶、吡嗪、哒嗪和嘧啶等。
[0129]
本文所用的术语“杂原子”是指除了碳和氢以外的所有元素的原子。优选的杂原子是氮、氧和硫。
[0130]
术语“杂环基、杂环和杂环的(heterocyclyl、heterocycle和heterocyclic)”是取代或未取代的非芳香环结构,优选3至10元环,更优选3至7元环,并且是指其中它的环结构包含至少一个杂原子,优选1至4个杂原子,更优选的是1或2个杂原子的那些杂环。术语“杂环基(heterocyclyl)和杂环的(heterocyclic)”还指包含两个或更多环的多环体系,其中,两个相邻环共享两个或更多个碳,其中,至少一个环是杂环,另外的环可以为例如环烷基、环烯基、环炔基、芳基、杂芳基和/或杂环基。杂环基包括例如哌啶、哌嗪、吡咯烷、吗啉、内酯、内酰胺等。
[0131]
当与例如酰基、酰氧基、烷基、烯基、炔基或烷氧基一起使用时,表述“低级”旨在包括例如其中存在10个以下、优选6个以下的除氢以外的原子的官能团。例如,“低级烷基”是指具有10个以下,优选6个以下碳原子的烷基。
[0132]
术语“共价键”是指原子之间的化学键,它不是离子键。
[0133]
术语“包含x的官能团”是指包含x原子的化学官能团。例如,作为典型的包含o的官能团,有酮、羟基、烷氧基烷基、羧基等。
[0134]“hsp90”是指体内存在的伴侣蛋白hsp90(hsp90-α1、hsp90-α2、hsp90-β)和旁系同源物(grp94、trap-1)。除非另有说明,“hsp90”整体来说是指hsp90-α1、hsp90-α2、hsp90-β,它们是细胞质中存在的hsp90。
[0135]“结合能力”是指由两个分子之间的静电力和/或疏水作用引起的生化和/或物理现象,并不旨在将该术语限制为具有特定水平以上强度的相互作用。
[0136]“前药”是指在体内给药后转化为感兴趣的药学材料的药物,此处,所述转化可以
是酶促的或非酶促的。活性化合物的前药可以通过对该活性化合物中的官能团进行修饰而制备,所述修饰以允许活性母体化合物的修饰通过常规操作断裂或体内断裂的方式进行。前药包括其中羟基、氨基或巯基基团与任何基团结合的化合物,也就是说,当给予受试者时,将活性化合物的前药断裂以分别形成游离羟基、游离氨基或游离巯基基团。前药的实例包括活性化合物中的醇的乙酸酯/盐衍生物、甲酸酯/盐衍生物和苯甲酸酯/盐衍生物,以及氨官能团的乙酰胺衍生物、甲酰胺衍生物和苯甲酰胺衍生物等,但并不限于此。
[0137]“药学上可接受的盐”是指源自各种生理学上可接受的有机抗衡离子和无机抗衡离子的化合物的盐。抗衡离子在本领域中是众所周知的,当分子包含酸性官能团时,包括例如钠、钾、钙、镁、铝、锂和铵,例如,四烷基铵等;当该分子含有碱性官能团时,包括有机酸或无机酸的盐,例如盐酸盐、硫酸盐、磷酸盐、二磷酸盐、硝酸盐、氢溴酸盐、酒石酸盐、甲磺酸盐、乙酸盐、苹果酸盐、马来酸盐、富马酸盐、酒石酸盐、琥珀酸盐、柠檬酸盐、乳酸盐、帕莫酸盐、水杨酸盐、硬脂酸盐、甲烷磺酸盐、对甲苯磺酸盐、草酸盐等。其合适的药学上可接受的盐包括文献[remington's pharmaceutical sciences,17th edition,pg.1418(1985)和p.heinrich stahl,camille g.wermuth(eds.),handbook of pharmaceutical salts properties,selection,and use;2002]中列出的盐。酸加成盐的实例包括由以下酸形成的盐,例如氢碘酸、磷酸、偏磷酸、硝酸和硫酸;以及有机酸,例如海藻酸、抗坏血酸、邻氨基苯甲酸、苯甲酸、樟脑磺酸、柠檬酸、扑酸(帕莫酸)、乙磺酸、甲酸、富马酸、糠酸、半乳糖醛酸、龙胆酸、葡糖酸、葡萄糖醛酸、谷氨酸、乙醇酸、异烟酸、羟乙磺酸、乳酸、苹果酸、扁桃酸、甲磺酸、粘酸、泛酸、苯乙酸、丙酸、葡糖二酸、水杨酸、硬脂酸、琥珀酸、磺胺酸、三氟乙酸和芳基磺酸(例如苯磺酸和对甲苯磺酸)。碱金属、碱土金属和有机碱形成的碱加成盐的实例不仅包括氯普鲁卡因、胆碱、n,n-二苄基乙二胺、二乙醇胺、乙二胺、赖氨酸、葡甲胺(n-甲基葡糖胺)和克罗卡因,还包括内成盐。具有非生理学上可接受的阴离子或阳离子的盐在本发明的范围内,是制备生理学上可接受的盐和/或用于非治疗性(例如体外情况)的有用中间体。根据本技术的药学上可接受的盐包括卤素盐,即氟盐、溴盐、碘盐等。
[0138]
2.本技术的化合物结构
[0139]
本技术提供了用于抑制trap-1的药物分子。
[0140]
根据本技术的药物分子是具有以下[化学式7]表示的结构的化合物,
[0141]
[化学式7]
[0142]
b-l-c
+
[0143]
其中,
[0144]
b是对trap-1原聚体具有亲和力的结合部分,l是连接结合部分和阳离子部分的连接部分,而c
+
是具有正电荷的阳离子部分。
[0145]
在这种情况下,结合部分包括环,或者进一步包括4至10元单环,或者进一步可以包含5至7元单环。此外,所述环可以是碳环或可以是杂环。在这种情况下,构成杂环的杂原子可以示例性地是氮、氧、硫或磷。另外,环可以是芳基。此外,环可以是多环,例如双环。
[0146]
或者,环可以用一个或多个取代基取代。在这种情况下,取代基可以是烷基、烯基或炔基。或者,取代基可以是包含杂原子的官能团。
[0147]
在这种情况下,包含杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0148]
同样,在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基或亚甲基二氧基。
[0149]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0150]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐(thiolate)或二硫羟酸酯/盐(dithioate)。
[0151]
在这种情况下,结合部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0152]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0153]
或者,在这种情况下,结合部分可以包括烯基或炔基。
[0154]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团结合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的结合部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0155]
或者,结合部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上的整数和0。
[0156]
或者,结合部分可以包括烷基和亚乙氧基单元。
[0157]
或者,结合部分可具有特定范围的长度。在这种情况下,结合部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0158]
c是具有正电荷的阳离子部分或其还原形式。
[0159]
在这种情况下,阳离子部分可以具有正电荷。
[0160]
在这种情况下,阳离子部分可以包括氧化原子。此外,氧化原子可以是n
+
或p
+
。
[0161]
或者,在这种情况下,阳离子部分可以包括包含氧化原子的官能团。
[0162]
在这种情况下,该官能团可以是胺基或磷烷基(phosphane group)。
[0163]
或者,在这种情况下,阳离子部分可以是其还原的形式。
[0164]
或者,阳离子部分包括环,或者进一步包括4至10元单环,或者进一步地可以包括5至7元单环。此外,环可以是碳环或可以是杂环。在这种情况下,构成杂环的杂原子可以是氮、氧、硫或磷。另外,环可以是芳基。此外,环可以是多环,例如双环。
[0165]
或者,环可以用一个或多个取代基取代。在这种情况下,取代基可以是烷基、烯基或炔基。或者,取代基可以是包括杂原子的官能团。
[0166]
或者,当阳离子部分包括氧化原子时,环可以是与氧化原子结合的取代基。
[0167]
或者,在整个分子的位置关系中,结合部分与阳离子部分之间的距离可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0168]
根据本技术的药物分子是具有以下[化学式8]所示的结构的化合物,
[0169]
[化学式8]
[0170][0171]
在这种情况下,构成左侧结合部分的6元环可以是碳环或杂环。该6元环内所示的虚线和“?”表示在允许的结合范围内,在构成上述环的原子之间可任意形成单键或多键。在这种情况下,构成杂环的杂原子可以是氮、氧、硫或磷。另外,该6元环可以是芳基。
[0172]
在这种情况下,r1至r5可以各自独立地为h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1至r5中的任何一个都可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0173]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0174]
在优选的实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羰基。
[0175]
在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0176]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0177]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0178]
在化学式8中,l是连接结合部分和阳离子部分的连接部分。
[0179]
在这种情况下,连接部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0180]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0181]
或者,在这种情况下,连接部分可以包括烯基或炔基。
[0182]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团结合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的连接部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0183]
或者,连接部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上的整数和0。
[0184]
或者,连接部分可以包括烷基和亚乙氧基单元。
[0185]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0186]
根据本技术的药物分子是具有以下[化学式9]所示的结构的化合物,
[0187]
[化学式9]
[0188][0189]
在这种情况下,r1至r3可以各自独立地为h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1至r3中的任何一个都可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0190]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。
[0191]
在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0192]
在优选的实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羰基。
[0193]
或者,在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0194]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0195]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0196]
在化学式9中,l是连接结合部分和阳离子部分的连接部分。
[0197]
在这种情况下,连接部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0198]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0199]
或者,在这种情况下,连接部分可以包括烯基或炔基。
[0200]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团键合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的连接部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0201]
或者,连接部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上的整数和0。
[0202]
或者,连接部分可以包括烷基和亚乙氧基单元。
[0203]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0204]
或者,根据本技术的药物分子可以是具有化学式9结构的化合物的还原形式的化合物。
[0205]
根据本技术的药物分子是具有以下[化学式10]所示的结构的化合物,
[0206]
[化学式10]
[0207][0208]
在这种情况下,r1至r3可以各自独立地为h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1至r3中的任何一个都可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0209]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0210]
在优选的实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羰基。
[0211]
在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0212]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0213]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0214]
在化学式10中,l是连接结合部分和阳离子部分的连接部分。
[0215]
在这种情况下,连接部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0216]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0217]
或者,在这种情况下,连接部分可以包括烯基或炔基。
[0218]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团键合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的连接部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0219]
或者,连接部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上的整数和0。
[0220]
或者,连接部分可以包括烷基和亚乙氧基单元。
[0221]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0222]
根据本技术的药物分子是具有以下[化学式6]所示的结构的化合物,
[0223]
[化学式6]
[0224][0225]
在这种情况下,r1可以是h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0226]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0227]
在优选的实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羰基。
[0228]
在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0229]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0230]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0231]
在化学式6中,l是连接结合部分和阳离子部分的连接部分。
[0232]
在这种情况下,连接部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0233]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0234]
或者,在这种情况下,连接部分可以包括烯基或炔基。
[0235]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团键合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的连接部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0236]
或者,连接部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上的整数和0。
[0237]
或者,连接部分可以包括烷基和亚乙氧基单元。
[0238]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0239]
或者,根据本技术的药物分子可以是具有化学式6结构的化合物的还原形式的化合物。
[0240]
根据本技术的药物分子是具有以下[化学式11]所示的结构的化合物,
[0241]
[化学式11]
[0242][0243]
在这种情况下,r1至r3可以各自独立地是h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1至r3中的任一个可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0244]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0245]
在优选的实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羰基。
[0246]
在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0247]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0248]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0249]
另外,在连接部分的结构中,n是1以上的整数。在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。在这种情况下,烃部分在允许范围内可以包含多个键。
[0250]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0251]
示例性地,根据化学式6的化合物可以是具有以下结构的化合物。
[0252][0253]
此外,根据本技术的化合物分子是具有由以下化学式1表示的结构的新型化合物,
[0254]
[化学式1]
[0255][0256]
在这种情况下,r1至r5可以各自独立地为h、烷基、烯基、炔基或包含杂原子的官能团。示例性地,r1至r5中的任何一个都可以是烷基,进一步地,烷基可以是c
1-5
烷基,或者,更进一步地,烷基可以是甲基。
[0257]
在这种情况下,包含杂原子的官能团可以是包含氧的官能团。在这种情况下,包含
氧的官能团可以示例性地是羟基、羰基、甲酰基、烷氧羰基氧基、羧基、烷氧羰基、烷氧基、烷氧基烷基或亚甲基二氧基。
[0258]
在实施方式中,包含氧的官能团可以是c
1-5
烷氧基,或进一步地是甲氧基。在另一个优选实施方式中,包含氧的官能团可以是羟基或羟烷基。
[0259]
在这种情况下,包括杂原子的官能团可以是包含卤素的官能团。在这种情况下,包含卤素的官能团可以示例性地是f、cl、br或i。
[0260]
或者,在这种情况下,包含杂原子的官能团可以是包含氮的官能团。在这种情况下,包含氮的官能团可以示例性地是酰胺、胺、铵、亚胺、酰亚胺、氰酸酯/盐、硝酸酯/盐、腈、吡啶、叠氮或氨基甲酸酯/盐。
[0261]
或者,在这种情况下,包含杂原子的官能团可以是包含硫的官能团。在这种情况下,包含硫的官能团可以示例性地是硫醇、硫化物、二硫化物、亚砜、砜、磺酸、磺酸酯/盐、硫酮、硫醛、硫羟酸酯/盐或二硫羟酸酯/盐。
[0262]
例如,r1、r2、r3、r4和r5可以选自于h、卤素、羟基、羟基c
1~5
烷基、c
1~5
烷基、c
1~5
烯基、c
1~5
炔基和c
1~5
烷氧基。
[0263]
在这种情况下,当r1、r2、r3、r4和r5中的任何一个是羟基时,位于正对着羟基的方向上的取代基可以不是羟基。在这里,位于正对着羟基的方向的取代基可以是例如(r1和r4)或(r2和r5)。具体而言,当r1和r4中的任何一个是羟基时,另一个可以不是羟基,或者,当r2和r5的任何一个是羟基时,另一个可以不是羟基。在这里,“(x和y)”是指由x和y组成的一组。
[0264]
当r1、r2、r3、r4和r5相对的取代基均为羟基时,例如,在r1和r4均为羟基、或r2和r5均为羟基的化合物的情况下,该化合物本身的元素的电磁交换可以导致氧化/还原反应。因此,当化学式2的化合物的相对的取代基之一是羟基时,由于另一个不是羟基,则可能对化合物本身中发生的氧化/还原反应具有灭活作用。
[0265]
具体而言,在化学式1中,r1可以为h、c
1~5
烷基、c
1~5
烯基或c
1~5
炔基,进一步地,可以是c
1~5
烷基,更进一步地,可以是甲基。
[0266]
或者,r3和r4各自可以是c
1~5
烷氧基。
[0267]
或者,r2和r5各自是羟基、烷氧基或卤素,在这种情况下,r2和r5中的至少一个可以是卤素。
[0268]
此外,化学式1中的l是连接部分,将包括r1至r5取代基的结合部分(左)和三苯基膦(右)相连接。
[0269]
在这种情况下,连接部分可以包括脂肪族官能团,例如烷基((ch2)n)。
[0270]
在这种情况下,n可以是1以上且20以下的整数。此外,n可以是选自于1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19和20的两个数之间的整数。此外,n可以是选自于5、6、7、8、9、10、11和12的两个数之间的整数。此外,n还可以是5以上且12以下的整数。优选地,n可以是10。
[0271]
或者,在这种情况下,连接部分可以包括烯基或炔基。
[0272]
或者,在这种情况下,脂肪族官能团可以通过杂原子与结合部分、阳离子部分或另一个脂肪族官能团键合。在这种情况下,杂原子可以示例性地是氧、氮或硫。例如,一个示例性的连接部分可以具有-(ch2)
x-o-(ch2)
y-(x和y是0以上的整数)的结构。
[0273]
或者,连接部分可以包括亚乙氧基单元((c2h4o)m)。在这种情况下,m可以是1以上
的整数和0。
[0274]
或者,连接部分可以包括烷基和亚乙氧基单元。
[0275]
或者,连接部分可具有特定范围的长度。在这种情况下,连接部分的长度可以为5埃至60埃,进一步地,长度为10埃至50埃,更进一步地,长度为20埃至40埃,或者,更进一步地,长度为25埃至35埃。
[0276]
在优选实施方式中,根据化学式1的化合物可以是具有以下化学式2所示的结构的化合物。
[0277]
[化学式2]
[0278][0279]
在另一个实施方式中,根据化学式1的化合物可以是具有以下化学式3所示的结构的化合物。
[0280]
[化学式3]
[0281][0282]
在另一个实施方式中,根据化学式1的化合物可以是具有以下化学式4所示的结构的化合物。
[0283]
[化学式4]
[0284][0285]
在另一个实施方式中,根据化学式1的化合物可以是具有以下化学式5所示的结构的化合物。
[0286]
[化学式5]
[0287][0288]
根据本技术由化学式1至11表示的化合物可以以其药学上可接受的盐或前药的形式提供。在示例性的实施方式中,药学上可接受的盐可以是酸加成盐或卤素盐。优选地,药学上可接受的盐可以是溴盐。
[0289]
3.血管新生因子与血管新生性疾病之间的关系
[0290]
血管新生性疾病是由异常的血管新生引起的。血管新生最初是正常的内稳态维持动作,用于血液向组织的平稳供给。但是,当这种血管新生比正常组织中过多时,这种异常的血管新生会导致血管新生性疾病。尽管血管新生性疾病的原因具有某些共同点,但不能认为它们具有相同的病理机制,总体而言,详细的病理机制尚不清楚。但是,通常,血管新生因子参与了大多数血管新生性疾病,是其主要原因。
[0291]
在血管新生性疾病中,与正常细胞相比,异常细胞过多产生血管新生因子。重要的血管新生因子的代表是血管内皮生长因子(vegf)。已知对vegf配体、vegf受体(vegfr)及参与其信号转导的因子的遏制对这种异常的血管新生进行抑制。
[0292]
此外,最近的研究成果已经公布,缺氧诱导因子1-α(hif-1α)参与血管新生。例如,结果是hif-1α会诱导眼睛中的视网膜和视网膜下的血管新生[5]。已知hif-1α是在缺氧状态下过表达的因子,并参与细胞的生长和存活。另外,hif-1α参与了血管新生,是vegf的亲代调节物之一。hif-1α的血管新生信号通路如图1所示。尽管研究了hif-1α的直接抑制治疗的可能性,但由于不期待的毒性,迄今为止尚未产生任何有重大意义的结果[6]。
[0293]
此外,在hif-1α的子代调节物中,epo、pgf、angptl4、sdf-1、vegfr1、vegfr2、pdgfr
β和cxcr4也充当血管新生因子。
[0294]
4.通过遏制trap-1治疗血管新生性疾病
[0295]
本技术的发明人研究了仅在异常细胞中表达的因子,即vegf和hif-1α的亲代调节物,而非vegf和hif-1α(这些因子已被用作相关领域中的主要靶标)。本技术包括抑制这些因子的方法,用于抑制这些因子的有效材料,以及包含这些因子的配方产品和治疗方法。
[0296]
肿瘤坏死因子受体相关蛋白-1(trap-1)是热休克蛋白90(hsp90)的旁系同源物,它是仅在线粒体中存在的伴侣蛋白和线粒体蛋白。
[0297]
hsp90是与人体蛋白质的很大一部分有关的伴侣蛋白,并且已知参与调节细胞稳态的功能,因此已被作为抗癌靶标进行研究[7]。然而,由于hsp90是不区分异常细胞和正常细胞而表达的常见的伴侣蛋白,因此在许多情况下,通过抑制hsp90产生不期待的副作用。因此,自1999年以来,有18种hsp90抑制剂已经进入了临床试验阶段,但直到2018年,只有5种hsp90抑制剂仍在进行临床试验[8]。
[0298]
包括trap-1在内的hsp90具有如下结构,在该结构中,两个原聚体相结合。在这种情况下,各原聚体被称为第一原聚体和第二原聚体。当特定的客户蛋白与hsp90结合时,由于间隔排列的原聚体靠近,hsp90具有结合和降解atp、形成客户蛋白的立体结构的功能。各原聚体由n-末端结构域、中间结构域和c-末端结构域组成。n-末端结构域是结合atp以产生hsp90活性的能量的结构域。中间结构域是客户蛋白特异性地与各hsp90结合的结构域。c-末端结构域是两个原聚体被连接的结构域。示例性地,trap-1的结构和运作过程如图3所示。
[0299]
hsp90的特征是旁系同源物之间的n-末端结构域的同源性很高,但中间结构域同源性低。现有的hsp90抑制剂包含与atp同源的腺苷骨架,因此,通过竞争性地抑制atp与hsp90的n-末端的结合来遏制hsp90。这引起的问题在于现有的hsp90抑制剂非选择性地作用于所有hsp90[9]。虽然上述问题提高了人们对靶向各旁系同源物的具有低同源性的位点的选择性抑制剂的需求意识,但并未导致实际药物分子的开发[10]。
[0300]
在对hsp90及其抑制剂的研究中,本技术的发明人证实,线粒体hsp90而非细胞质hsp90积极参与血管新生性疾病中异常细胞的代谢。基于此,本技术的发明人开发了gamitrinib,其中将具有线粒体膜通透性的官能团连接至被称为hsp90抑制剂的格尔德霉素(geldanamycin),并证实gamitrinib具有出色的治疗作用。然而,gamitrinib仍表现出对细胞质hsp90的遏制能力,因此,存在显示强毒性的问题(请参阅韩国专利申请公开号kr20150109540a)。
[0301]
因此,进一步地,本技术的发明人对发生血管新生性疾病的组织进行分析,发现线粒体hsp90中的trap-1仅在异常组织部位中表达。因此,认识到存在选择性遏制trap-1并进行实验以遏制trap-1的需求,结果证实了trap-1是治疗血管新生性疾病的有效靶点。因此,随后在继续研究能够选择性遏制trap-1的抑制剂之后,trap-1的选择性抑制剂得以首次开发。通过结合至trap-1的原聚体中具有低同源性的中间结构域,根据本技术的(其中之一的)化合物可以选择性遏制trap-1。
[0302]
trap-1是hif-1α的亲代调节物,确认的trap-1和hif-1α的信号传导系统如图2所示。
[0303]
5.通过根据本技术的化合物遏制trap-1
[0304]
本部分的目的是阐明根据本技术的化合物的结构和trap-1之间的相互作用,并在此方面公开该化合物的结构特征。
[0305]
根据本技术的药物分子是具有化学式7的结构的化合物,
[0306]
[化学式7]
[0307]
b-l-c
+
[0308]
其中,
[0309]
b是对trap-1原聚体具有亲和力的结合部分,l是连接结合部分和阳离子部分的连接部分,而c
+
是具有正电荷的阳离子部分。
[0310]
在这种情况下,结合部分可具有与trap-1原聚体的n-末端结构域、中间结构域或c-末端结构域相结合的能力。在这种情况下,结合部分需要具有与trap-1原聚体的活性位点结合的能力,并且希望结合部分具有与中间部分结合的能力。
[0311]
另外,优选结合部分具有特定的体积。这是因为希望它具有合适的体积以使其具有与trap-1原聚体结合的能力。在某些优选的示例性实施方式中,结合部分可以包含烷氧基作为取代基。进一步地,结合部分可以包含两个或更多个烷氧基作为取代基。在某些优选的示例性实施方式中,结合部分可以包含羰基作为取代基。进一步地,结合部分可以包含两个或更多个羰基作为取代基。在某些优选的示例性实施方式中,结合部分可以包含烷基作为取代基。在某些优选的示例性实施方式中,结合部分可以包含烷氧基和羰基基团作为取代基。进一步地,结合部分可以包含两个或更多个烷氧基和两个或更多个羰基作为取代基。
[0312]
在这种情况下,连接部分的长度可能会影响trap-1与本技术的药物分子之间的相互作用。trap-1与药物分子之间的相互作用可以是:(1)当药物分子与trap-1的两个原聚体的特定部分结合时,(2)当单个药物分子的全部与trap-1的两个原聚体的特定部分结合时,以及(3)当药物分子无法与trap-1原聚体的特定部分结合时。除了(3)的情况外,例如当连接部分的长度小于特定长度时,(1)的比例可能高于(2)的比例。此外,当连接部分的长度等于或超过特定长度时,(2)的比例可能高于(1)的比例。另外,当连接部分的长度是特定长度时,可以优化(2)的比例以使其最高。
[0313]
此外,在trap-1不与atp结合的状态下,连接部分的长度可以是接近于第一原聚体的特定部分与第二原聚体的特定部分之间的距离的值。在这种情况下,各原聚体的特定部分可以分别是n-末端结构域、中间结构域或c-末端结构域。进一步地,在这种情况下,各原聚体的至少一个特定部分可以是中间结构域。或者,在这种情况下,在trap-1与atp结合的状态下,连接部分的长度可以是接近于第一原聚体的特定部分与第二原聚体的特定部分之间的距离的值。在这种情况下,各原聚体的特定部分可以分别是n-末端结构域、中间结构域或c-末端结构域。进一步地,在这种情况下,各原聚体的至少一个特定部分可以是中间结构域。
[0314]
或者,阳离子部分可以具有高亲脂性。示例性地,具有高亲脂性的分子具有大的非极性部分、具有被该非极性部分包围的极性部分、或者可具有大的分子量。阳离子部分透过线粒体膜以使药物分子被递送到线粒体中。
[0315]
另外,阳离子部分可具有与原聚体的n-末端结构域、中间结构域或c-末端结构域结合的能力。这是因为当阳离子部分显示出与原聚体结合的能力时,可以有效降低治疗有效的药物分子的最小量。然而,阳离子部分与原聚体结合的能力不需要跟结合部分与原聚
体结合的能力相同。
[0316]
另外,优选结合部分的特定部分具有特定的体积。这是因为需要合适的体积以具有与原聚体的特定部分结合的能力。在这种情况下,特定部分可能具有环结构。
[0317]
6.眼部疾病的分类
[0318]
眼部疾病可以根据各种方式分类。首先,可以根据异常发生部位的眼部结构对眼部疾病进行分类。
[0319]
在这种情况下,异常发生部位的眼部结构可以是围绕眼球的眼球周边结构。眼球周边结构可以是眼睑或泪腺。
[0320]
或者,异常发生部位的眼部结构可以是眼球的组成部分,该组成部分构成眼球。眼球的组成部分可以是结膜、巩膜、角膜、虹膜、睫状体、晶状体、脉络膜、视网膜、玻璃体、视神经或眼肌。在这种情况下,在视网膜中发生的眼部疾病称为视网膜变性或视网膜病变。
[0321]
另外,可以根据血管新生的存在与否对眼部疾病进行分类。
[0322]
由特定眼部结构或整个眼球的血管新生引起的眼部疾病称为血管新生性眼部疾病。血管新生指的是在先前存在的血管周围形成新的血管这样的生理现象,在本技术中,血管新生包括血管生成,这是从先前存在的血管生长出新的血管的现象。异常的血管变弱、缺血或血管新生因子的过度产生可能导致眼部结构中的异常血管新生。因此,由于血管结构变得密集,并且血管无法生长地足够厚,会发生异常症状,例如血管压力的增加以及血管与眼部结构的分离。
[0323]
血管新生性眼部疾病可以根据发生血管新生的眼部结构进行分类。在这种情况下,血管新生性眼部疾病可以是脉络膜血管新生、视网膜血管新生、视网膜下血管新生、角膜血管新生或虹膜血管新生(虹膜红变)。
[0324]
其中,当血管新生从视网膜内部开始延伸至视网膜和玻璃体之间的边界时,发生视网膜血管新生。由于新的血管中发生缺血,出现诸如玻璃体污染和视网膜脱落等症状。由此引起的眼部疾病包括糖尿病性视网膜病变、早产儿视网膜病变、视网膜静脉阻塞等。视网膜血管新生性疾病伴有视网膜血管的缺血性症状,因此也称为缺血性视网膜病变。
[0325]
在视网膜下血管新生中,从视网膜的下部结构开始的血管新生延伸至下部结构和视网膜之间的边界。在这种情况下,视网膜下血管新生可能是由脉络膜血管新生引起的。由此引起的代表性眼部疾病包括湿性年龄相关性黄斑变性(湿性amd)等。在湿性年龄相关性黄斑变性的情况下,渗透到黄斑之下的新的血管中发生的缺血会引起黄斑的光感受器和黄斑细胞的变性。
[0326]
或者,血管新生性眼部疾病可以根据症状而不针对特定眼部结构进行分类。例如,血管新生性青光眼就属于这样的类别。
[0327]
在特定的眼部结构或整个眼睛中未观察到血管新生的眼部疾病称为非血管新生性眼部疾病。干性年龄相关性黄斑变性(干性amd)就属于这样的类别。
[0328]
在本技术中要治疗的眼部疾病对应于血管新生性眼部疾病。
[0329]
7.根据本技术的药物组合物
[0330]
本技术可以提供包含一种或多种选自于根据本技术的化合物作为活性成分的药物组合物。在这种情况下,根据本技术的化合物是指化学式1至11表示的化合物,并且“2.本技术的化合物结构”的内容加以必要的变更将适用于化学式1至11。
[0331]
在特定的示例性实施方式中,根据本技术的药物组合物可用于治疗血管新生性眼部疾病。进一步地,血管新生性眼部疾病可以是脉络膜血管新生性疾病、视网膜血管新生性疾病、视网膜下血管新生性疾病、角膜血管新生性疾病、虹膜血管新生性疾病(虹膜红变)或血管新生性青光眼。在一个实施方式中,血管新生性眼部疾病可以是视网膜血管新生性疾病。进一步地,视网膜血管新生性疾病可以是糖尿病性视网膜病变、早产儿视网膜病变或视网膜静脉阻塞。优选地,视网膜血管新生性疾病可以是糖尿病性视网膜病变。在另一个示例性实施方式中,血管新生性眼部疾病可以是脉络膜血管新生性疾病。进一步地,脉络膜血管新生性疾病可以是湿性年龄相关性黄斑变性(湿性amd)。
[0332]
在特定的示例性实施方式中,根据本技术的药物组合物可以用于口服或滴眼给药。优选地,根据本技术的药物组合物可以用于滴眼给药。
[0333]
本技术可以提供治疗血管新生性眼部疾病的方法,所述方法包括给予一种或多种选自于根据本技术的化合物。在这种情况下,根据本技术的化合物是指化学式1至11表示的化合物,并且“2.本技术的化合物结构”的内容加以必要的变更将适用于化学式1至11。
[0334]
在特定的示例性实施方式中,血管新生性眼部疾病可以是脉络膜血管新生性疾病、视网膜血管新生性疾病、视网膜下血管新生性疾病、角膜血管新生性疾病、虹膜血管新生性疾病(虹膜红变)或血管新生性青光眼。在一个实施方式中,血管新生性眼部疾病可以是视网膜血管新生性疾病。进一步地,视网膜血管新生性疾病可以是糖尿病性视网膜病变、早产儿视网膜病变或视网膜静脉阻塞。优选地,视网膜血管新生性疾病可以是糖尿病性视网膜病变。在另一个示例性实施方式中,血管新生性眼部疾病可以是脉络膜血管新生性疾病。进一步地,脉络膜血管新生性疾病可以是湿性年龄相关性黄斑变性(湿性amd)。
[0335]
在特定的示例性实施方式中,根据本技术的治疗血管新生性眼部疾病的方法可以包括通过口服或滴眼途径给予根据本技术的化合物。优选地,根据本技术的治疗血管新生性眼部疾病的方法可以包括通过滴眼途径给予根据本技术的化合物。
[0336]
本技术的组合物和发明可用于对有此需要的受试者进行治疗。在特定的示例性实施方式中,受试者是哺乳动物,例如人类或非人类哺乳动物。当对受试者(例如人类)给药时,组合物或化合物优选作为例如药物组合物进行给药,所述药物组合物包含本技术的化合物和药学上可接受的载体。药学上可接受的载体在本领域中是众所周知的,例如,包括水或生理缓冲盐水等水溶液,或乙二醇、丙三醇、橄榄油等油,或诸如可注射有机酯等其他溶剂或溶媒。在优选的示例性实施方式中,当将这样的药物组合物给予给人类时,尤其是当通过侵入性途径给药(即,避免通过上皮顶壁运输或扩散的途径,例如,注射或移植)时,水溶液是无热原或基本上无热原的。可以对添加剂进行选择,例如,有助于制剂的持续释放或选择性地靶向一个或多个细胞、组织或器官。药物组合物可以片剂、胶囊剂(包括撒粉胶囊剂(sprinkle capsule)和明胶胶囊剂)、颗粒剂、可复溶的冻干剂、散剂、溶液剂、糖浆剂、栓剂或注射剂或其他单位剂型提供。组合物还可作为透皮给药系统(例如,皮肤贴剂)提供。组合物也可以作为适合局部给药的溶液剂(例如滴眼剂)提供。
[0337]
药学上可接受的载体可以包括生理学上可接受的试剂,例如用作使化合物(例如本技术的化合物)稳定、增加溶解度或增加吸收的试剂。生理学上可接受的试剂包括例如诸如葡萄糖、蔗糖或葡聚糖等碳水化合物,诸如抗坏血酸或谷胱甘肽等抗氧化剂,螯合剂,低分子量蛋白或其他稳定剂或添加剂。药学上可接受的载体(例如生理学上可接受的试剂)的
选择取决于例如组合物的给药途径。药物组合物的配方产品可以是自乳化药物递送系统或自微乳化药物递送系统。药物组合物(配方产品)可以是例如将本技术的化合物包含在脂质体或其他聚合物基质中的药物组合物。脂质体例如由磷脂或其他脂质组成,是无毒的、生理学上可接受的且可代谢的载体,它们相对易于制备和给予。
[0338]
本文所用的术语“药学上可接受的”是指在合理的医学判断范围内,未表现出过度毒性、刺激、过敏反应或其他问题或副作用,因此适合与受试者组织接触并具有合理的效益/风险比的化合物、材料、组合物和/或剂型。
[0339]
本文所用的术语“药学上可接受的载体”是指药学上可接受的材料、组合物或载体,例如液体或固体填充剂、稀释剂、添加剂、溶剂或包封材料。每种载体应为“可接受的”,这意味着它与剂型的其他成分相容,并且不会损害受试者。能够用作药学上可接受的载体的材料的一些实例包括:(1)糖,诸如乳糖、葡萄糖和蔗糖;(2)淀粉,诸如玉米淀粉或土豆淀粉;(3)纤维素,例如羧甲基纤维素钠、乙基纤维素和醋酸纤维素及其衍生物;(4)西黄蓍胶粉末;(5)麦芽;(6)明胶;(7)滑石;(8)添加剂,诸如可可黄油和栓剂蜡;(9)油,诸如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆油;(10)二醇类,诸如丙二醇;(11)多元醇,诸如甘油、山梨糖醇、甘露醇和聚乙二醇;(12)酯,诸如油酸乙酯和月桂酸乙酯;(13)琼脂;(14)缓冲剂,诸如氢氧化镁和氢氧化铝;(15)海藻酸;(16)无热原的水;(17)等渗蒸馏水;(18)林格溶液;(19)乙醇;(20)磷酸盐缓冲溶液;以及(21)药物剂型中包含的其他无毒相容的材料。
[0340]
药物组合物(配方产品)可以通过各种给药途径进行给药,给药途径包括口服(例如,诸如水溶液或非水溶液等浸渍液、或悬浮液剂、片剂、胶囊剂(包括撒粉胶囊剂和明胶胶囊剂)、大丸剂、散剂、颗粒剂、施用于舌头的糊剂);通过口腔粘膜吸收(例如:舌下);肛门、直肠或阴道(例如,作为阴道栓剂、乳剂或泡沫剂);肠胃外(肌肉内、静脉内、皮下或鞘内注射,例如无菌溶液或悬浮液);鼻;腹膜内;皮下;透皮(例如,作为施用于皮肤上的贴剂);和局部(例如,作为施用于皮肤的乳剂、软膏剂或喷雾剂,或者作为滴眼剂)给药。化合物也可以配制用于吸入。在某些示例性的实施方式中,可以简单地将化合物溶解或悬浮在无菌水中。适当的给药途径和适用于该途径的组合物的具体实例可以在例如美国专利号6,110,973、5,763,493、5,731,000、5,541,231、5,427,798、5,358,970和4,172,896以及其中引用的专利中查到。
[0341]
该配方产品可以方便地以单位剂型形式提供,并且可以通过制药学领域中众所周知的任意方法制备。可与载体材料混合以产生单一剂型的活性材料的量会因待治疗的受试者和特定的给药方式而有所不同。可与载体材料混合以产生单一剂型的活性材料的量通常是产生治疗作用的化合物的量。通常,基于100%,该量为约1%至约99%的活性材料,优选约5%至约70%,最优选约10%至约30%。
[0342]
制备这些配方产品或组合物的方法包括将活性化合物(例如,本技术的化合物)与载体以及一种或多种辅助成分组合在一起。通常,配方产品是通过将本技术的化合物与液体载体或细碎的固体载体或两者均匀紧密地合并在一起而制备的,必要时,将产物模制成型。
[0343]
适合口服给药的本技术的配方产品可以采用胶囊剂(包括撒粉胶囊剂和明胶胶囊剂)、扁囊剂、丸剂、片剂、含片剂(使用风味基料,通常是蔗糖和阿拉伯胶或西黄蓍胶)、冻干
剂、散剂、颗粒剂、或在水性或非水性液体中的溶液剂或悬浮液剂、或水包油液态乳剂或油包水液态乳剂、或酏剂或糖浆剂、或锭剂(使用惰性基料,例如明胶和甘油、蔗糖和/或阿拉伯胶)和/或漱口水等的形式,每种配方产品含有预定量的本技术的化合物作为活性材料。组合物或化合物也可以丸、软膏或糊的形式施用。
[0344]
为了制备固体剂型(胶囊剂(包括撒粉胶囊剂和明胶胶囊剂)、片剂、丸剂、糖衣丸剂、散剂、颗粒剂等),将活性成分与一种或多种药学上可接受的载体混合,所述载体例如柠檬酸钠或磷酸氢钙,和/或以下任一种:(1)填充剂或增量剂,诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和/或硅酸;(2)粘合剂,诸如羧甲基纤维素、海藻酸、明胶、聚乙烯基吡咯酮、蔗糖和/或阿拉伯胶;(3)保湿剂,诸如甘油;(4)崩解剂,例如琼脂、碳酸钙、土豆淀粉或木薯淀粉、海藻酸、某些硅酸盐和碳酸钠;(5)溶液阻滞剂,例如石蜡;(6)吸收加速剂,诸如季铵盐化合物;(7)润湿剂,诸如鲸蜡醇和单硬脂酸甘油酯;(8)吸附剂,诸如高岭土和膨润土;(9)润滑剂,诸如滑石、硬脂酸钾、硬脂酸镁、固体聚乙二醇、十二烷基硫酸钠及其混合物;(10)络合剂,诸如修饰的和未修饰的环糊精;以及(11)着色剂。对于胶囊剂(包括撒粉胶囊剂和明胶胶囊剂)、片剂和丸剂,药物组合物也可以包含缓冲剂。类似类型的固体组合物也可以使用乳糖或奶糖以及高分子量聚乙二醇等作为软填充明胶胶囊和硬填充明胶胶囊中的填充剂。
[0345]
可以通过压制或模制成片剂,并任选含有一种或多种其他成分。压制片剂可以使用粘合剂(例如,明胶或羟丙基甲基纤维素)、润滑剂、惰性稀释剂、防腐剂、崩解剂(例如,淀粉乙醇酸钠或交联羧甲基纤维素钠)、表面活性剂或分散剂来制备。模制片剂可以通过在合适的机器中对用惰性液体稀释剂浸泡的粉状化合物的混合物进行模制来制备。
[0346]
药物组合物的片剂和其他固体剂型例如糖衣丸剂、胶囊剂(包括撒粉胶囊剂和明胶胶囊剂)、丸剂和颗粒剂,可任选地用包衣或壳(诸如药品配制领域熟知的肠溶包衣和其他包衣)包围或制备。这些配方产品也可以:使用例如以不同比例的羟丙基甲基纤维素来制备,以提供所需的释放曲线;使用其他聚合物基质、脂质体和/或微球来制备,以提供其中的活性成分的缓慢释放或受控释放。这些配方产品可以通过以下方式灭菌:例如经细菌保留过滤器过滤;或以无菌固体组合物的形式与灭菌剂结合,所述无菌固体组合物能够溶于无菌水;或在即将使用前混入适量的无菌可注射材料。这些组合物也可以任选地包含乳浊剂,并且可以是组合物的形式,即这些组合物必然或优先地(任选地,以延迟的方式)在胃肠道中以一定量释放活性成分。可以使用的包埋组合物的实例包括聚合物材料和蜡。如果适当的话,活性材料也可以与一种或多种上述添加剂一起处于微囊密封的形式。
[0347]
对口服给药有用的液体剂型包括药学上可接受的乳剂、复溶冻干剂、微乳剂、溶液剂、悬浮液剂、糖浆剂和酏剂。除活性成分外,液体剂型还可以包含本领域中常用的惰性稀释剂,例如水或其他溶剂、环糊精及其衍生物、助溶剂和乳化剂,诸如乙醇、异丙醇、碳酸乙酯、乙酸乙酯、苄醇、苯甲酸苄酯、丙二醇、1,3-丁二醇、油(特别是棉籽油、花生油、玉米油、胚芽油、橄榄油、蓖麻油和芝麻油)、甘油、四氢糠醇、聚乙二醇和失水山梨糖醇的脂肪酸酯以及它们的混合物。
[0348]
除了惰性稀释剂外,口服组合物还可以包含佐剂,诸如润湿剂、乳化或悬浮剂、甜味剂、调味剂、着色剂、芳香剂和防腐剂。
[0349]
除活性材料外,悬浮液剂还可以包含悬浮剂,例如乙氧基化的异十八醇、聚氧乙烯山梨糖醇和失水山梨醇酯、微晶纤维素、氢氧化铝氧化物、膨润土、琼脂、西黄蓍胶及它们的
混合物。
[0350]
用于直肠、阴道或尿道给药的药物组合物的配方产品可以作为栓剂提供,并且可以通过将一种或多种活性化合物与一种或多种合适的无刺激的添加剂或载体混合在一起来制备,所述无刺激的添加剂或载体例如可可黄油、聚乙二醇、栓剂蜡或水杨酸酯/盐、以及在室温下为固体但在体温下为液体因而会在直肠或阴道腔中融化以释放活性化合物的材料。
[0351]
用于口服给药的药物组合物的配方产品可以作为漱口水、口腔喷雾或口服软膏的形式提供。
[0352]
或者或另外地,可以配制组合物用于通过导管、支架、线或其他管腔内装置来递送。通过此类装置的递送可能对膀胱、尿道、输尿管、直肠或肠道的递送是特别有用的。
[0353]
适合阴道给药的配方产品还包括阴道栓剂、棉塞、乳霜、凝胶、糊剂、泡沫剂或喷雾配方产品,其中含有本领域中已知的适当的载体。
[0354]
局部或透皮给药的剂型包括散剂、喷雾剂、软膏剂、糊剂、乳霜剂、乳液剂、凝胶剂、溶液剂、贴剂和吸入剂。活性化合物可以在无菌环境中与药学上可接受的载体混合,并在必要时与任何防腐剂、缓冲剂或喷射剂混合。
[0355]
除活性化合物,软膏剂、糊剂、乳霜剂和凝胶剂可以包含添加剂,诸如动物脂肪和植物脂肪、油、蜡、石蜡、淀粉、西黄蓍胶、纤维素衍生物、聚乙二醇、硅酮、膨润土、硅酸、滑石和氧化锌或它们的混合物。
[0356]
除活性化合物外,散剂和喷雾剂还可以包含添加剂,诸如乳糖、滑石、硅酸、氢氧化铝、硅酸钙和聚酰胺粉末或这些材料的混合物等。喷雾剂可以还包含常规的喷射剂,诸如氯氟烃以及挥发性的未取代烃诸如丁烷和丙烷。
[0357]
透皮贴剂具有将本技术的化合物的受控地递送至身体的附加优势。这种剂型可以通过将活性化合物溶解或分散在适当的溶媒中来制备。吸收增强剂可用于增加化合物穿透皮肤的流动。这种流动的速率可以通过提供速率控制膜或将化合物分散在聚合物基质或凝胶中来控制。
[0358]
眼部配方产品、眼用软膏剂、散剂、溶液剂等也被认为是在本技术的范围内。在美国公开号2005/0080056、2005/0059744、2005/0031697和2005/004074和美国专利号6,583,124中对示例性的眼部配方产品进行描述,通过引用将其内容并入本文。如果需要,液体眼部配方产品具有类似于泪液、房水或玻璃体液的特性或与此类流体相容的特性。优选的给药途径是局部给药(例如,诸如通过滴眼或通过植入物给药等局部给药)。
[0359]
本文所用的术语“非肠道给药”是指除肠内和局部给药外的给药方式,一般通过注射给药,包括静脉内、肌肉内、动脉内、鞘内、包膜内、眶内、心内、皮内、腹腔内、经气管、皮下、角质层下、关节内、包膜下、蛛网膜下、椎管内和胸骨内注射和输注,但不限于此。
[0360]
适用于肠胃外给药的药物组合物包含与一种或多种药学上可接受的无菌等渗水溶液或非水溶液、分散剂、悬浮液或乳液或无菌粉末混合的一种或多种活性化合物,或者在即将使用前可复溶于无菌可注射溶液或分散液的无菌粉末,并且,所述组合物可以包含抗氧化剂、缓冲剂、抑菌剂、当将配方产品给予预定接受者的血液时使该配方产品保持等渗的溶质、或悬浮或增稠剂。
[0361]
可以在本技术的药物组合物中使用的合适的水性或非水性载体的实例包括水、乙
醇、多元醇(例如甘油、丙二醇、聚乙二醇等)、它们的适宜的混合物、植物油(诸如橄榄油)和可注射的有机酯(例如油酸乙酯)。可以通过例如使用包衣材料(例如卵磷脂)、通过在分散剂和使用表面活性剂的情况下维持所需的粒径来维持适当的流动性。
[0362]
这些组合物还可以包含佐剂,诸如防腐剂、润湿剂、乳化剂和分散剂。可以通过包含各种抗菌剂和抗真菌剂,诸如对羟基苯甲酸酯、氯丁醇、苯酚山梨酸等来确保遏制微生物的活性。另外,组合物中包含等渗剂诸如糖、氯化钠等可能是有利的。另外,可注射药物配方产品的延长吸收时间可以通过包含延长吸收的试剂(诸如单硬脂酸铝和明胶)来实现。
[0363]
在某些情况下,为了延长药物的作用,减慢从皮下和肌肉内注射剂的药物吸收是有利的。这可以通过使用具有水溶性差的结晶或无定形材料的液体悬浮液来实现。在这种情况下,药物的吸收速率取决于该材料的溶解速率,即可取决于晶体的大小和形状。或者,肠胃外给予的药物配方产品的延迟吸收可以通过将该药物溶解或悬浮在油溶媒中实现。
[0364]
可注射的储库形式是通过在可生物降解聚合物(例如聚乳酸-聚乙醇酸)中形成主体化合物的微包封基质制成的。根据药物与聚合物的比率以及所使用的特定聚合物的性质,可以对药物的释放速率进行调节。其他可生物降解聚合物的实例包括聚原酸酯和聚酸酐。可注射的储库配方产品也可以通过将药物包封在与身体组织相容的脂质体或微乳液中来制备。
[0365]
为了在本技术的方法中使用,活性化合物可以由其本身提供或作为含有例如0.1%至99.5%的有效成分(更优选地0.5%至90%)与药学上可接受的载体相结合的药物组合物提供。
[0366]
引入方法也可以由可重复负载或可生物降解的装置提供。最近已经开发各种缓释聚合物器件,并在体内进行测试以调整包含蛋白质生物药物在内的药物的释放。可以使用各种生物相容性聚合物(包括水凝胶),包括可生物降解和不可降解的聚合物,以用于制造使化合物在特定目标位点持续释放的植入物。
[0367]
药物组合物中活性成分的实际剂量水平可能会发生变化,以便获得一定量的活性成分,该量可有效地实现对特定患者、组合物和给药方式的所需治疗响应而不会表现出对患者的毒性。
[0368]
所选剂量水平将取决于多种因素,包括:使用的特定化合物、或化合物或其酯、盐或酰胺的组合的活性;给药途径;给药时间;使用的特定化合物的排泄速率;治疗持续时间;与使用特定化合物的组合中使用的其他药物、化合物和/或材料;待治疗的患者的年龄、性别、体重、病症、全身健康情况和先前用药史;以及医疗领域众所周知的其他因素。
[0369]
具有本领域普通技能的医师或兽医可以容易地确定并开具出所需的治疗有效量的药物组合物。例如,医师或兽医可以低于达到所需治疗效果的水平开始给予药物组合物或化合物,并缓慢增加剂量,直到达到所需的效果。“治疗有效量”是指足以引起所需治疗作用的化合物的浓度。通常可以理解,该化合物的有效量会根据受试者的体重、性别、年龄和病史而变化。影响该有效量的其他因素包括受试者病症的严重程度、所治疗的紊乱、化合物的稳定性以及(如果需要的话)与本技术的化合物一起给药的另一类型的治疗剂,但不限于此。较大的总剂量可以由制剂的多次给予而递送。确定疗效和剂量的方法是本领域技术人员已知的方法(isselbacher等(1996年),harrison’s principles of internal medicine 13ed.,1814-1882,并入本文作为参考)。
[0370]
通常,在本技术的组合物和方法中使用的活性化合物的适当每日剂量是有效产生治疗作用的该化合物的最低剂量的量。这样的有效剂量通常取决于上述因素。
[0371]
在某些示例性实施方式中,本技术的化合物可以单独使用或与另一类型的治疗剂联合给药。本文所用的术语“联合给药”是指两种或更多种不同的治疗化合物的任何给药方式,该给药方式使得在先前给药的治疗化合物在体内仍然有效时给予第二种化合物(例如,两种化合物同时对受试者有效,并且可以包含两种化合物的协同作用)。例如,不同的治疗化合物可以同一配方产品或单独的配方产品(同时或顺序地)给药。在某些示例性的实施方式中,可以在1小时、12小时、24小时、36小时、48小时、72小时或一周内给予不同的治疗化合物。因此,接受这种治疗的受试者可以从不同治疗化合物的联合作用中受益。
[0372]
在某些示例性的实施方式中,相较于本技术的化合物或一种或多种另外的治疗剂的各自单独给药,本技术的化合物与一种或多种另外的治疗剂(例如一种或多种另外的化疗药剂)的联合给药提供了改善的疗效。在某些此类的示例性实施方式中,联合给药提供了累加作用,其中累加作用是指本技术化合物以及一种或多种另外的治疗剂单独给予的各自作用的总和。
[0373]
本技术包括本技术的化合物的药学上可接受的盐在本技术的组合物和方法中的用途。在某些示例性的实施方式中,本技术的预期盐包括烷基、二烷基、三烷基或四烷基铵盐,但不限于此。在某些示例性的实施方式中,本技术的预期盐包括如下的盐:l-精氨酸、苯乙苄胺、苄星青霉素、甜菜碱、氢氧化钙、胆碱、二甲基乙醇胺、二乙醇胺、二乙胺、2-(二乙基氨基)乙醇、乙醇胺、乙二胺、n-甲基葡糖胺、海巴明、1h-咪唑、锂、l-赖氨酸、镁、4-(2-羟基乙基)吗啉、哌嗪、钾、l-(2-羟基乙基)吡咯烷、钠、三乙醇胺、氨丁三醇和锌,但不限于此。在某些示例性的实施方式中,本技术的预期盐包括na、ca、k、mg、zn或其他金属的盐,但不限于此。
[0374]
药学上可接受的酸加成盐也可以作为水、甲醇、乙醇、二甲基甲酰胺和其他各种溶剂化物的形式存在。也可以制备这种溶剂化物的混合物。这种溶剂化物的来源可以来自进行结晶的溶剂,这可能是由于要进行制备或要进行结晶的溶剂的内在特征,或者是由于溶剂的意外原因所致。
[0375]
根据本技术的组合物可以包含润湿剂(诸如十二烷基硫酸钠和硬脂酸镁)、乳化剂和润滑剂、着色剂、释放剂、包衣剂、甜味剂、调味和芳香剂、防腐剂和抗氧化剂。
[0376]
药学上可接受的抗氧化剂的实例包括:(1)水溶性抗氧化剂,诸如抗坏血酸、半胱氨酸盐酸盐、硫酸氢钠、焦亚硫酸钠、亚硫酸钠等;(2)油溶性抗氧化剂,诸如抗坏血酸棕榈酸酯、丁基化羟基茴香醚(bha)、丁基化羟基甲苯(bht)、卵磷脂、没食子酸丙酯、α-生育酚等;(3)金属螯合剂,例如柠檬酸、乙二胺四乙酸(edta)、山梨糖醇、酒石酸、磷酸等。
[0377]
具体实施例
[0378]
实施例1.以下实验的实验方法
[0379]-氧诱导的视网膜病变小鼠模型和smx眼球注射
[0380]
出生后7日龄(p7)的c57bl/6j(hyochang science inc.)幼龄小鼠在高氧室(coy lab.,体内室,75%o2)中饲养5天(p12),然后在常氧环境中这些小鼠饲养5天(p17),诱导氧诱导的视网膜病变小鼠模型。在将幼龄小鼠从高氧室中移出后(p12),以0.15mm的浓度(0.1%dmso)将1μl的smx进行眼内注射一次。用于眼注射的smx在磷酸盐缓冲盐水(1
×
pbs)
中稀释至0.1%后使用,并将在1
×
pbs中的0.1%dmso用于对照小鼠。在eylea(阿柏西普,抗vegfab)的情况下,将40μg在1μl中稀释,将1μl进行眼内注射。
[0381]-视网膜血管分析
[0382]
形成氧诱导的视网膜病变小鼠模型,然后用1
×
pbs和4%pfa灌注。摘取小鼠眼睛并将其固定在4%pfa中后,分离视网膜。将分离的视网膜用1
×
pbs洗涤,然后在室温下封闭(在1
×
pbs中的0.1%bsa、0.1%triton x-100)1小时。在4℃下用cd31血管染色抗体(cd31,细胞信号转导,1:100)处理视网膜过夜。次日,用1
×
pbs洗涤视网膜,然后用第二抗体(alexa fluor 594-anti兔抗体,1:500)在4℃染色过夜。次日,用1
×
pbs洗涤视网膜,然后固定。在荧光显微镜(zen,axio zoom)下对血管染色进行分析。使用zen软件进行分析。
[0383]-细胞培养
[0384]
muller细胞(mio-m1)在37℃和5%co2条件下培养,使用包含10%胎牛血清和1%青霉素和链霉素的dmem-低葡萄糖(sigma)作为培养基。缺氧室保持在37℃、5%co2和1%o2条件下。
[0385]-蛋白质印迹
[0386]
将muller细胞(mio-m1)以4
×
105的数量接种在60mm板上,并在3ml培养基中培养24小时。次日,根据图中所示的浓度,用smx(线粒体热休克蛋白90(trap1)抑制剂)对细胞进行处理,并在缺氧室中培养6小时。全细胞裂解物由培养的细胞制成,电泳,转移到pvdf膜上,然后在4℃下用第一抗体处理18小时。次日,用第二抗体对全细胞裂解物处理1小时,并使用蛋白质印迹检测试剂对蛋白质表达进行分析。
[0387]-总rna提取和rna水平分析
[0388]
muller细胞用gamitrinib和smx以3μm进行处理,并在1%o2环境中培养8小时,然后提取总rna(qiagen,总rna提取试剂盒)。由1μg总rna合成cdna(neb,cdna合成试剂盒)。使用pcr合成血管生成因子后,通过琼脂糖电泳对rna水平进行分析。
[0389]-血管形成试验(tube formation assay)
[0390]
使用sirna将trap1从muller细胞中敲除。更换培养基后,通过将细胞在1%o2环境中培养24小时制成条件培养基,并在-86℃下储存。用基质胶对96孔板进行涂覆,并将huve细胞和条件培养基混合并等分。将细胞在37℃孵化器中培养4小时后,对血管形成进行分析。使用bio image navigator显微镜捕获图像,并通过image j程序进行分析。
[0391]-atp酶活性试验
[0392]
根据制造商的手册,通过picolorlock gold磷酸盐检测试剂盒(innova biosciences)测量无机磷酸盐的释放,对atp酶活性进行测量。在ph 7.0和37℃的条件下,用在100mm tris、20mm kcl和6mm mgcl2中的0.2mm atp培养trap1(0.5μm)3小时。此后,将picolorlock gold试剂和加速剂(100:1)添加到100μl atp水解产物样品中。在25℃培养5分钟后,通过向其中添加10μl的终止溶液停止颜色的变化,并通过synergy neo酶标仪(biotek instruments)测量620nm处的吸光度。
[0393]
为了抑制能力的分析,用预先确定浓度(0.520μm)的抑制剂培养trap1 30分钟,然后与atp搅拌。将吸光度值归一化至dmso对照,并将数据表示为atp酶活性%。
[0394]
实施例2.trap1的遏制对血管新生性眼部疾病具有治疗作用。
[0395]
实施例2.1.使用trap1+/+、+/
–
小鼠制备氧诱导的视网膜病变小鼠模型
[0396]
采用trap1+/
–
(雌性)和trap1+/
–
(雄性)杂交的方法,获得来自同一胚胎的trap1+/+和trap1+/
–
小鼠。
[0397]
通过将野生或杂化trap1幼龄小鼠在出生后第7天至第12天(p7-p12)在高氧室(coy lab.,体内室,75%o2)饲养,然后从第12天到第17天在常氧环境中饲养这些小鼠,建立视网膜病变小鼠模型。
[0398]
实施例2.2.trap-1+/+、+/
–
氧诱导的视网膜病变小鼠模型的视网膜血管分析
[0399]
形成氧诱导的视网膜病变小鼠模型,然后用1
×
pbs和4%pfa灌注。摘取小鼠眼睛并将其固定在4%pfa中后,分离视网膜。将分离的视网膜用1
×
pbs洗涤,然后在室温下封闭(在1
×
pbs中的0.1%bsa、0.1%triton x-100)1小时。在4℃下用cd31血管染色抗体(cd31,细胞信号转导,1:100)处理视网膜过夜。次日,用1
×
pbs洗涤视网膜,然后用第二抗体(alexa fluor 594-anti兔抗体,1:500)在4℃染色过夜。次日,用1
×
pbs洗涤视网膜,然后固定。在荧光显微镜(zen,axio zoom)下对血管染色进行分析。使用zen软件进行分析。
[0400]
视网膜血管分析的结果是,在trap-1+/+氧诱导的视网膜病变小鼠模型中,观察到视网膜的无血管区域增加,同时视网膜的血管新生区域增加,因此清楚地观察到了氧诱导的视网膜病变的症状。相反,在trap-1+/
–
氧诱导的视网膜病变小鼠模型中,观察到视网膜的无血管区域和血管新生区域的变化明显更小,因此可以证实,氧诱导的视网膜病变得到了显著改善(图4和图5)。此外,在trap1+/
–
氧诱导的视网膜病变小鼠模型中,除了氧诱导的视网膜病变的改善之外,未观察到与野生型小鼠不同的表型,因此可以证实,没有依据trap-1遏制的副作用。总而言之,可能得出结论,即trap-1是治疗或预防血管新生性疾病的有效且安全的靶标。
[0401]
实施例2.3.trap-1的遏制遏制了血管新生因子的产生
[0402]
先前提到,trap-1既是hif-1α的亲代调节物,也是血管新生因子(诸如vegf)的亲代调节物(图2)。由于糖尿病性视网膜病变是典型的视网膜血管新生性疾病,因此trap-1的遏制会遏制血管新生因子,还将治疗糖尿病性视网膜病变。
[0403]
进行血管形成试验,以验证trap-1是血管新生因子的亲代调节物。
[0404]
muller细胞(mio-m1)在37℃和5%co2条件下培养,并使用包含10%胎牛血清、1%青霉素和链霉素的dmem-低葡萄糖(sigma)作为培养基。缺氧室保持在37℃、5%co2和1%o2条件下。使用sirna将trap1从muller细胞中敲除。更换培养基后,通过将细胞在1%o2环境中培养24小时制成条件培养基,并在-86℃下储存。用基质胶对96孔板进行涂覆,并将huve细胞和条件培养基混合并等分。将细胞在37℃孵化器中培养4小时后,对血管形成进行分析。使用bio image navigator显微镜捕获图像,并通过image j程序进行分析。
[0405]
血管形成试验的结果是(图6和图7),可以证实血管长度、分支数量和交叉点减少,因此,在使用由敲除了trap1的muller细胞制成的条件培养基的组中,血管新生得到遏制。
[0406]
实施例3.smx分子具有遏制trap-1的能力。
[0407]
实施例3.1.实验中使用的材料和来源
[0408]
为了进行实验,获得了具有以下结构式的smx分子。smx分子购自medchemexpress(cas no.845959-50-4)。
[0409][0410]
此外,为了与smx进行比较,获得pu-h71(hsp90的n-末端结构域抑制剂)和gamitrinib(先前由发明人开发)。通过tocris获得pu-h71,通过legochem biosciences,inc.获得gamitrinib。
[0411]
实施例3.2.trap-1和smx的结合结构
[0412]
对trap-1和smx的结合结构进行x射线衍射(xrd)测量,以确认smx在trap-1上结合的位置。对于trap-1,使用斑马鱼的trap-1。观察trap-1和smx的结合结构的结果如图12所示。图12的右侧示出了smx在trap-1上结合所涉及的主要氨基酸残基。为了了解残基的功能,对相应残基的种间保守进行分析。分析的结果是,由于位点具有极高的物种间保守性,可以预测相应的残基在trap-1的功能中发挥着重要作用(图13)。
[0413]
为了更好地理解它们的作用,对上述结合结构进行比较,并将其与hsp90(trap-1的同源物)的结构进行了分析。从以前的论文中,推导出hsp90和cdk4(hsp90的客户蛋白)的结合结构[11]。对两种结构比较分析的结果是,可以通过结构比较来证实,smx在trap-1上的结合位置与客户蛋白在hsp90上的结合位置一致。对应的位置是属于trap-1中间结构域的残基,可以预测smx将与该客户蛋白竞争性地结合至trap-1(图11)。
[0414]
实施例3.3.smx与客户蛋白竞争性地结合至trap-1
[0415]
进行pull-down试验,以确认smx是否竞争性地与已知为trap-1的客户蛋白的sirt3和sdhb结合。从细菌细胞中纯化处于gst融合形式的trap1蛋白,然后与谷胱甘肽珠结合以制成trap1-珠形式,然后trap1-珠形式在4℃下用药物与从哺乳动物细胞分离的线粒体(thermo scientific线粒体分离试剂盒)结合18小时。根据图中所示的浓度,用smx、gamitrinib和pu-h71(先前开发的hsp90的n-末端结构域抑制剂)作为药物对细胞进行处理。pull-down试验的结果(图14)是,证实了随着smx浓度的增加,客户蛋白的结合显著降低。这是smx具有与trap-1中间结构域结合能力的有力证据。这一事实支持了,在用gamitrinib和pu-h71的处理的结果中,sirt3和sdhb的表达无显著改变。
[0416]
另外,在制备突变体(其在实施例2.2中证实的smx在trap-1上的结合部分被修饰)的trap1突变体-珠形式之后,进行pull-down试验(图15,左侧)。实验的结果是,观察到相应位置被修饰的突变体无法正确地结合至客户蛋白。这交叉验证了smx与trap-1结合的位置与trap-1与客户蛋白结合的位点相同。
[0417]
实施例3.4.smx降低了血管新生因子的表达。
[0418]
muller细胞(mio-m1)在37℃和5%co2条件下培养,使用包含10%胎牛血清和1%青霉素和链霉素的dmem-低葡萄糖(sigma)作为培养基。缺氧室保持在37℃、5%co2和1%o2条件下。将muller细胞(mio-m1)以4
×
105的数量接种在60mm板上,并在3ml培养基中培养24
小时。次日,根据图中所示的浓度,用smx(线粒体热休克蛋白90(trap1)抑制剂)和pu-h71(先前开发的hsp90的n-末端结构域抑制剂)对细胞进行处理,并在缺氧室中培养24小时。全细胞裂解物由培养的细胞制成,电泳,转移到pvdf膜上,然后在4℃下用第一抗体处理18小时。次日,用第二抗体对全细胞裂解物处理1小时,并使用蛋白质印迹检测试剂对蛋白质表达进行分析。
[0419]
另外,蛋白质印迹和定量聚合酶链反应的结果是,可以证实,smx遏制了包括hif-1α(图8)在内的众所周知的血管新生因子的表达(图9)。实施例3.5.smx通过与现有hsp90抑制剂不同的新机制对trap1进行遏制。
[0420]
从实施例3.3的结果中,可以证实smx成功遏制了trap-1的功能。随后,为了验证smx通过与现有的hsp90抑制剂不同的新机制来抑制trap-1,通过用smx和pu-h71各自对trap-1进行处理,进行atp酶活性试验。
[0421]
现有的hsp90抑制剂(gamitrinib、pu-h71)具有与hsp90的n-末端结构域结合的特征,而hsp90的n-末端结构域作为atp酶起效,因此,具有降低trap-1的atp酶活性的特征。然而,由于hsp90的n-末端结构域在旁系同源物之间具有很高的同源性,因此存在一个问题,即这些抑制剂非选择性地遏制hsp90。当hsp90被非选择性遏制时可能发生的问题是在前面“4.通过遏制trap-1治疗血管新生性疾病”中提到的问题。
[0422]
因此,当smx可以如前所述遏制trap-1而无需与trap-1的n-末端结构域结合,则可能创新地解决现有hsp90的问题。此外,这将通过测量用smx处理后的trap-1的atp酶活性来验证。
[0423]
根据制造商的手册,通过picolorlock gold磷酸盐检测试剂盒(innova biosciences)测量无机磷酸盐的释放,对atp酶活性进行测量。在ph 7.0和37℃的条件下,用在100mm tris、20mm kcl和6mm mgcl2中的0.2mm atp培养trap1(0.5μm)3小时。此后,将picolorlock gold试剂和加速剂(100:1)添加到100μl atp水解产物样品中。在25℃培养5分钟后,通过向其中添加10μl的终止溶液停止颜色的变化,并通过synergy neo酶标仪(biotek instruments)测量620nm处的吸光度。
[0424]
为了抑制能力的分析,用预先确定浓度(0.520μm)的抑制剂培养trap1 30分钟,然后与atp搅拌。将吸光度值归一化至dmso对照,并将数据表示为atp酶活性%。
[0425]
atp酶活性试验的结果是,观察到在用pu-h71处理trap-1时,随着pu-h71浓度的增加,trap-1的atp酶活性显著降低,而当用smx处理trap1时,随着smx浓度的增加,trap-1的atp酶活性显著增加(图16)。这表明smx不与trap1的n-末端结构域结合,并强烈表明smx通过与客户蛋白的结合位置结合而同时促进atp的结合。
[0426]
实施例4.smx分子对血管新生性眼部疾病具有治疗功效。
[0427]
出生后7日龄(p7)的c57bl/6j(hyochang science inc.)幼龄小鼠在高氧室(coy lab.,体内室,75%o2)中饲养5天(p12),然后,在常氧环境中饲养这些小鼠5天(p17),诱导氧诱导的视网膜病变小鼠模型。在将幼龄小鼠从高氧室中移出后(p12),以0.15mm的浓度(0.1%dmso)将1μl的smx进行眼内注射一次。用于眼注射的smx在磷酸盐缓冲盐水(1
×
pbs)中稀释至0.1%后使用,并将在1
×
pbs中的0.1%dmso用于对照小鼠。在eylea(阿柏西普,抗vegfab,其在相关领域中已知对糖尿病性视网膜病变具有治疗功效)的情况下,将40μg在1μl中稀释,将1μl进行眼内注射。
[0428]
形成氧诱导的视网膜病变小鼠模型,然后用1
×
pbs和4%pfa灌注。摘取小鼠眼睛并将其固定在4%pfa中后,分离视网膜。将分离的视网膜用1
×
pbs洗涤,然后在室温下封闭(在1
×
pbs中的0.1%bsa、0.1%triton x-100)1小时。在4℃下用cd31血管染色抗体(cd31,细胞信号转导,1:100)处理视网膜过夜。次日,用1
×
pbs洗涤视网膜,然后用第二抗体(alexa fluor 594-anti兔抗体,1:500)在4℃染色过夜。次日,用1
×
pbs洗涤视网膜,然后固定。在荧光显微镜(zen,axio zoom)下对血管染色进行分析。使用zen软件进行分析。
[0429]
对照小鼠、注射eylea的小鼠和注射smx的小鼠的视网膜血管分析的结果是(图17),首先,注射smx的小鼠视网膜的血管新生区域降低到与注射eylea的小鼠视网膜相同的水平,因此可以证实smx对糖尿病性视网膜病变具有治疗功效。此外,特别地,证实与注射eylea的小鼠视网膜相比,注射smx的小鼠视网膜的无血管区域显著降低,由此可以证实,与eylea不同,smx具有将视网膜的血管新生模式正常化的作用。
[0430]
实施例5.本技术的分子是可以通过口腔/滴眼来施用的小分子。
[0431]
现有的糖尿病性视网膜病变的治疗剂是针对vegf的抗体药物,由于其大的分子量,对组织的侵入性较小,因此只有眼部注射处方是可能的。由于smx和根据本技术的新型化合物分子作为小分子药物被认为在递送方面得到显著改善,因此,将滴眼剂给予小鼠模型。
[0432]
出生后7日龄(p7)的c57bl/6j(hyochang science inc.)幼龄小鼠在高氧室(coy lab.,体内室,75%o2)中饲养5天(p12),然后,在常氧环境中饲养这些小鼠5天(p17),诱导氧诱导的视网膜病变小鼠模型。
[0433]
出生后7日龄(p7)的c57bl/6j(hyochang science inc.)幼龄小鼠在高氧室(coy lab.,体内室,75%o2)中饲养5天(p12),然后,在常氧环境中饲养这些小鼠5天(p17),诱导氧诱导的视网膜病变小鼠模型。在p12至p17期间(5天),即由正常氧所诱导的氧诱导视网膜病变的时间期间,用liposic(溶剂)将smx稀释至1mm,然后,滴眼给药,每天3次。右眼给予smx,左眼给予对照liposic。
[0434]
形成氧诱导的视网膜病变小鼠模型,然后用1
×
pbs和4%pfa灌注。摘取小鼠眼睛并将其固定在4%pfa中后,分离视网膜。将分离的视网膜用1
×
pbs洗涤,然后在室温下封闭(在1
×
pbs中的0.1%bsa、0.1%triton x-100)1小时。在4℃下用cd31血管染色抗体(cd31,细胞信号,1:100)处理视网膜过夜。次日,将视网膜用1
×
pbs洗涤,然后,用第二抗体(alexa fluor 594-anti兔抗体,1:500)在4℃染色过夜。次日,视网膜用1
×
pbs洗涤,然后固定。在荧光显微镜(zen,axio zoom)下对血管染色进行分析。使用zen软件进行分析。
[0435]
滴眼给药后通过视网膜血管分析获得的照片和程序分析结果如图18和图19所示。smx被滴注到每个个体的右眼中,左眼未给予smx并用作对照。结果证实,在注射smx的眼睛中,血管新生区域和无血管区域显著降低,因此改善了视网膜血管新生性疾病。
[0436]
实施例6.合成本技术新型化合物的方法
[0437]
提供的是用于制备本技术的由[化学式2]至[化学式5]表示的新型分子的方法。
[0438]
用于制备所述化合物的方法不限于下面描述的特定实施例,并且可以使用本领域技术人员广泛知晓的方法来生产该化合物。
[0439]
实施例6.1.制备(10-(2-溴-5-羟基-3,4-二甲氧基-6-甲基苯基)癸基)三苯基膦甲酸盐(sb-u009)的方法
[0440]
在本技术中,将对制备由[化学式2]表示的化合物的方法进行描述(图20)。制备化学式2的化合物的方法包括以下制备步骤1至7。
[0441]
步骤1.5-(10-溴癸基)-1,2,3-三甲氧基-苯的制备
[0442]
将正丁基锂(n-buli,2.5m,2.10ml,1eq)在
–
78℃下滴加于溶解在四氢呋喃(thf,20ml)中的5-溴-1,2,3-三甲氧基-苯的溶液。
[0443]
添加后,将混合物在相同的温度下搅拌1小时,然后在
–
78℃下滴加1,10-二溴代癸烷(3.16g,10.52mmol,2eq)的thf溶液(10ml)。然后将所得的混合物在20℃下搅拌11小时。
[0444]
通过液相色谱质谱仪(lcms)证实,检测到50.6%的期望的质量。
[0445]
残余物用饱和nh4cl(10ml)稀释,并用etoac(50ml
×
3)萃取。
[0446]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0447]
残余物通过柱色谱法(sio2,石油醚/乙酸乙酯=100/0至95/5)纯化以获得化合物5-(10-溴癸基)-1,2,3-三甲氧基-苯(580mg,1.02mmol,产率为19.35%,纯度为68%),无色油。
[0448]1h nmr(400mhz,cdcl3)δ=6.40(s,2h),3.86(s,6h),3.83(s,3h),3.42(t,j=6.8hz,2h),2.59-2.52(m,2h),1.86(quin,j=7.2hz,2h),1.60(br d,j=5.5hz,2h),1.48-1.38(m,2h),1.38-1.26(m,10h).
[0449]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h31
bro3)的质量进行测量,确认m/z测量值387.4、387.1[m+h]
+
。
[0450]
步骤2.6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛的制备
[0451]
在0℃下,将溶解于无水二氯甲烷(干燥的ch2cl2,2ml)中的5-(10-溴癸基)-1,2,3-三甲氧基-苯(580mg,1.02mmol,1eq)的溶液滴加至alcl3的干燥ch2cl2溶液(8ml)中。
[0452]
将混合物在相同的温度下搅拌45分钟,用10分钟逐渐滴加二氯(甲氧基)甲烷(188.97mg,1.64mmol,145.36μl,1.61eq,收率68%)的干燥的ch2cl2(2ml)溶液,将混合物在0℃下搅拌5分钟。
[0453]
这时,通过lcms证实反应已完成。
[0454]
将反应混合物倒入30ml冰水中,分离二氯甲烷(亚甲基氯)相,然后使用二氯甲烷(亚甲基氯,50ml)萃取水相两次。
[0455]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0456]
粗产物不经进一步纯化直接用于下一步。
[0457]
获得化合物6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛(510mg,858.27μmol,产率为84.29%,纯度为69.9%),无色油。
[0458]1h nmr(400mhz,cdcl3)δ=10.41(s,1h),6.53(s,1h),4.00(s,3h),3.95(s,3h),3.89(s,3h),3.43(t,j=6.9hz,2h),2.99-2.92(m,2h),1.93-1.82(m,2h),1.49-1.39(m,4h),1.32(br s,10h).
[0459]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h31
bro4)的质量进行测量,确认m/z测量值415.4、415.1[m+h]
+
。
[0460]
步骤3.6-(10-溴癸基)-2-羟基-3,4-二甲氧基-苯甲醛的制备
[0461]
在0℃下,将三氯化硼(bcl3,1m,1.9ml,2.21eq)滴加至溶解于ch2cl2(10ml)的6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛(510mg,858.27μmol,1eq,纯度为69.9%)的溶液中。
[0462]
将混合物在0℃下搅拌30分钟,然后在20℃下搅拌30分钟。
[0463]
这时,经lcms证实反应已完成。
[0464]
将残余物倒入冰水(30ml)中,并使用ch2cl2(50ml
×
3)萃取。
[0465]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0466]
残余物使用柱色谱法(sio2,石油醚/乙酸乙酯=100/0至95/5)纯化以获得化合物6-(10-溴癸基)-2-羟基-3,4-二甲氧基-苯甲醛(300mg,583.06μmol,产率为67.93%,纯度为78%),无色油。
[0467]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h29
bro4)的质量进行测量,确认m/z测量值401.3、401.1[m+h]
+
。
[0468]1h nmr(400mhz,cdcl3)δ=12.30-12.20(m,1h),10.24-10.03(m,1h),6.34(s,1h),3.96(s,3h),3.89(s,3h),3.43(t,j=6.9hz,2h),2.90-2.83(m,2h),1.88(quin,j=7.1hz,2h),1.70-1.60(m,2h),1.50-1.38(m,3h),1.49-1.29(m,1h).
[0469]
步骤4.3-溴-2-(10-溴癸基)-6-羟基-4,5-二甲氧基-苯甲醛的制备
[0470]
在0℃下,将溶解于氯仿(chcl3,2.5ml)和四氯化碳(ccl4,2.5ml)中的6-(10-溴癸基)-2-羟基-3,4-二甲氧基-苯甲醛(250mg,622.92μmol,1eq)的溶液加入至n-溴代琥珀酰亚胺(133.04mg,747.51μmol,1.2eq)。
[0471]
将混合物在0℃下搅拌1小时,然后在20℃下搅拌11小时。
[0472]
这时,经lcms证实反应已完成。
[0473]
使用碳酸钠(nahco3,10ml)和乙酸乙酯(etoac,20ml
×
3)对混合物进行萃取。
[0474]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0475]
残余物通过制备tlc(sio2,石油醚/乙酸乙酯=4:1)纯化以获得化合物3-溴-2-(10-溴癸基)-6-羟基-4,5-二甲氧基-苯甲醛(200mg,307.77μmol,产率为49.41%,纯度为73.9%),黄色油。
[0476]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h28
br2o4)的质量进行测量,确认m/z测量值480.2、481.0[m+h]
+
。
[0477]
步骤5.5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚的制备
[0478]
在0℃下,通过加料漏斗,将溶解于ch2cl2(4ml)中的3-溴-2-(10-溴癸基)-6-羟基-4,5-二甲氧基-苯甲醛(190mg,292.38μmol,1eq,纯度为73.9%)和三乙基硅烷(tes,et3sih,169.99mg,1.46mmol,233.50μl,5eq)的溶液用5分钟滴加至三氟乙酸(tfa,708.40mg,6.21mmol,460μl,21.25eq)中。
[0479]
将反应混合物在0℃下搅拌2小时。
[0480]
这时,经lcms证实反应已完成。
[0481]
将混合物缓慢倒入饱和的碳酸氢钠(nahco3,50ml)中,然后,使用100ml ch2cl2(100ml
×
3)萃取。
[0482]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0483]
残余物使用制备tlc(sio2,石油醚:乙酸乙酯=4:1)纯化以获得化合物5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚(130mg,241.64μmol,产率为82.65%,纯度为72%),无色油。
[0484]
步骤6.4-溴-5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚的制备
[0485]
将溶解于乙酸(acoh,5ml)并搅拌的5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚(130mg,241.64μmol,1eq,纯度为72%)和溴化钠(nabr,37.29mg,362.46μmol,11.65μl,1.5eq)的溶液加入到过氧化氢(h2o2,41.09mg,362.46μmol,34.82μl,纯度为30%,1.5eq)中,所得混合物在20℃下搅拌3小时。
[0486]
这时,经lcms证实反应已完成。
[0487]
用30ml饱和nahco3/na2s2o3以10:1的比例将残余物稀释,然后,使用etoac(30ml
×
3)萃取。
[0488]
将混合的有机层用盐水(10ml)洗涤,然后经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0489]
粗产物不经进一步纯化直接用于下一步。
[0490]
经上述操作,得到化合物4-溴-5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚(140mg,195.18μmol,产率为80.77%,纯度为65%),黄色油。
[0491]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h30
br2o3)的质量进行测量,确认m/z测量值466.3、466.9[m+h]
+
。
[0492]1h nmr(400mhz,cdcl3)δ=5.73(s,1h),3.86(s,3h),3.78(s,3h),3.34(t,j=6.9hz,2h),2.71-2.64(m,2h),2.14(s,3h),1.84-1.76(m,2h),1.37(br d,j=4.1hz,7h),1.24(br s,7h).
[0493]
步骤7.(10-(2-溴-5-羟基-3,4-二甲氧基-6-甲基苯基)癸基)三苯基膦甲酸盐的制备
[0494]
将溶解于甲苯(2ml)中的4-溴-5-(10-溴癸基)-2,3-二甲氧基-6-甲基-苯酚(140mg,195.18μmol,1eq,纯度为65%)和三苯基膦(pph3,255.96mg,975.88μmol,5eq)的搅拌溶液在n2下于125℃加热8小时。
[0495]
这时,经lcms证实反应已完成。
[0496]
在真空中去除溶剂获得残余物。
[0497]
残余物使用柱色谱法(sio2,石油醚/乙酸乙酯=100/0至0/100;乙酸乙酯:meoh=100/0至92/8)纯化。
[0498]
残余物使用制备hplc(fa条件;柱:xtimate c18 100
×
30mm
×
3μm;流动相:[水(0.225%fa)-acn];b%:40%-70%,8分钟)纯化。
[0499]
获得化合物(10-(2-溴-5-羟基-3,4-二甲氧基-6-甲基苯基)癸基)三苯基膦甲酸盐(6mg,8.61μmol,产率为4.41%,纯度为99.54%),无色胶状物。
[0500]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
37h45
bro3p
+
)的质量进行测量,确认m/z测量值648.6、649.2[m+h]
+
。
[0501]1h nmr(400mhz,cdcl3)δ=8.56(br s,1.309h),7.78-7.59(m,15h),3.84(s,3h),3.76(s,3h),3.44(br s,2h),2.70-2.59(m,2h),2.13(s,3h),1.50(br s,4h),1.40-1.12(m,12h).
[0502]
31
p nmr(162mhz,cdcl3)δ=24.17(s,1p).
[0503]
实施例6.2.制备(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物(sb-u005)的方法
[0504]
在本技术中,将对制备由[化学式3]代表的化合物的方法进行描述(图21)。制备化
学式3的化合物的方法包括以下制备步骤1至4。
[0505]
步骤1.10-溴-1-(2-羟基-3,4-二甲氧基-6-甲基-苯基)癸烷-1-酮的制备将新制成粉的alcl3(457.89mg,3.43mmol)加入至在干燥的dce(10ml)中的10-溴代癸酰基氯化物(0.536g,1.89mmol)和1,2,3-三甲氧基-5-甲基-苯(312.86mg,1.72mmol)的溶液中,将所得的混合物在25℃下搅拌40小时。
[0506]
这时,经lcms证实产生了所期望的材料作为主要成分。
[0507]
将混合物倒入冰水中,并使用ch2cl2(50ml
×
2)萃取。
[0508]
将混合的萃取物用水洗涤,然后用na2so4干燥,然后浓缩以获得油。获得的油通过柱色谱法(sio2,石油醚/etoac=10:0-10:1)纯化,得到无色油(520mg,产率为66.56%)。
[0509]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h29
bro4)的质量进行测量,确认m/z测量值400.12、402.8[m+h]
+
。
[0510]1h nmr(400mhz,cdcl3)δ1.21-1.55(m,10h),1.56-1.78(m,2h),1.85(m,2h),2.46(s,3h),2.89(t,j=7.4hz,2h),3.41(t,j=6.8hz,2h),3.88(d,j=12.3hz,6h),6.31(s,1h),10.38(s,1h).
[0511]
步骤2.2-(10-溴癸基)-5,6-二甲氧基-3-甲基-苯酚的制备
[0512]
将10-溴-1-(2-羟基-3,4-二甲氧基-6-甲基-苯基)癸烷-1-酮(520mg,1.14mmol)溶解在三氟乙酸(tfa,10ml)中之后,向其中加入et3sih(2ml),然后所得的混合物在80℃下搅拌12小时。
[0513]
这时,经lcms证实,作为起始原料的酮被消耗,形成新的峰。
[0514]
将反应混合物蒸发,干燥并通过柱色谱法(sio2,石油醚/etoac=5:0至5:1)纯化以获得无色油(410mg,产率为82.34%)。
[0515]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h31
bro3)的质量进行测量,确认m/z测量值386.15、388.9[m+h]
+
。
[0516]1h nmr(400mhz,cdcl3)δ1.22-1.55(m,14h),1.86(quin,j=7.1hz,2h),2.26(s,3h),2.51-2.65(m,2h),3.42(t,j=6.9hz,2h),3.86(m,6h),5.82(s,1h),6.29(s,1h).
[0517]
步骤3.4-溴-2-(10-溴癸基)-5,6-二甲氧基-3-甲基-苯酚的制备
[0518]
将2-(10-溴癸基)-5,6-二甲氧基-3-甲基-苯酚(410mg,940.98μmol)和nabr(145.23mg,1.41mmol)溶于乙酸(acoh,10ml)中,向其中加入过氧化氢(h2o2,160.04mg,1.41mmol,30%),然后,将所得混合物在25℃下搅拌2小时。
[0519]
这时,经lcms证实,起始材料被消耗,并形成一个新的峰。
[0520]
将反应混合物在50ml的水中淬灭,并使用etoac(40ml
×
2)萃取。
[0521]
将混合的有机相用饱和的nahco3洗涤,直至ph》7,然后经na2so4干燥,浓缩得到无色油(300mg,粗品)。
[0522]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h30
br2o3)的质量进行测量,确认m/z测量值464.06、466.9[m+h]
+
。
[0523]1h nmr(400mhz,cdcl3)δ1.22-1.55(m,14h),1.86(quin,j=7.1hz,2h),2.26(s,3h),2.51-2.65(m,2h),3.42(t,j=6.9hz,2h),3.85(s,3h),3.93(s,3h),5.77(s,1h).
[0524]
步骤4.(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物的制备
[0525]
将4-溴-2-(10-溴癸基)-5,6-二甲氧基-3-甲基-苯酚(300mg,597.11μmol)和三苯
基膦(pph3,939.68mg,3.58mmol)溶解在甲苯(1ml)中之后,将所得溶液在n2下于130℃搅拌18小时。
[0526]
通过薄层色谱(tlc,dcm:meoh=10:1,rf=0.2)证实,在opph3下形成了一个新峰。
[0527]
通过将反应混合物蒸发获得棕色残余物,并通过制备hplc(柱:3_phenomenex luna c18 75
×
30mm
×
3μm;流动相:[水(0.2%fa)-acn];b%:52%至82%,6分钟)纯化。
[0528]
将残余物冻干后,获得了所期望的产物,为白色固体(16mg,产率为12.24%,纯度为97.2%)。
[0529]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
37h45
bro3p
+
)的质量进行测量,确认m/z测量值647.23、649.3[m+h]
+
。
[0530]1h nmr(400mhz,氯仿-d)δ1.13-1.70(m,16h),2.34(s,3h),2.56-2.76(m,2h),3.65-3.79(m,2h),3.68-3.77(m,1h),3.83(s,3h),3.88(s,3h),7.61-7.93(m,15h),8.76(s,1h);
31
p nmr(162mhz,氯仿-d)δ24.47(s,1p).
[0531]
实施例6.3.制备(10-(2-溴-3,4,5-三甲氧基-6-甲基苯基)癸基)三苯基膦溴化物(sb-u012)的方法
[0532]
在本技术中,将对制备由[化学式4]表示的化合物的方法进行描述(图22)。制备化学式4的化合物的方法包括以下制备步骤1至5。
[0533]
步骤1.5-(10-溴癸基)-1,2,3-三甲氧基-苯的制备
[0534]
在
–
78℃下,将溶解于thf(30ml)中的5-溴-1,2,3-三甲氧基苯(2g,8.09mmol,1eq)的溶液滴加至n-buli(2.5m,3.24ml,1eq)中。
[0535]
添加后,将混合物在相同的温度下搅拌1小时,在78℃下,向其中滴加溶解于thf(10ml)中的1,10-二溴癸烷(4.86g,16.19mmol,2eq)的溶液。
[0536]
将产生的混合物在20℃下搅拌11小时。
[0537]
这时,经lcms证实,检测到20%的期望的质量。
[0538]
残余物用饱和的nh4cl(10ml)稀释,并用etoac(50ml
×
3)萃取。
[0539]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0540]
残余物使用柱色谱法(sio2,石油醚/乙酸乙酯=100/0至95/5)纯化以获得化合物5-(10-溴癸基)-1,2,3-三甲氧基-苯(430mg,395.86μmol,产率为4.89%,纯度为35.66%),无色油。
[0541]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
19h31
bro3)的质量进行测量,确认m/z测量值387.4、389.1[m+h]
+
。
[0542]
步骤2.6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛的制备
[0543]
在0℃,将溶解于无水二氯甲烷(干ch2cl2,2ml)中的5-(10-溴癸基)-1,2,3-三甲氧基-苯(430mg,395.86μmol,1eq,纯度为35.66%)的溶液滴加至alcl3(178mg,1.33mmol,72.95μl,3.37eq)的干燥ch2cl2(6ml)中。
[0544]
添加后,将混合物在相同的温度下搅拌45分钟,用10分钟向其中逐渐滴加二氯(甲氧基)甲烷(140mg,1.22mmol,107.69μl,3.08eq)的干燥ch2cl2(2ml)溶液。
[0545]
将反应混合物倒入30ml冰水中,分离二氯甲烷相,然后使用二氯甲烷(50ml
×
2)对水相萃取两次。
[0546]
将合并的有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0547]
粗产物不经进一步纯化直接用于下一步。
[0548]
获得无色油状化合物6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛(410mg,384.97μmol,产率为97.25%,纯度为39%)。
[0549]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h31
bro4)的质量进行测量,确认m/z测量值415.4、415.2[m+h]
+
。
[0550]
步骤3.1-(10-溴癸基)-3,4,5-三甲氧基-2-甲基-苯的制备
[0551]
将tfa(3ml)加入到6-(10-溴癸基)-2,3,4-三甲氧基-苯甲醛(410mg,384.97μmol,1eq,纯度为39%)和et3sih(447.64mg,3.85mmol,614.89μl,10eq)的混合物中。
[0552]
将混合物在20℃下搅拌12小时。
[0553]
这时,经lcms证实反应已完成。
[0554]
将混合物缓慢倒入饱和的nahco3(50ml)中,并使用ch2cl2(50ml
×
3)萃取。
[0555]
将混合物有机层经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0556]
残余物通过制备tlc(sio2,石油醚/乙酸乙酯=4:1)纯化以获得化合物1-(10-溴癸基)-3,4,5-三甲氧基-2-甲基-苯(120mg,152.18μmol,产率为39.53%,纯度为50.9%),无色油。
[0557]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h33
bro3)的质量进行测量,确认m/z测量值401.4、402.8[m+h]
+
。
[0558]
步骤4.1-溴-2-(10-溴癸基)-4,5,6-三甲氧基-3-甲基-苯的制备
[0559]
将h2o2(17.25mg,152.18μmol,14.62μl,纯度为30%,1eq)加入到溶解于acoh(4ml)中的1-(10-溴癸基)-3,4,5-三甲氧基-2-甲基-苯(120mg,152.18μmol,1eq,纯度为50.9%)和nabr(15.66mg,152.18μmol,4.89μl,1eq)的搅拌溶液中,将所得的混合物在20℃下搅拌12小时。
[0560]
这时,经lcms证实反应已完成。
[0561]
残余物用30ml饱和nahco3:na2s2o3=10:1稀释,并使用etoac(30ml
×
3)萃取。
[0562]
将合并的有机层用盐水(10ml)洗涤,经[na2so4]干燥、过滤,然后在减压下浓缩以获得残余物。
[0563]
粗产物不经进一步纯化而直接用于下一步。
[0564]
获得化合物1-溴-2-(10-溴癸基)-4,5,6-三甲氧基-3-甲基-苯(130mg,粗品),黄色油。
[0565]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h32
br2o3)的质量进行测量,确认m/z测量值480.3、480.9[m+h]
+
。
[0566]
步骤5.1-溴-2-(10-blah癸基)-4,5,6-三甲氧基-3-甲基-苯的制备
[0567]
将溶解于甲苯(2ml)中的1-溴-2-(10-溴癸基)-4,5,6-三甲氧基-3-甲基-苯(130mg,162.41μmol,1eq,纯度为60%)和pph3(212.99mg,812.04μmol,5eq)的搅拌溶液在n2下于125℃加热12小时。
[0568]
这时,经lcms证实反应已完成。
[0569]
在减压下除去溶剂以获得残余物。
[0570]
残余物通过柱色谱法(sio2,石油醚/乙酸乙酯=100/0至0/100;乙酸乙酯:meoh=100/0~92/8)纯化以获得化合物1-溴-2-(10-blah癸基)-4,5,6-三甲氧基-3-甲基-苯
(30mg,39.69μmol,产率为24.44%,纯度为98.234%),无色油。
[0571]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
38h47
bro3p
+
)的质量进行测量,确认m/z测量值662.7、663.2[m+h]
+
。
[0572]1h nmr(400mhz,cdcl3)δ=7.93-7.66(m,15h),3.94-3.78(m,11h),2.78-2.67(m,2h),2.22(s,3h),1.64(br s,4h),1.50-1.34(m,4h),1.25(br d,j=10.1hz,8h).
[0573]
31
p nmr(162mhz,cdcl3)δ=24.54(s,1p).
[0574]
实施例6.4.制备(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物(sb-u011)的方法。
[0575]
在本技术中,将对制备由[化学式5]表示的化合物的方法进行描述(图23)。制备化学式5的化合物的方法包括以下制备步骤1至4。
[0576]
步骤1.10-溴-1-(2,3,4-三甲氧基-6-甲基-苯基)癸烷-1-酮的制备
[0577]
将alcl3(206.41mg,1.55mmol)加入至溶解于dce(10ml)中的4-溴-1,2,3-三甲氧基-5-甲基-苯(449.11mg,1.72mmol)和o-溴癸酰氯(536.94mg,1.89mmol)的搅拌溶液中,将所得的混合物在25℃下搅拌18小时。
[0578]
这时,经lcms证实反应已完成。
[0579]
通过tlc(石油醚:etoac=3:1,rf=0.4)证实形成了一个新的主要的斑点。
[0580]
将反应混合物倒入30ml的冰水中,并使用dcm(30ml
×
3)对水相萃取,经[na2so4]干燥,然后在减压下浓缩。获得黄色油。
[0581]
黄色油通过基于硅胶快速柱色谱法(石油醚中的etoac为0%至100%,30分钟)纯化,以获得无色油(215mg,产率为29.1%)。
[0582]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h31
bro4)的质量进行测量,确认m/z测量值414.14、416.8[m+h]
+
。
[0583]1h nmr(400mhz,cdcl3)δ1.32(m,8h),1.38-1.49(m,2h),1.67(m,2h),1.86(quin,j=7.2hz,2h),2.19(s,3h),2.75(t,j=7.4hz,2h),3.41(t,j=6.9hz,2h),3.77-3.92(m,9h),6.48(s,1h).
[0584]
步骤2.4-(10-溴癸基)-1,2,3-三甲氧基-5-甲基-苯的制备
[0585]
在25℃下,将et3sih(1.46g,12.52mmol,2ml)加入至溶解于tfa(10ml)中的10-溴-1-(2,3,4-三甲氧基-6-甲基-苯基)癸烷-1-酮(210mg,455.03μmol)的搅拌溶液中,将所得的混合物在80℃下搅拌2小时。
[0586]
这时,经lcms证实产生了所期望的产物作为主要成分。
[0587]
由tlc(石油醚:etoac=4:1,rf=0.45)证实,形成了一个新的主要斑点。
[0588]
将反应混合物蒸发并在真空下干燥以获得无色油,并使用基于硅胶快速柱色谱法(25g,石油醚中的etoa为0-50%的,30分钟)对无色油进一步纯化以获得所期望的产物4-(10-溴癸基)-1,2,3-三甲氧基-5-甲基-苯(118mg,250.93μmol,产率为55.15%),无色油。
[0589]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h33
bro3)的质量进行测量,确认m/z测量值400.16、403.0[m+h]
+
。
[0590]1h nmr(400mhz,cdcl3)δ1.20-1.54(m,14h),1.77-1.96(m,2h),2.27(s,3h),2.46-2.64(m,2h),3.42(t,j=6.8hz,2h),3.76-3.97(m,9h),6.49(s,1h).
[0591]
步骤3.1-溴-5-(10-溴癸基)-2,3,4-三甲氧基-6-甲基-苯的制备
[0592]
将h2o2(42.68mg,376.39μmol)滴加至溶解于acoh(5ml)中的4-(10-溴癸基)-1,2,3-三甲氧基-5-甲基-苯(118mg,250.93μmol)和nabr(38.73mg,376.39μmol)的溶液中,并在25℃下搅拌1小时。
[0593]
这时,经lcms证实形成了所期望的产物作为主要组分。
[0594]
将反应混合物在etoac/h2o(80ml/60ml)之间分配。
[0595]
有机层使用nahco3饱和水溶液(60ml)洗涤,直至ph》7。
[0596]
将合并的有机层经[na2so4]干燥,减压下浓缩,然后获得黄色油(140mg,粗品)。
[0597]
这时,经hnmr证实黄色油是所期望的产物,其纯度足以用于下一步。
[0598]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
20h32
bro3)的质量进行测量,确认m/z测量值478.07、481.0[m+h]
+
。
[0599]1h nmr(400mhz,cdcl3)δ1.20-1.52(m,14h),1.78-1.94(m,2h),2.36(s,3h),2.62(m,2h),3.42(t,j=6.9hz,2h),3.81-3.98(m,9h).
[0600]
步骤4.(10-(3-溴-4,5,6-三甲氧基-2-甲基苯基)癸基)三苯基膦溴化物的制备
[0601]
将溶解于甲苯(1ml)中的1-溴-5-(10-溴癸基)-2,3,4-三甲氧基-6-甲基-苯(140mg,279.13μmol)和pph3(366.06mg,1.40mmol)的搅拌溶液在n2下于130℃加热18小时。
[0602]
这时,经lcms确认形成了所期望的产物。
[0603]
此外,由tlc(dcm:meoh=10:1,rf=0.2)证实,在opph3下方形成了一个主要的新峰。
[0604]
将反应混合物干燥并获得棕色残余物,并使用基于硅胶快速柱色谱法(25g,在dcm中的0-15%meoh,30分钟)纯化。
[0605]
使用冷冻干燥法获得所期望的产物,其为白色固体(108.5mg,产率为51.41%,纯度为98.2%)。
[0606]
通过质谱-电喷雾电离(ms-esi)对获得的化合物(c
38h47
bro3p
+
)的质量进行测量,确认m/z测量值661.24、663.3[m+h]
+
。
[0607]1h nmr(400mhz,氯仿-d)δ1.12-1.50(m,12h),1.64(m,4h),2.34(s,3h),2.52-2.71(m,2h),3.77-3.97(m,11h),7.60-7.97(m,15h);
31
p nmr(162mhz,氯仿-d)δ24.53(s,1p).
[0608]
实施例7.smx和本技术的新型化合物分子通过与客户蛋白竞争性地与trap1结合来遏制trap1。
[0609]-细胞培养
[0610]
22rv1细胞系在37℃和5%co2条件下培养,使用包含10%胎牛血清和1%青霉素和链霉素的dmem-低葡萄糖(sigma)作为培养基。
[0611]-蛋白质印迹
[0612]
将22rv1细胞系以5
×
105的数量接种于60mm板上,并在3ml培养基中培养24小时。次日,细胞用5μm smx和sm系列(sb-u005、sb-u009、sb-u011、sb-u012)、线粒体热休克蛋白90(trap1)进行处理,并培养2小时。全细胞裂解物由培养的细胞制成,电泳,转移到pvdf膜上,然后在4℃下用第一抗体处理18小时。次日,用第二抗体体处理全细胞裂解物1小时,并使用蛋白质印迹检测试剂对蛋白质表达进行分析(图24)。
[0613]
用smx分子和在实施例6中合成的新型化合物分子sb-u005、sb-u009、sb-u011和
sb-u012作为药物对培养的细胞进行处理,并用dmso对治疗结果进行归一化。
[0614]
作为pull-down试验的结果(图24),可以证实,如同在实施例3.3中用smx对培养的细胞进行处理时的结果那样,当用各新型化合物分子对培养的细胞进行处理时,客户蛋白(sirt3、sdhb)的浓度显著降低(图24)。也就是说,这些结果可以有力的证明,本技术的新型化合物分子通过与客户蛋白竞争性地与trap1的中间结构域结合来遏制trap1。
[0615]
实施例8.smx和本技术的新型化合物分子具有糖尿病性视网膜病变(dr)的治疗功效。
[0616]-使用mio-m1 hre细胞系对药物活性进行分析
[0617]
在用5hre/gfp质粒(addgene#46926)((jetprime试剂盒)转染mio-m1 muller细胞后,使用1mg/ml g418(新霉素)(一种选择性标记物)选择质粒转染的细胞。通过选择从单个细胞以菌落形式生长的细胞来制造稳定的细胞系。所制造的稳定的mio-m1-hre/gfp细胞系被等分进96孔,并在次日用药物在各种浓度下进行处理。在细胞系暴露于缺氧环境(1%o2)24小时后,通过synergy neo酶标仪(biotek instrument)对gfp(ex/em:488/507)荧光信号进行测量。使用dmso作为阴性对照,dmso是在其中溶解药物的溶剂,基于以阴性对照作为100%来进行计算。
[0618]
可以证实,当用smx和新型化合物分子处理mio-m1细胞系时,血管新生因子hif1-α受到遏制(图25)。通过这种结果可以看出,smx和本技术的新型化合物分子具有糖尿病性视网膜病变(dr)的治疗功效。实施例9.smx和本技术的新型化合物分子具有湿性年龄相关性黄斑变性(湿性amd)的治疗功效。
[0619]-细胞培养
[0620]
将人视网膜色素上皮arpe-19细胞以3
×
105的数量接种在60mm板上,并在37℃和5%co2下培养,使用dmem/f-12(包含10%的胎牛血清和1%青霉素&链霉素)作为培养基,培养2天。两天后,将细胞用dmso(0.5%)、smx、sb-u005、sb-u009、sb-u011和sb-u012以1μm进行处理,并在缺氧室中1%的氧条件下培养6个小时,然后进行蛋白质印迹实验。
[0621]-蛋白质印迹
[0622]
细胞培养实验完成后,除去细胞培养溶液,用冷pbs将细胞洗涤一次,向其中加入ripa溶液(50mm tris-hcl ph 7.4,150mm nacl,0.25%脱氧胆酸钠,1%np-40),使用细胞刮匙收获细胞。将细胞充分裂解后,将6倍样品的缓冲液与通过低温离心获得的细胞悬浮液混合,将所得的混合物在95℃下煮沸5分钟,然后进行12%sds-page。用1小时20分钟的时间,在350ma下将电泳后的凝胶蛋白转移至pvdf膜,然后封闭(10%脱脂牛奶),并在室温下反应1小时以防止非特异性抗体结合。用抗体溶液(具有0.02%叠氮化钠的tbs-t,1mg/ml bsa)稀释第一抗体hif-1α(1:1000)和肌动蛋白(1:3000),并在4℃下与膜反应过夜。
[0623]
此后,用tbs-t(tris-缓冲的生理盐水+吐温-20)将膜洗涤两次,然后,在室温下用第二抗体(以1:5000稀释)反应1小时。用tbs-t将膜洗涤两次后,使用clarity western ecl底物(bio-rad)通过chemidoc系统(ge,las 4000)测量蛋白质表达水平的变化。
[0624]
结果是,可以证实smx和本技术的新型化合物分子有效地遏制了arpe-19细胞系中的hif-1α(图26)。由此可知,本技术的化合物对湿性年龄相关性黄斑变性(湿性amd)具有治疗功效。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1