病原物种特异PCR引物优化设计方法与流程
本发明涉及生物信息学分析领域,特别是涉及一种特异pcr引物的设计优化方法。
背景技术:
聚合酶链式反应(polymerasechainreaction,pcr)作为最基础的分子生物学实验手段之一,能够在体外成百万倍地扩增目标dna的拷贝数,因而被广泛应用于基因工程、基因诊断、目标序列富集等领域。其中引物设计的优劣是影响pcr试验的重要参数之一。
在临床微生物诊断中,多重pcr检测、illumina和三代nanopore高通量平台已成为检测临床样本未知感染的重要检测手段。且近年已有通过pcr方法靶向富集病原区域来提升检测精准度的趋势。在这些检测手段中,设计灵敏、高效、物种特异的pcr引物成为了关键。
针对微生物的pcr引物设计,现有方法主要为:1.基于16ribosomalrna(rrna)的引物设计;2.基于多重序列比对,获得同源性保守序列作兼并引物设计。然而这两种方法都有其局限性:1.不同微生物之间16srrna基因序列的高度相似性。基于16srrna只能在较高的分类水平(例如属和科)而不能在物种/菌株水平上可靠地鉴定微生物。即使在属级,许多研究者也报告了16srrna基因序列的分辨率问题,因此16srrna设计的引物并不适用于感染样本这种复杂环境中对菌株/物种水平上的微生物进行鉴定。2.基于多重序列比对进行兼并引物设计,1)前期需要大量有关目的微生物的背景知识调研积累;2)重复劳动,针对每一个目的物种,都需要重复构建多序列比对及同源性保守区域筛选;3)获得序列保守性的高低,会严重影响引物设计的灵敏度和特异性,4)兼并引物不能直接进行引物间相互作用的自由能评估,影响湿试验作多重pcr组合反应性能。因此基于多重序列比对设计兼并引物也不适用于大批量感染样本作多重pcr反应进行靶向富集。
鉴于此,提出本发明。
技术实现要素:
本发明要解决的技术问题是提供一种便捷的特异性pcr引物的设计方法,该方法的核心设计思路是:
1.从目标物种多个基因组使用k-mer方法获取物种共有k-mer集合,避免使用单一基因组造成的菌株特异性;
2.基于ncbirefseq库使用k-mer方法构建背景比对库,并对目标物种共有k-mer集合作比对,获取不包含在背景比对库中的物种特异k-mer集合,根据k-mer位置信息获取特异区间序列;
3.对物种特异序列作pcr引物设计,引入引物间相互作用的结合自由能阈值δg等进行筛选过滤;
4.引物经过与1600个临床微生物基因组作模板结合能力评估,确保引物与目标物种优先结合;
5.构建microbial_nthash比对库,使用ncbi-epcr软件获取引物的扩增区间target,提取序列作blast比对,确保引物的物种特异性。
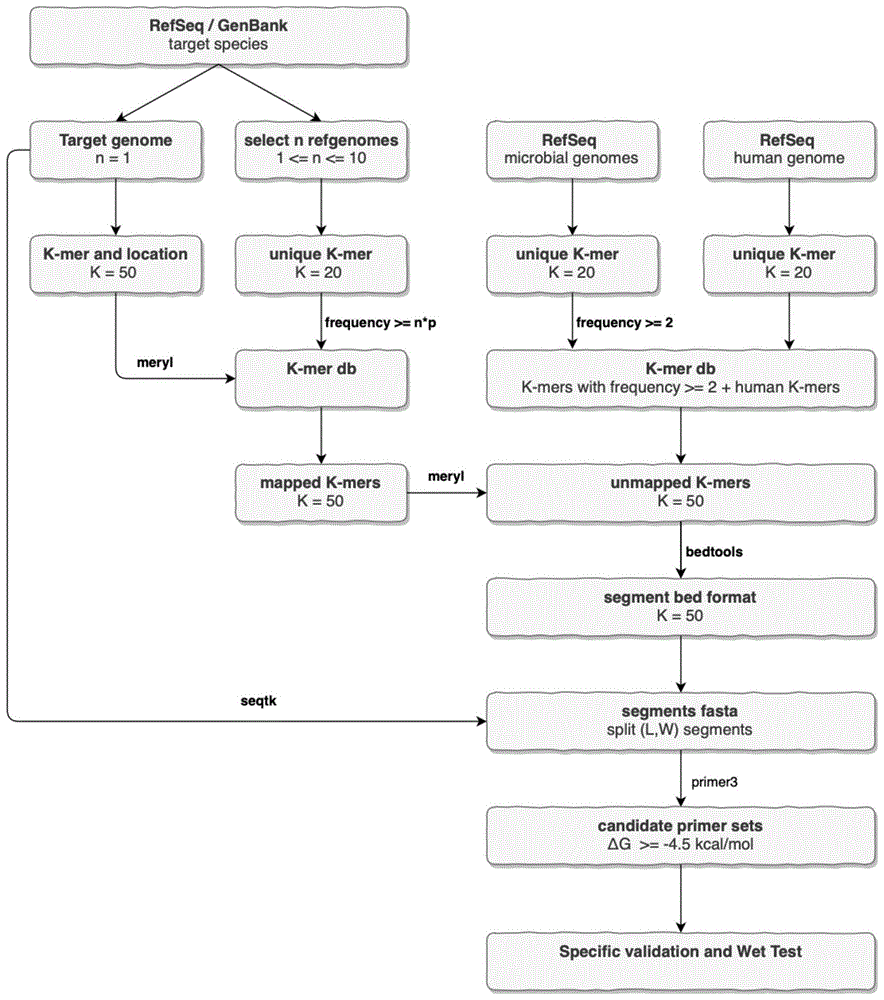
具体的,本发明首先提供了一种病原物种特异pcr引物优化设计方法,所述方法包括:
1)从ncbirefseq/genbank库中挑选n个目标物种的基因组;
2)对步骤1)中的每一个基因组划分k-mer,分别作uniquek-mer集合;
3)对步骤2)中所有uniquek-mer集合合并,挑选出频率大于等于n*p的k-mer,构建k-mer比对库;所述n取值1-10,优选10;所述p为概率值,取值0.5-1,优选0.8;
4)从步骤1)中的基因组按最佳参考基因组的筛选规则顺序挑选目标物种的参考基因组,并将基因组划分k-mer集合,并记录所有k-mer的位置信息;优选的,所述最佳参考基因组的筛选规则顺序为“referencegenome>representativegenome>completegenome>chromosome>scaffold>contig”;
5)对步骤4)中的k-mer集合和步骤3)中的比对库作比对,得到有比对结果的k-mer集合,此集合为物种水平共有序列k-mer集合;
6)步骤5)中的k-mer集合与背景比对库作比对,获取未比对上背景库的k-mer集合,制备物种特异的k-mer集合;
7)对物种特异k-mer集合根据步骤4)中记录的k-mer位置信息进行合并处理,整理成bed格式,并使用seqtk软件进行特异片段segments序列提取,制备物种特异序列集合;所述bed格式为基因组注释文件格式;
8)对物种特异序列集合作长度l和窗口w片段序列切分,得到(l,w)segments序列集合;
9)调用primer3软件对步骤8中的segments序列作引物设计。
在一些方式中,所述步骤1)中,所述n可取值1-10,优选10;
在一些方式中,所述步骤2)-3)中所述k-mer中的k可取值为18-20,优选为20。
在一些方式中,所述步骤4)-7)中所述k-mer中的k取值为40-60,优选为50。
在一些方式中,所述步骤8)中,当针对illumina平台、微流控pcr平台作多重pcr反应时,l取值100-1000bp,优选200bp;w取值范围为100-1000p,优选100bp;
进一步的,当针对nanopore平台作多重pcr靶向富集时,l取值1500-3000bp,优选2000bp;w取值范围为1500-3000bp,优选1500bp。
在一些方式中,所述步骤6)中背景比对库制备方法如下:
d)获取ncbirefseq中所有微生物物种水平的参考基因组,对每一个基因组划分k-mer,形成uniquek-mer集合,所述k取值为18-20;
e)从微生物物种水平的uniquek-mer集合中挑选出频率>=2的k-mer,作为候选k-mer集合,所述k取值为18-20;
f)人基因组按步骤a)切分uniquek-mer集合,与步骤b)的微生物物种水平的候选k-mer集合合并,用于构建k-merdb,获得背景比对库;所述k取值为18-20。
在一些方式中,所述步骤9)中,所述引物设计满足以下任一条件或其组合:1)引物长度在18-25nt;2)gc含量在40-65%之间;3)退火温度tm值在59-65之间;4)模板扩增长度在80-3000bp之间;5)引物间相互作用的结合自由能阈值δg为-4--9kcal/mol;6)引物不能出现超过4个连续重复碱基;7)引物间不能出现超过5个碱基的连续互补。
本发明还提供了一种病原物种特异pcr引物优化设计装置,其特征在于,包括:至少一个存储器,用于存储程序;至少一个处理器,用于加载所述程序以执行上述的方法。
本发明进一步提供了一种存储介质,其中存储有处理器可执行的指令,其特征在于,所述处理器可执行的指令在由处理器执行时用于实现如上述的方法。
本发明最后提供了一种上述检测装置及存储介质在病原物种特异pcr引物设计的应用,尤其是在病原物种宏基因组测序领域中的特异pcr引物设计的应用。
本发明有益的技术效果:
1)本发明使用k-mer方法鉴定物种的特异区间,基于特异片段作引物设计确保了引物的物种特异性,避免了常规使用单一基因组进行特异引物设计所造成的菌株特异性问题,在病原微生物尤其病原微生物宏基因组的检测分析中具有显著优势。
2)本发明制定了一套引物的评估体系,包括获取引物的理论扩增区间,与目标物种的模板结合能力评估,及物种特异性评估等。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1:物种特异pcr引物设计流程图;
图2:细菌肺炎支原体/真菌黄曲霉/病毒ebv选择的基因组;
图3:细菌肺炎支原体/真菌黄曲霉/病毒ebvprimer3引物设计结果;
图4:细菌肺炎支原体/真菌黄曲霉/病毒ebv引物与模板结合能力评估;
图5:细菌肺炎支原体/真菌黄曲霉/病毒ebv引物物种特异性验证结果;
图6:细菌肺炎支原体引物primerblast验证结果;
图7:真菌黄曲霉引物primerblast验证结果;
图8:病毒ebv引物primerblast验证结果;
图9:真菌黄曲霉引物湿试验验证结果。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围,并且所述实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
部分术语定义
除非在下文中另有定义,本发明具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
如本发明中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由…组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。
本发明中的术语“大约”、“大体”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的±10%,优选±5%。
在提及单数形式名词时使用的不定冠词或定冠词例如“一个”或“一种”,“所述”,包括该名词的复数形式。
此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本发明描述的实施方案能以不同于本发明描述或举例说明的其它顺序实施。
以下术语或定义仅仅是为了帮助理解本发明而提供。这些定义不应被理解为具有小于本领域技术人员所理解的范围。
本发明所述的“k-mer”是指将一条序列连续切割,逐个碱基划动得到k个碱基的子字符串,例如reads长度为l,k-mer长度设为k,则产生的k-mers数目为:l-k+1,再例如序列aactgact,设置k为3,则可以将其分割为aac、act、ctg、tga、gac、act共6个k-mers。
本发明所述的“位置信息”是指所选k-mer在参考基因组中的位置。
如下实施例进一步描述,所述实施例只是为了例证本发明的特定实施方案,不应理解为以任何方式限制本发明范围之意。除非另外说明,本发明中所公开的实验方法均采用本技术领域常规技术,实施例中所用的试剂和原材料均可由市场购得。
实施例1特异性引物设计方法的优化开发
本发明经大量生信分析优化试验,最终确立开发出特异性引物设计方法,具体方案步骤如下:
一、背景比对库的构建:
1.获取ncbirefseq中所有微生物物种水平的参考基因组,对每一个基因组划分k-mer,形成uniquek-mer集合。所述k取值为18-20,优选20。
2.从微生物物种水平的uniquek-mer集合中挑选出频率>=2的k-mer,作为候选k-mer集合。所述k取值为18-20,优选20。
3.人基因组按方法1切分uniquek-mer集合,与方法2的微生物物种水平的候选k-mer集合合并,用于构建k-merdb,获得背景比对库。所述k取值为18-20,优选20。
二、物种特异pcr引物设计:
1.从ncbirefseq/genbank库中挑选出n个目标物种的基因组。所述n取值1-10,优选10。
2.对方法1中的基因组分别作uniquek-mer集合。所述k取值为18-20,优选20。
3.对方法2中的所有uniquek-mer集合合并,挑选出现频率大于等于n*p的k-mer,并对这些k-mer构建k-mer比对库。所述k取值为18-20,优选20;所述n取值1-10,优选10;所述p为概率值,取值0.5-1,优选0.8。
4.从方法1中的基因组按ncbirefseq/genbank最佳基因组筛选规则挑选目标物种的最佳参考基因组,并将基因组划分k-mer集合,并记录所有k-mer的位置信息。所述k取值为40-60,优选50;所述最佳参考基因组的筛选顺序为“referencegenome>representativegenome>completegenome>chromosome>scaffold>contig”,参考信息分别出自“refseq_category”和“assembly_level”。
5.使用meryl软件对k-mer集合和方法3中的比对库作比对,得到有比对结果的k-mer集合,此集合为物种水平共有序列k-mer集合。所述k取值为40-60,优选50。
6.方法5中的k-mer集合与背景比对库作比对,获取未比对上背景库的k-mer集合,此集合为物种特异的k-mer集合。所述k取值为40-60,优选50。
7.物种特异k-mer集合根据方法4中记录的k-mer位置信息进行合并处理,整理成bed格式,并使用seqtk软件进行特异片段segments序列提取,为物种特异序列集合。所述k取值为40-60,优选50;所述bed格式为基因组注释文件格式。
8.对物种特异序列集合作长度l和窗口w片段序列切分,得到(l,w)segments序列集合。当针对illumina平台、微流控pcr平台作多重pcr反应时,l取值100-1000bp,优选200bp;w取值范围为100-1000bp,优选100bp。当针对nanopore平台作多重pcr靶向富集时,l取值1500-3000bp,优选2000bp;w取值范围为1500-3000bp,优选1500bp。
9.调用primer3软件对方法8中的segments序列作引物设计。所述引物设计须满足以下条件:1)引物长度在18-25nt;2)gc含量在40%-65%之间;3)退火温度tm值在59-65之间;4)模板扩增长度在80-3000bp之间;5)引物间相互作用的结合自由能阈值δg为-4--9kcal/mol;6)引物不能出现超过4个连续重复碱基;7)引物间不能出现超过5个碱基的连续互补等等。
10.引物作干试验和湿试验验证。干试验包括:1)引物与目标物种结合能力评估;2)引物的物种特异性评估;3)ncbi在线primerblast评估。湿试验为引物与目标物种、非目标物种和人源gdna作pcr反应验证试验(具体参见图1)。
实施例2细菌/真菌/病毒物种特异pcr引物设计和评估
一、以细菌肺炎支原体/真菌黄曲霉/病毒ebv为例分别作pcr引物设计:
1.从ncbirefseq/genbank库中筛选n个目标物种基因组。所述n取值为10,见图2。
2.将筛选的基因组分别作k-mer划分得到uniquek-mer集合。所述k取值20。
3.使用meryl软件对方法2中的uniquek-mer集合构建k-merdb,从中抽取出现频率大于n*p的k-mer,作为该物种水平共有的k-mer集合,并构建物种共有k-mer比对库。所述k取值20,n取值10,p取值0.8。
4.以ncbirefseq/genbank最佳参考基因组筛选规则筛选目标物种的参考基因组,划分k-mer,并保留所有k-mer的位置信息,所述k取值50。所选参考基因组见图2。
5.使用meryl对k-mer集合与方法3中的物种共有比对库作比对,获取能够比对上库的k-mer集合。所述k取值50。
6.方法5中的k-mer集合与预先构建的背景比对库作比对,获取未比对上背景库的k-mer集合,此集合为物种特异的k-mer集合。所述k取值为50。
7.根据记录的k-mer位置信息对物种特异的k-mer集合进行bed格式合并整理成segment区间,使用seqtk软件提取segments序列,为物种特异序列集合。所述k取值50。
8.方法7中的物种特异序列作(l,w)窗口片段序列切分,得到候选的segments序列集合。所述l取值为2000,w取值1500。
9.调用primer3软件对方法8中的segments序列作引物设计。所述引物设计须满足以下条件:1)引物长度在18-25nt;2)gc含量在40%-65%之间;3)退火温度tm值在59-65之间;4)模板扩增长度在80-3000bp之间;5)引物间相互作用的结合自由能阈值δg>=-4.5kcal/mol;6)引物不能出现超过4个连续重复碱基;7)引物间不能出现超过5个碱基的连续互补等。
10.细菌肺炎支原体/真菌黄曲霉/病毒ebv引物设计结果,引物格式为“物种_f_r”(见图3)。
二、细菌肺炎支原体/真菌黄曲霉/病毒ebv引物与模板结合能力评估
1.临床关注微生物物种关联基因组:从公司自建的数据库中整理出1670个临床关注的微生物物种,按照最佳参考基因组筛选规则从refseq/genbank库中关联物种参考基因组,并获取序列id与物种taxid的对应关系。所述最佳参考基因组的筛选顺序为“referencegenome>representativegenome>completegenome>chromosome>scaffold>contig”,参考信息分别出自“refseq_category”和“assembly_level”。
2.构建基因组hash比对库:使用ncbi-epcr软件对方法1中的临床微生物基因组和人基因组作hash比对库,硬盘使用空间约180g。
3.物种特异pcr引物作模板结合自由能计算:
1)引物对分别与方法2构建的hash比对库比对,产物阈值为50-5000,最多允许有10个mismatch,获取引物的匹配结果。
2)引物对的匹配结果整理成bed格式,使用seqtk软件提取匹配的target序列。
3)首先计算引物f/r与目标物种target序列的结合自由能δg值,然后依次计算与其他物种的自由能δg值,若其他物种的δg值小于目标物种的δg值,则未通过模板结合评估,应过滤掉。所述自由能δg为使用primer3中的calcendstability函数计算引物与target序列结合的自由能,以最小δg代表物种水平;引物f须反向互补后与模板target序列进行评估。
4.细菌肺炎支原体/真菌黄曲霉/病毒ebv引物与模板结合能力评估结果(见图4)。可见,引物通过模板结合能力验证,本发明能够满足细菌/真菌/病毒微生物的特异引物设计。
三、细菌肺炎支原体/真菌黄曲霉/病毒ebv引物特异性评估
1.构建microbial_nthash比对库:
1)根据ncbitaxonomy库从nt库中分别抽取细菌/真菌/病毒微生物物种的序列,获取microbial_nt序列集。
2)使用ncbi-epcr软件对microbial_nt序列和人基因组作hash比对库,硬盘使用空间约2t。
2.匹配引物扩增区间:
1)将上述通过模板结合能力验证的引物对f/r分别与microbial_nthash比对库作比对,产物阈值为50-5000,最多允许有2个mismatch,获取引物的匹配结果。
2)引物对的匹配结果整理成bed格式,使用seqtk软件提取匹配的target序列,作为target产物集合。
3.引物物种特异性验证结果:
1)将上述target产物集合与nt库作blast比对,根据bitscore阈值进行比对物种结果统计。所述bitscore阈值为max(bitscore)*0.95,即同一序列只保留满足阈值条件的比对结果。
2)对保留的比对结果作物种统计,过滤掉有非特异性比对的引物,即获得通过物种特异性验证的引物集合。
3)细菌肺炎支原体/真菌黄曲霉/病毒ebv引物特异性验证结果(见图5)。可见,引物通过物种特异性验证,本发明能够满足细菌/真菌/病毒微生物的特异引物设计。
四、细菌肺炎支原体/真菌黄曲霉/病毒ebv引物与ncbiprimerblast评估
1.以上述验证通过的引物例,使用ncbiprimerblast在线网站(https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi)作物种特异性验证。
2.在网页窗口分别输入f和r引物序列,database选择nr库,organism选择人homosapiens、细菌bacteria(taxid:2)、真菌fungi(taxid:4751)、病毒viruses(taxid:10239),其他项选择默认,配置后提交作特异性验证。
3.结果表明,引物验证结果与目标物种一致:
1)细菌肺炎支原体引物验证结果(见图6);
2)真菌黄曲霉引物验证结果(见图7);
3)病毒ebv引物验证结果(见图8)。
实施例3物种特异pcr引物的湿试验验证
本实施例进一步以黄曲霉物种特异pcr引物(f:5’-ccctcttgcctgttccagag-3’(seqidno.1),r:5’-catgggtgggtgctcttcat-3’(seqidno.2))的湿试验验证为例,来说明基于本发明思路设计的特异性引物的有效性。
1)试剂耗材
无酶无菌水:thermofisher,nuclease-freewater(notdepc-treated)(货号:am9937);qubit荧光定量仪dna检测试剂盒:qubit1xdsdnahsassaykit(货号:q33231);pcr扩增酶:
2)引物验证
以黄曲霉标准品提取的核酸为模板进行pcr反应验证,同时pcr反应体系平行增加gdna、zymo菌dna模板模拟真实的临床样本进行特异性验证。pcr配置体系和反应条件如下表:
3)pcr产物浓度结果
4)pcr产物4200电泳结果
以退火温度58℃的pcr产物为例,4200电泳结果如图9所示。
a1(l):gdnamarker
b1:黄曲霉58℃pcr产物模板目的条带1194bp,条带单一且与预期大小一致;
c1:阴性对照gdna,使用人源gdna为模板,无扩增反应;
d1:阴性对照zymo,使用zymo菌dna为模板,无扩增反应;
其中zymo菌包含大肠杆菌、肠道沙门氏菌、金黄色葡萄球菌、铜绿假单胞菌、李斯特菌、发酵乳酸杆菌、粪肠球菌、枯草芽孢杆菌8种细菌和啤酒酵母、新型隐球菌2种真菌。
5)结果
黄曲霉引物测试了在不同模板gdna、zymo菌(8种细菌和2种真菌)以及水里不存在非特异性扩增,在退火温度58℃条件下阳性标准品达到了有效的扩增效果。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
序列表
<110>江苏先声医疗器械有限公司
江苏先声诊断技术有限公司
<120>病原物种特异pcr引物优化设计方法
<160>2
<170>siposequencelisting1.0
<210>1
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>1
ccctcttgcctgttccagag20
<210>2
<211>20
<212>dna
<213>人工序列(artificialsequence)
<400>2
catgggtgggtgctcttcat20
- 还没有人留言评论。精彩留言会获得点赞!