一种基于二代测序的微单倍型检测分型系统和方法与流程
本发明属于生物信息
技术领域:
,具体地,涉及一种基于二代测序的微单倍型检测分型系统和方法。
背景技术:
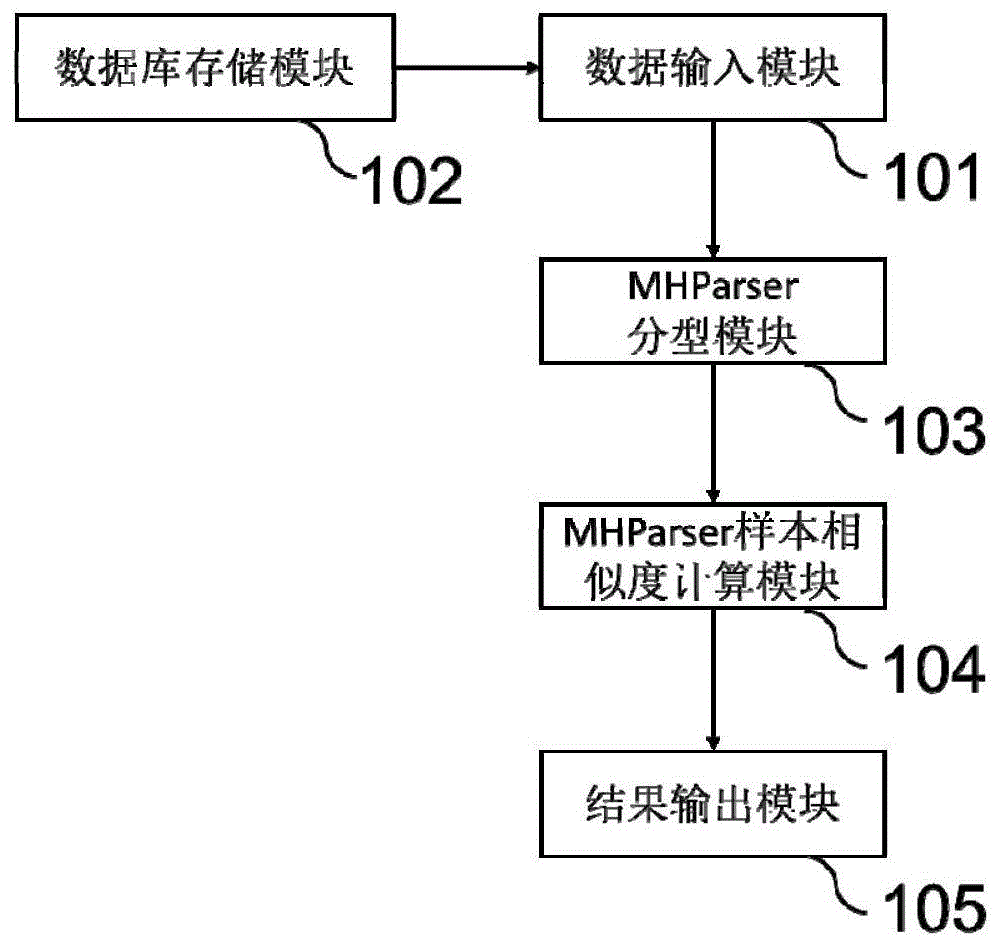
:单倍型(haplotype)是指在一条染色体或线粒体上,紧密连锁的多个等位基因的线性组合,每一种组合方式即为一种单倍型。近年来单倍型在法医学中的应用越来越广泛,主要涉及y染色体、x染色体及线粒体上str和snp多态性的研究。微单倍型(microhaplotype)的片段长度更短,其是在200bp范围内2~5个snp的组合。由于微单倍型基因座内含有多个snp位点,所以微单倍型是多等位基因遗传标记,包含更加丰富的遗传信息。微单倍型是近年来国际法医遗传学界广泛关注的一类新型遗传标记,在混合dna分型领域展现出巨大潜力,且支持人类种族地域推断、复杂亲缘关系鉴定、微量降解检材检验等法医学应用。它兼具str和snp遗传标记的优势:(1)高度多态性。通常snp位点仅有2个等位基因,若n个二等位基因的snp构成一个微单倍型位点,那么该微单倍型位点理论上最多有2n种snp的组合,即最多有2n种基因型。(2)低突变率。微单倍型的突变率相当于snp的突变率,为10-9~10-8/代,是str突变率的百万分之一到十万分之一,在亲权鉴定中有独特优势。(3)检测无阴影带。基于电泳技术分型的str会产生阴影带,不利于复杂混合dna样本的分析。微单倍型通过测序手段检测,无阴影带,且二代测序具有高通量、高灵敏度的优点,在定量分析复杂混合dna中有很大潜力。(4)长度优势。str基因座的等位基因长度跨度很大,因此会产生扩增不平衡问题,而较长的等位基因在降解检材中极可能被破坏,得不到准确的分型结果,微单倍型长度相对均一,可减少因长度差异带来的扩增不平衡问题。微单倍型检测方法包括二代测序、高分辨熔解曲线、单链构象多态性分析等,其中二代测序,也称下一代测序(nextgenerationsequencing,ngs)或大规模并行测序(massivelyparallelsequencing,mps),在科研和临床诊断领域已得到广泛应用。由于微单倍型为近几年才兴起的一种检测技术,基于二代测序数据进行微单倍型分析的方法较少,且功能比较单一,例如一些基于扩增子平台的微单倍型检测软件,只能进行微单倍型的单一计数。技术实现要素:为了解决上述技术问题中的至少一个,本发明采用的技术方案如下:一种基于二代测序的微单倍型检测分型系统,包括:数据输入模块,用于接受样本二代测序比对文件输入;数据库存储模块,用于存储检测位点列表;mhparser分型模块,分别与所述数据输入模块和所述数据库存储模块连接,用于基于所述检测位点列表,根据输入的样本二代测序比对文件计算微单倍型位点,并输出微单倍型分型结果;其中,所述微单倍型分型结果包括单倍型信息及计数信息;mhparser样本相似度计算模块,与所述mhparser分型模块连接,用于接受所述mhparser分型模块输出的微单倍型分型结果,并根据微单倍型分型结果计算任意两个样本之间的相似性分值;结果输出模块,用于输出所述相似性分值。在本发明中,mhparser是自定义名称,对相应模块没有限定作用。在本发明的一些实施方案中,所述位点包括微单倍型和单独snp/indel位点。在本发明的一些实施方案中,所述检测位点列表格式下表所示:在本发明的一个具体实施方案中,所述检测位点列表如下:在本发明的一些实施方案中,所述计数信息是指支持相应位点的reads数目信息。在本发明的一些实施方案中,所述mhparser样本相似度计算模块还输出样本整合微单倍型序列信息;所述微单倍型检测分型系统还包括:mhparser多序列比对模块,与所述mhparser样本相似度计算模块连接,用于接受mhparser样本相似度计算模块输出的样本整合微单倍型序列信息,并根据样本整合微单倍型序列信息进行多序列比对,得到样本多序列比对结果;mhparser样本聚类模块,与所述mhparser多序列比对模块连接,用于接受mhparser多序列比对模块输出的多序列比对结果,并根据所述样本多序列比对结果进行聚类分析;所述结果输出模块还与所述mhparser样本聚类模块连接,用于样本输出聚类分析结果。在本发明的一些实施方案中,所述整合微单倍型序列信息是指将检测位点的序列拼接在一起得到的序列。在本发明的一些实施方案如此,所述数据库存储模块还用于存储测序变异检测信息。进一步地,所述mhparser分型模块输出位点的基因型。更进一步地,所述位点为snp/indel位点。本发明的第二方面提供一种基于二代测序的微单倍型检测分型方法,包括以下步骤:s1,获得检测位点列表和样本二代测序比对文件;s2,利用检测位点列表,根据样本二代测序比对文件计算样本的微单倍型位点,得到微单倍型分型结果,所述微单倍型分型结果包括单倍型信息及计数信息;s3,根据步骤s2的微单倍型分型结果计算任意两个样本之间的相似性分值。在本发明的一些实施方案中,步骤s3还获得样本整合微单倍型序列信息。进一步地,所述方法还包括以下步骤:s4,根据样本整合微单倍型序列信息进行多序列比对,得到多序列比对结果;s5,根据多序列比对结果进行聚类分析,得到聚类分析结果。在本发明的一些实施方案中,所述步骤s2中,具体利用以下步骤获得所述微单倍型分型结果:s21,针对检测位点列表中的任意微单倍型位点,遍历样本二代测序比对文件中的每条序列,根据位点的坐标信息提取reads的碱基组合,得到单倍型,并对具有相同单倍型的reads进行计数;s22,针对相同单倍型,按照reads数目对不同碱基组合进行排序;由此得到检测位点列表中所述微单倍型分型结果。在本发明的一些实施方案中,所述步骤s3中,具体利用以下公式获得所述相似性分值:s31,按照步骤s2的方法获得多个样本的微单倍型分型结果s32,按照下面公式计算任意两个样本间的相似性分值:其中,s为相似性分值;m为位点个数;c1为常量,取值1,为两个样本某个位点碱基型相同时的打分值;c2为常量,取值-1,为两个样本某个位点碱基型不同时的打分值;g1和g2分别为某个位点样本1和样本2的碱基组合数目,其中,样本1为该位点上碱基组合数目较少的样本,样本2为该位点上碱基组合数目较多的样本;g1∩g2表示该位点上两个样本具有相同的碱基组合的数目。在本发明的一些具体实施方案中,针对某个位点,如果两个样本在该位点上碱基组合数目相同,则任意一个样本为样本1,另外一个样本为样本2。针对碱基组合,例如对于某个位点,参考位点为t,g和a;但某个样本该位点有t,a和a;g,g和a;t,g和c;t,-(表示缺失)和g;-,g和c。则该样本的在该位点的碱基组合为5。例如,针对某个位点,如果两个样本的碱基组合分别为5和6,则碱基组合为6的为样本1,碱基组合为5的为样本2。如果两个样本该位点相同的碱基组合为4,即为4,则针对该位点的相似性为c1*(g1∩g2)+c2*(g1-g1∩g2)=1*4-1*(6-4)=2。在本发明的一些实施方案中,在步骤s4中,利用mafft软件进行多序列比对。在本发明的一些实施方案中,在步骤s5中,利用fasttree软件进行样本聚类。在本发明的一些实施方案中,进一步包括利用可视化软件识别步骤s5获得的聚类分析结果。在本发明的一些具体实施方案中,所述可视化软件为mega软件。本发明的有益效果相对于现有技术,本发明具有以下有益效果:本发明的系统和方法采用二代测序技术,相比传统的dna测序技术,二代测序具有高通量、高速度、集成化、低成本等显著优势,在法医遗传学领域也具有重要应用前景。测序是序列多态性遗传标记最好的检测手段,微单倍型是snp的线性组合,其本质仍然是snp,二代测序能够一次性获得复合体系中snp位点的全部基因分型,也同时获得全部微单倍型遗传标记的准确分型,本发明有助于推动二代测序技术成为微单倍型检测的金标准。本发明的系统和方法能适用于不同捕获方式产生的测序数据,不论是液相捕获还是多重pcr扩增产生的数据都适用,因此具有广泛的应用场景,易于推广。本发明的系统和方法除了对微单倍型进行检测计数外,还能进行snp/indel的检测计数分型。本发明的系统和方法除了能检测由多个snp构成的微单倍型之外,还能容忍微单倍型中包含indel位点的情况,检测范围更广,算法更通用。本发明的系统和方法可以利用样本的微单倍型分型结果,进一步判断样本的相似度情况,对样本聚类,进一步对同批次样本进行严格质控,保证检测过程的可靠性。附图说明图1示出了本发明实施例1的微单倍型检测分型系统的示意图。图2示出了本发明实施例2的微单倍型检测分型系统的示意图。图3示出了本发明实施例4微单倍型检测分型方法的流程示意图。图4示出了本发明实施例6对11例样本进行微单倍型检测分型的聚类分析结果图。具体实施方式为了使本发明所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。实施例以下例子在此用于示范本发明的优选实施方案。本领域内的技术人员会明白,下述例子中披露的技术代表发明人发现的可以用于实施本发明的技术,因此可以视为实施本发明的优选方案。但是本领域内的技术人员根据本说明书应该明白,这里所公开的特定实施例可以做很多修改,仍然能得到相同的或者类似的结果,而非背离本发明的精神或范围。除非另有定义,所有在此使用的技术和科学的术语,和本发明所属领域内的技术人员所通常理解的意思相同,在此公开引用及他们引用的材料都将以引用的方式被并入。那些本领域内的技术人员将意识到或者通过常规试验就能了解许多这里所描述的发明的特定实施方案的许多等同技术。这些等同将被包含在权利要求书中。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的仪器设备,如无特殊说明,均为实验室常规仪器设备;下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。实施例1基于二代测序的微单倍型检测分型系统结合图1,本实施例提供一种基于二代测序的微单倍型检测分型系统,其包括:数据输入模块101,用于接受样本二代测序比对文件输入;数据库存储模块102,用于存储检测位点列表;mhparser分型模块103,分别与数据输入模块101和数据库存储模块102连接,用于根据输入的样本二代测序比对文件计算微单倍型位点,并输出微单倍型分型结果,该微单倍型分型结果包括单倍型信息及计数信息;mhparser样本相似度计算模块104,与mhparser分型模块103连接,用于接受mhparser分型模块103输出的微单倍型分型结果,并根据微单倍型分型结果计算任意两个样本之间的相似性分值;结果输出模块105,用于输出所述相似性分值。其中,数据库存储模块102存储的检测位点列表格式如表1所示:表1检测位点列表格式说明mhparser分型模块103输出的微单倍型分型结果格式如表2所示:表2微单倍型分型结果格式mhparser样本相似度计算模块104的输出结果格式如表3所示:表3样本相似度结果格式实施例2实施例1中的微单倍型检测分型系统的改进本实施例1提供另一种基于二代测序的的微单倍型检测分型系统,如图2所示,其包括实施例1描述的微单倍型检测分型系统的全部模块,相当于对实施例1的微单倍型检测分型系统的改进,与实施例1描述的微单倍型检测分型系统的区别是:mhparser样本相似度计算模块104还可以输出样本整合微单倍型序列信息;进一步,该微单倍型检测分型系统还包括:mhparser多序列比对模块106,与mhparser样本相似度计算模块104连接,用于接受mhparser样本相似度计算模块104输出的样本整合微单倍型序列信息,并根据样本整合微单倍型序列信息进行多序列比对,得到多序列比对结果;mhparser样本聚类模块107,与mhparser多序列比对模块106连接,用于接受mhparser多序列比对模块106输出的多序列比对结果,并根据多序列比对结果进行聚类分析;结果输出模块105还与mhparser样本聚类模块107连接,并用于输出样本聚类结果。实施例3实施例1或实施例2的微单倍型检测分型系统的改进本实施例提供实施例1或实施例2的微单倍型检测分型系统的改进,其包含的模块与实施例1或实施例2的微单倍型检测分型系统完全相同,区别在于:数据库存储模块102还用于存储测序变异检测信息,是为了判断snp/indel位点的基因型信息。实施例4一种基于二代测序的微单倍型检测分型方法本实施例提供一种基于二代测序的微单倍型检测分型方法,为实施例1的微单倍型检测分型系统的配套使用方法,如图3所示,包括以下步骤:s1,获得检测位点列表和样本二代测序比对文件;s2,利用检测位点列表,根据样本二代测序比对文件计算微单倍型位点,得到微单倍型分型结果,该微单倍型分型结果包括单倍型信息及计数信息;s3,根据步骤s2的微单倍型分型结果计算任意两个样本之间的相似性分值。实施例5一种基于二代测序的微单倍型检测分型的改进方法本实施例提供一种实施例4描述的微单倍型检测分型的方法的改进,为实施例2的微单倍型检测分型系统的配套使用方法,如图4所示,其区别在于:步骤s3可获得样本的整合微单倍型序列信息;相应地,该改进方法除上述步骤s1-s3外,还包括以下步骤:s4,根据样本整合微单倍型序列信息进行多序列比对,得到多序列比对结果;s5,根据多序列比对结果进行聚类分析,得到聚类分析结果。实施例6微单倍型检测分型系统的应用本实施例提供实施例2的微单倍型检测分型系统的应用,或实施例3对实施例2改进的微单倍型检测分型系统的应用。区别在于数据库存储模块102是否存储有测序变异检测信息。1.检测位点列表本实施例检测位点列表信息如表4所示:表4检测位点列表2.测序变异检测信息设置测序变异检测信息(实施例3对实施例2的改进系统),是为了判断snp/indel位点的基因型。在该系统中,测序变异检测信息的存储文件为vcf格式,由胚系突变检测软件gatkhaplotyper检测得到。与vcf文件伴随存储的还有后缀为tbi的索引文件。本实施例的测序变异检测信息如表5所示:表5测序变异检测信息vcf文件最前面头部注释信息,每行以‘##’开始,示例如下:以上vcf格式信息是一种示例,本领域技术人员熟知其含义。如果没有该测序变异检测信息(实施例2的系统),系统不会输出snp/indel变异的基因型信息,但也会输出snp/indel变异的碱基深度和计数信息。3.样本二代测序比对文件本实施例共获得11例待比对样本的二代测序信息,11例待比对样本的信息如表7所示:表711例待比对样本的信息序号样本名样本组别样本来源1o1o组人源edta抗凝全血2o2o组人源edta抗凝全血3o3o组人源edta抗凝全血4o4o组人源edta抗凝全血5o5o组人源edta抗凝全血6mix_1mix组,混有o5人源edta抗凝全血7mix_2mix组,混有o5人源edta抗凝全血8mix_3mix组,混有o5人源edta抗凝全血9mix_4mix组,混有o5人源edta抗凝全血10na12878_01na12878组细胞系基因组dna11na12878_02na12878组细胞系基因组dna输入系统的待比对样本二代测序基因组比对文件为bam格式文件,是由样本二代测序结果(fastq格式)比对到人参考基因组后得到的比对文件;同时还伴随输入一个对应的bai格式索引文件。bam格式文件为sam格式的二进制形式。本实施例sam示例文件如表6所示:表6sam示例文件4.mhparser位点分型以样本mix_1为例,利用以下步骤获得snp/indel变异位点的微单倍型分型结果:1)以检测位点列表、样本二代测序比对文件和测序变异检测信息为输入,计算输出初始的微单倍型分型结果,具体计算方法为:对检测位点列表中的某个微单倍型或snp/indel位点,通过遍历样本二代测序比对文件中的每条序列,根据位点的坐标信息提取reads的碱基组合,得到单倍型信息,并对具有相同单倍型的reads进行计数,针对snp/indel位点,如果有变异检测的vcf文件,还可从中读取到位点的基因型信息。2)针对相同单倍型,按照reads数目对不同碱基组合从高到低排序,得到排序结果;3)将排序后的分型文本文件转换成excel文件,方便查阅。样本mix_1的微单倍型分型结果如表7所示:表7样本mix_1的微单倍型分型结果部分同样的方法可以获得其他样本的微单倍型分型结果。5.mhparser样本相似度计算在得到多个样本的微单倍型分型结果后,可以计算样本相似度,输出单倍型序列,具体步骤如下:1)将多个样本微单倍型分型的结果列表作为输入文件;2)利用相似性计算程序,输出任意两个样本间的相似性分值,具体计算方法为:其中,s为相似性分值;m为位点个数;c1为常量,取值1,为两个样本某个位点碱基型相同时的打分值;c2为常量,取值-1,为两个样本某个位点碱基型不同时的打分值;g1和g2分别为某个位点样本1和样本2的碱基组合数目,其中,样本1为该位点上碱基组合数目较少的样本,样本2为该位点上碱基组合数目较多的样本;如果针对某个位点,两个样本的碱基组合数目相同,则任意一个样本为样本1,另外一个样本为样本2。g1∩g2表示该位点上两个样本具有相同的碱基组合的数目。输出由此得到样本的相似性分值如表8所示:表8样本的相似性信息注:数值为similarity_score,即相似性分值,分值越高,越相似。。同时,该步骤还输出样本单倍型序列信息(样本微单倍型位点合并序列文件),如下所示:>o1atgcgcggtcttccaggcctgatctgaagcaactgataatgttactgggtggtccgtcacc>o2atgcgcggtcttccaggagatctgaagcaactaatgttgggtggttcaccgcc>o3atgcacggtccagatttggcctgaagaaactggtactgttggttgggccgctgcc>o4atgcgcggtcttccttcagatctgacctgaagcaactaatggtgttgggccgtcacc>o5acggttttccaggcctgaaactgataatacgaagtgggccactgcc>mix_1acggacgctcctttttcaggcctgatctgaaacagatgatgataatggtactgtgaagtgggtggtccactgtcacctt>mix_2acggacgctcctttaggcctgatctgaaacagatgatgataatggtactgtgaagtgggtggtccactgtcacctt>mix_3acggacgctcctttaggcctgaaacagatgatgataatacgaagtgggtggtccactgtcacctt>mix_4acggtcctttaggcctgaaactgataatacgaagtgggtggtccactgcc>na12878_01acgcgccgtcttcaagaaggtttgattttaaactgataatattgttgggtggtccgccacctt>na12878_02acgcgccgtcttcatatagaaggtttgattttaaactgataatattgttgggtggtccgccacctt6.mhparser多序列比对根据上述样本单倍型序列,利用mafft软件进行多序列比对。该步骤会做一个对齐的工作,可以得到fasta格式的多序列比对文件,如下所示:>o1atgcgcggtcttcc------aggcctgatctgaagcaactga------taatgttac----tgggtggtccgtcacc----->o2atgcgcggtcttcc------agg--agatctgaagcaactaa------------tgt----tgggtggttcaccgcc----->o3atgcacggtcc---------agatttggcctgaagaaactgg---------tactgt----tggttgggccgctgcc----->o4atgcgcggtcttcc---ttcagatctgacctgaagcaactaa---------tggtgt----tggg----ccgtcacc----->o5----acggttttcc-----------aggcctgaaac------------tgataatacgaagtggg----ccactgcc----->mix_1acggacgctccttt---ttcaggcctgatctgaaacagatgatgataatggtactgtgaagtgggtggtccactgtcacctt>mix_2acggacgctccttt------aggcctgatctgaaacagatgatgataatggtactgtgaagtgggtggtccactgtcacctt>mix_3acggacgctccttt-----------aggcctgaaacagatga------tgataatacgaagtgggtggtccactgtcacctt>mix_4----acggtccttt-----------aggcctgaaac------------tgataatacgaagtgggtggtccactgcc----->na12878_01acgcgccgtcttca---agaaggtttgattttaaactgataa---------tattgt----tgggtggtccgccacctt--->na12878_02acgcgccgtcttcatatagaaggtttgattttaaactgataa---------tattgt----tgggtggtccgccacctt---7.mhparser样本聚类利用fasttree软件,根据样本单倍型多序列比对结果进行样本聚类,得到nwk格式文件的样本聚类结果,利用可视化软件mega识别,以便进一步查看样本聚类的情况,导入可视化后结果如图4所示。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1