具有细胞渗透性的BCL-XL抑制剂的抗体药物缀合物的制作方法
本申请是申请日为2015年12月9日,申请号为201580075744.2,名称为“具有细胞渗透性的bcl-xl抑制剂的抗体药物缀合物”的发明专利申请的分案申请。
1.发明领域
本公开涉及抑制bcl-xl抗凋亡蛋白的活性的化合物、含有这些抑制剂的抗体药物缀合物、用于合成这些抑制剂和抗体药物缀合物的方法、含有这些抑制剂和抗体药物缀合物的组合物以及治疗其中抗凋亡bcl-xl蛋白被表达的疾病的方法。
2.背景
细胞程序死亡被认为是所有活物种的组织动态平衡的主要生化过程。尤其是在哺乳动物中,已经表明它可调节早期胚胎发育。在生命的后期,细胞死亡是除去潜在危险细胞(例如,携带癌性缺陷的细胞)的默认机构。已经发现了一些凋亡途径,一个最重要的途径与蛋白的bcl-2家族有关,其是细胞程序死亡的线粒体(还称为“固有的”)途径的关键调节剂。参见danial&korsmeyer,2004,cell116:205-219。
凋亡途径失调涉及许多重要疾病的病变,例如,神经变性病症(细胞程序死亡上调),例如,阿尔茨海默氏病;和增殖疾病(细胞程序死亡下调),例如,癌症、自身免疫疾病和促血栓形成的病症。
在一方面,下调的细胞程序死亡(且更尤其是蛋白的bcl-2家族)与癌性恶性肿瘤的发病有关的密切关系已经表明了靶向这种疑难病的新方法。研究已经表明,例如,抗凋亡蛋白bcl-2和bcl-xl在许多癌细胞类型中过度表达。参见,zhang,2002,naturereviews/drugdiscovery1:101;kirkin等人,2004,biochimicabiophysicaacta1644:229-249;以及amundson等人,2000,cancerresearch60:6101-6110。这种失调的结果是被改变的细胞的存活,这在正常情况下将会另外进行细胞程序死亡。与未调节的增殖有关的这些缺陷的重复,被认为是癌性演变的起点。
这些发现以及许多其它发现,使得在靶向癌症的药物研发中,可能出现新的策略。如果小分子能够进入细胞,并且克服抗凋亡蛋白的过度表达,则它可能重新设定凋亡过程。这种策略的好处是,它可以减轻耐药性的问题,这种问题通常是凋亡失调(异常存活)的结果。
研究人员还已经证明,为了通过固有的凋亡途径来执行编程性细胞死亡,血小板也含有必要的凋亡机构(例如,bax、bak、bcl-xl、bcl-2、细胞色素c、半胱天冬酶-9、半胱天冬酶-3和apaf-1)。虽然循环血小板产生是正常生理过程,但过量的血小板或血小板的非所要求的活化导致或加重许多疾病。以上说明,能够在哺乳动物中抑制血小板中的抗凋亡蛋白和减少血小板数量的治疗剂,可以用于治疗促血栓形成病症和以过量的血小板或血小板的非所要求的活化为特征的疾病。
已经研发了许多bcl-xl抑制剂,用于治疗与凋亡途径失调有关的疾病(例如,癌症)。然而,bcl-xl抑制剂可以作用于除了靶细胞(例如,癌细胞)以外的细胞。例如,临床前研究表明,bcl-xl的药理学失活降低血小板半衰期,并且引起血小板减少(参见mason等人,2007,cell128:1173-1186)。
由于bcl-xl在调节细胞程序死亡中的重要性,所以,本领域还需要选择性地或非选择性地抑制bcl-xl活性的药剂,作为治疗其中细胞程序死亡的失调通过抗凋亡bcl-2家族蛋白(例如,bcl-xl)表达或过度表达的疾病的途径。相应地,需要剂量限制性毒性降低的新的bcl-xl抑制剂。
另外,还需要递送限制毒性的bcl-xl抑制剂的新方法。将药物递送到细胞中的一个可能的方法是通过使用抗体药物缀合物(adc)来递送药物,这对于bcl-xl抑制剂来说,还没有进行探索。通过连接子,将细胞毒类药物与单克隆抗体进行化学连接,形成adc。adc的单克隆抗体选择性地与细胞(例如,癌细胞)的靶点抗原结合,并将药物释放到细胞中。adc具有治疗潜力,这是由于它们将抗体的特异性和药物的细胞毒素潜力结合在一起。尽管如此,到目前为止,由于各种因素,例如,不利的毒性特性、低的效果和差的药理学参数,因此研发adc作为治疗剂的成果很有限。相应地,研发能够克服这些问题并且可以选择性地将bcl-xl递送到靶向癌细胞中的新的adc,将成为有重大意义的发现。
3.概述
现在已经发现,当给药形式是与细胞表面上表达的抗原结合的抗体药物缀合物(adc;也称为免疫缀合物)形式时,bcl-xl的小分子抑制剂是有效的,其中抑制bcl-xl和后续的诱导细胞程序死亡是有益的。这个发现首次提供了对于感兴趣的具体细胞和/或组织的靶向bcl-xl抑制治疗、潜在地降低了达到目标治疗益处所必需的血清水平和/或避免和/或改善了与全身性给予小分子bcl-xl抑制剂本身有关的潜在副作用。
相应地,在一方面,本公开提供了包含bcl-xl抑制剂的adc,尤其可用于抑制抗凋亡的bcl-xl蛋白,作为治疗与细胞程序死亡途径失调有关的疾病(例如,癌症)的治疗途径。adc通常包含bcl-xl的小分子抑制剂(本文称为bcl-xl抑制剂),它通过连接子与抗体连接,所述抗体特异性地结合感兴趣的靶细胞上表达的抗原。
adc的抗体可以是通常(但不一定是特异性)与感兴趣的靶细胞的表面上表达的抗原结合的任何抗体。感兴趣的靶细胞通常包括:需要通过抑制抗凋亡bcl-xl蛋白来诱导细胞程序死亡的细胞,包括,例如(没有限制性),表达或过度表达bcl-xl的肿瘤细胞。靶点抗原可以是感兴趣的靶细胞上表达的任何蛋白,但通常是在靶细胞上独特表达并且在正常或健康细胞上没有表达的蛋白;或与正常或健康细胞相比较,在靶细胞上过度表达的蛋白,以便adc选择性地靶向感兴趣的具体细胞,例如,肿瘤细胞。正如本领域所公认的那样,与内化所结合的adc的某些细胞表面抗原结合的adc具有某些优越性。相应地,在一些实施方案中,抗体靶向的抗原是能够使其结合的adc内化的抗原。然而,adc靶向的抗原无需是使结合的adc内化的抗原。在靶细胞或组织外部释放的bcl-xl抑制剂可以通过被动扩散或其它机制进入细胞,抑制bcl-xl。
本领域技术人员会理解,具体抗原以及由此选择的抗体取决于感兴趣的目标靶细胞的特征。在某些具体治疗实施方案中,adc的抗体的靶点抗原是在已知的正常或健康细胞类型上没有表达的抗原,或被怀疑至少部分地依赖于bcl-xl存活的抗原。在其它某些具体治疗实施方案中,adc的抗体是适合于给予人类的抗体。
用作治疗靶点的种类繁多的细胞特异性抗原,以及结合这些抗原的抗体是本领域已知的,获得适合于靶向已知的细胞特异性抗原的其它抗体的技术或后来发现的细胞特异性抗原也是本领域已知的。这些不同抗体中的任何抗体可以包括在本文描述的adc中。
在性质上,连接adc的bcl-xl抑制剂与抗体的连接子可以是长的、短的、柔性、刚性、疏水性或亲水性的连接子,或可以包含不同特征的片段,例如,柔性片段、刚性片段等等。连接子可以是对于胞外环境化学上稳定的连接子,例如,在血流中化学稳定,或可以包括在胞外环境中不稳定并且释放bcl-xl抑制剂的连接基。在一些实施方案中,连接子包括被设计为adc在细胞中内化之时释放bcl-xl抑制剂的连接基。在一些具体实施方案中,连接子包括被设计为在细胞内部特异性或非特异性断裂和/或裂解或分解的连接基。在adc背景下用于连接药物与抗体的多种连接子在本领域是已知的。这些连接子中的任何连接子以及其它连接子,可以用于连接本文描述的adc中的bcl-xl抑制剂与抗体。
与adc的抗体连接的bcl-xl抑制剂的数量可以变化(称为“药物对抗体比例”或“dar”),并且仅仅受到抗体上的可供的连接位点的数量以及与单一连接子连接的抑制剂的数量的限制。通常,连接子连接单一bcl-xl抑制剂与adc的抗体。在使用和/或储存条件下,只要adc没有显示出不能接受的聚集水平,考虑包括dar为20或dar更高的adc。在一些实施方案中,本文描述的adc可以具有大约1-10、1-8、1-6或1-4范围内的dar。在某些具体实施方案中,adc可以具有2、3或4的dar。在一些实施方案中,选择bcl-xl抑制剂、连接子和dar的组合,使得所得到的adc在使用和/或储存条件下不会过度聚集。
含有本文所描述的adc的bcl-xl抑制剂通常是按照下面结构式(iia)的化合物,和/或其可药用盐,其中各个取代基ar、z1、z2、r1、r2、r4、r10a、r10b、r10c、r11a、r11b和n如详细说明部分所定义∶
在式(iia)中,#代表与连接子的连接点。在不是adc部分的抑制剂中,#代表氢原子。
在一些实施方案中,本文所描述的adc通常是按照结构式(i)的化合物∶
其中,ab代表抗体,d代表药物(本文中,代表bcl-xl抑制剂),l代表连接药物d与抗体ab的连接子,lk代表在连接子l上的官能团和抗体ab上的互补官能团之间形成的连接基,和m代表连接到抗体上的连接子-药物单元的数目。
在某些具体实施方案中,adc是按照下面结构式(ia)的化合物,其中各个取代基ar、z1、z2、r1、r2、r4、r10a、r10b、r10c、r11a、r11b和n分别如先前式(iia)所定义,ab和l如结构式(i)所定义,m是1至20的整数,和在一些实施方案中,m是2至8的整数,和在一些实施方案中,m是1至8的整数,和在一些实施方案中,m是2、3或4∶
在另一个方面,本公开提供了用于合成本文所描述的adc的中间体合成子,以及合成adc的方法。所述中间体合成子通常包含与连接子部分连接的bcl-xl抑制剂,所述连接子部分包括能够连接合成子与抗体的官能团。合成子通常是按照下面结构式(iii)的化合物,或其盐,其中d是本文先前所描述的bcl-xl抑制剂,l是先前所描述的连接子,rx含有能够使合成子与抗体上的互补官能团缀合的官能团∶
在某些具体实施方案中,中间体合成子是按照下面结构式(iiia)的化合物,或其盐,其中,各个取代基ar、z1、z2、r1、r2、r4、r10a、r10b、r10c、r11a、r11b和n如先前结构式(iia)所定义,rx含有上述官能团∶
为了合成adc,在官能团rx与抗体上的互补官能团反应形成共价键的条件下,使按照结构式(iii)或(iiia)-(iiid)的中间体合成子或其盐与感兴趣的抗体接触。基团rx的特征取决于目标偶合化学,以及合成子连接的抗体上的互补基团。适合于将分子与抗体结合的许多基团在本领域是已知的。这些基团中的任何基团可以适合于rx。非限制性的示例性官能团(rx)包括nhs-酯、马来酰亚胺、卤代乙酰基、异硫氰酸酯、乙烯基砜和乙烯基磺酰胺。
在另一个方面,本公开提供了包括本文所描述的adc的组合物。所述组合物通常包含一或多种本文所描述的adc和/或其盐,以及一或多种赋形剂、载体或稀释剂。可以将所述组合物配制为用于药物用途,或其它用途。在一个具体实施方案中,将所述组合物配制为用于药物用途,并且含有按照结构式(ia)的adc或其可药用盐,以及一或多种可药用赋形剂、载体或稀释剂。
配制为用于药物用途组合物可以是适合于多次给药的散装形式,或可以是适合于单一给药的单位剂量包装形式。无论是散装或单位剂量形式,所述组合物可以是干燥组合物,例如冷冻干燥物,或液体组合物。可以将单位剂量液体组合物方便地包装为预先充满适合于单次给药数量的adc的注射器形式。
在另一个方面,本公开提供了抑制抗凋亡bcl-xl蛋白的方法。所述方法通常包括:在抗体结合靶细胞上的抗原的条件下,使本文所描述的adc,例如,按照结构式(ia)的adc,与表达或超表达bcl-xl的靶细胞以及adc的抗体的抗原接触。取决于抗原,adc可以被内化于靶细胞中。可以在抑制bcl-xl活性的细胞试验中体外进行所述方法,或作为治疗其中需要抑制bcl-xl活性的疾病的治疗途径来体内进行。
在另一个方面,本公开提供了在细胞中诱导细胞程序死亡的方法。所述方法通常包括:在抗体结合靶细胞上的抗原的条件下,使本文所描述的adc,例如,按照结构式(ia)的adc,或其盐,与表达或超表达bcl-xl的靶细胞以及adc的抗体的抗原接触。取决于抗原,adc可以被内化于靶细胞中。可以在诱导细胞程序死亡的细胞试验中体外进行所述方法,或作为治疗疾病的治疗途径来体内进行,其中,在具体细胞中诱导细胞程序死亡有益于治疗所述疾病。在一个实施方案中,本文所描述的adc的抗体结合肿瘤细胞上表达的细胞表面受体或肿瘤相关抗原。在另一个实施方案中,本文所描述的adc的抗体结合选自egfr、epcam和ncam1的细胞表面受体或肿瘤相关抗原中的一种。在另一个实施方案中,本文所描述的adc的抗体结合egfr、epcam或ncam1。在另一个实施方案中,本文所描述的adc的抗体结合epcam或ncam1。在另一个实施方案中,本文所描述的adc的抗体结合epcam。在另一个实施方案中,本文所描述的adc的抗体结合ncam1。在另一个实施方案中,本文所描述的adc的抗体结合egfr。
在又一个方面,本公开提供了治疗疾病的方法,在所述疾病中,需要抑制bcl-xl和/或诱导细胞程序死亡。正如在详细说明部分中更充分讨论的那样,多种疾病至少部分地由失调的细胞程序死亡所介导、至少部分地由抗凋亡bcl-xl蛋白的表达和/或超表达所介导。可以用本文所描述的adc来治疗或改善这些疾病中的任何疾病。
所述方法包括:给予患有至少部分地由bcl-xl的表达或超表达所介导的疾病的患者能够有效提供治疗益处数量的adc。所给予的adc的抗体的特征取决于所治疗的疾病。本文所描述的adc获得的治疗益处也取决于所治疗的疾病。在某些情况下,当以单一疗法给予时,adc可以治疗或改善具体疾病。在其它情况下,adc可以是整个治疗方案的一部分,所述治疗方案包括其它药剂,以及adc,用于治疗或改善疾病。
例如,bcl-xl的表达水平升高,与癌症中的化学疗法和放射疗法的耐受性有关(datta等人,1995,cellgrowthdiffer6:363-370;amundson等人,2000,cancerres60:6101-6110;haura等人,2004,clinlungcancer6:113-122)。在治疗癌症的背景下,本文公开的数据证明,adc可以有效作为单一疗法,或当辅助其它靶向或非靶向的化学治疗剂和/或放射治疗给予或与它们一起给予时,adc也是有效的。尽管不受任何工作原理束缚,但人们相信,在对于靶向或非靶向化学和/或放射治疗具有耐受性的肿瘤中,用本文所描述的adc抑制bcl-xl活性,可以使肿瘤“敏感”,这样就可以使肿瘤再次对化学治疗剂和/或放射疗法敏感。
相应地,在治疗癌症的背景下,“治疗益处”包括给予还没有开始化学和/或放射疗法治疗方案或对于化学和/或放射疗法治疗方案显示出耐受性(或怀疑或变得耐受性)的患者本文所描述的adc作为靶向或非靶向化学治疗剂和/或放射治疗的辅助治疗,或与它们一起给予所述患者,作为使肿瘤对于化学和/或放射治疗敏感的方法。一个实施方案涉及使肿瘤对于标准细胞毒素药剂和/或放射疗法敏感的方法,所述方法包括:使肿瘤与有效使肿瘤细胞对于标准细胞毒素药剂和/或放射疗法敏感数量的、能够结合肿瘤的本文所描述的adc接触。另一个实施方案涉及使肿瘤对于标准细胞毒素药剂和/或放射疗法敏感的方法,所述方法包括:使肿瘤与有效使肿瘤细胞对于标准细胞毒素药剂和/或放射疗法敏感数量的、能够结合肿瘤的本文所描述的adc接触,其中,肿瘤对于标准细胞毒素药剂和/或放射疗法变得耐受。另一个实施方案涉及使肿瘤对于标准细胞毒素药剂和/或放射疗法敏感的方法,所述方法包括:使肿瘤与有效使肿瘤细胞对于标准细胞毒素药剂和/或放射疗法敏感数量的、能够结合肿瘤的本文所描述的adc接触,其中,肿瘤以前没有接触过标准细胞毒素药剂和/或放射治疗。
4.附图的简要说明
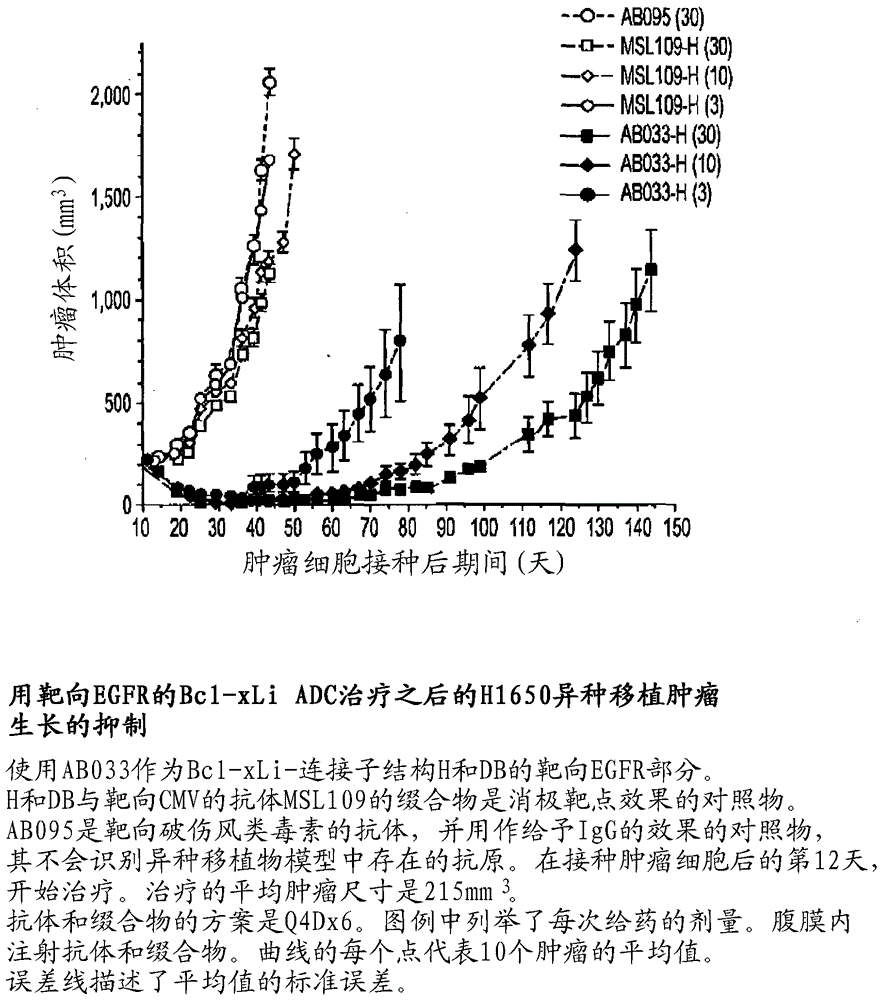
图1描述了用靶向egfr的bcl-xl抑制性adc(bcl-xliadc)或与多西他赛(dtx)联用药治疗之后的h1650异种移植肿瘤生长的抑制。
图2描述了用各种剂量的靶向egfr的bcl-xliadc治疗之后的h1650异种移植肿瘤生长的抑制。
图3描述了作为单一药剂或与多西他赛(dtx)联合的靶向egfr的bcl-xliadc治疗之后的ebc-1异种移植肿瘤生长的抑制。
图4描述了用靶向α-ncam1的、以单一药剂形式给予的或与选择性的bcl-2抑制剂abt-199联合给予的bcl-xliadc治疗后的h146异种移植肿瘤生长的抑制。
图5描述了用靶向α-ncam1的、以单一药剂形式给予的或与选择性的bcl-2抑制剂abt-199联合给予的bcl-xliadc治疗后的h1963.fp5异种移植肿瘤生长的抑制。
图6描述了含有细胞渗透性bcl-xl抑制剂的bcl-xliadc对于循环血小板数量的影响。
5.详细说明
本公开涉及adc、用于合成所述adc的合成子、含有所述adc的组合物和使用所述adc的各种方法。
正如技术人员所理解的那样,本文公开的adc性质上是“模块化的”。在整个本发明公开中,描述了包含adc的各个“模块”以及用于合成adc的合成子的各个具体实施方案。作为具体非限制性实施例,描述了抗体、连接子以及可以包含adc和合成子的bcl-xl抑制剂的具体实施方案。所描述的所有具体实施方案可以彼此组合,就好像单独明确地描述每个具体组合一样。
技术人员也可以理解,本文描述的各种bcl-xl抑制剂、adc和/或adc合成子可以是盐形式,在某些实施方案中,尤其是可药用盐。具有充分酸性官能团、充分碱性官能团或两种官能团的本公开的化合物,可以与许多无机碱、无机酸和有机酸反应,形成盐。或者,固有带电荷的化合物,例如具有季氮的那些化合物,可以与合适的反离子(例如,卤素,例如,溴、氯或氟)形成盐。
形成酸加成盐通常所使用的酸是无机酸,例如盐酸、氢溴酸、氢碘酸、硫酸、磷酸,等等;以及有机酸,例如对甲苯磺酸、甲磺酸、草酸、对溴苯基-磺酸、碳酸、琥珀酸、柠檬酸,等等。碱加成盐包括衍生自无机碱的那些盐,例如铵和碱金属或碱土金属氢氧化物、碳酸盐、碳酸氢盐,等等。
在下面的公开内容中,如果包括结构图和命名两者,并且如果命名与结构图不一致,则以结构图为准。
5.1.定义
除非本文另外定义,否则,与本公开结合使用的科学和技术术语具有本领域普通技术人员所通常理解的含义。
下面定义了各种化学取代基。在有些情况下,取代基(例如,烃基、烷基、烯基、炔基、环烃基、杂环基、杂芳基和芳基)中的碳原子数目是通过前缀“cx-cy”表示的,其中,x是取代基中的碳原子的最小数目,y是取代基中的碳原子的最大数目。由此,例如,“c1-c6烃基”是指含有1至6个碳原子的烃基。进一步举例说明,“c3-c8环烷基”是指含有3至8个碳环原子的饱和烃基环。如果取代基被描述为“取代的”,则碳或氮上的氢原子被非氢基团代替。例如,取代的烃基取代基是烃基上的至少一个氢原子被非氢基团代替。举例说明,单氟烃基是被氟基团取代的烃基,二氟烃基是被两个氟基团取代的烃基。应该认识到,如果在取代基上有一个以上的取代,则每个取代可以相同或不同(除非另有说明)。如果取代基被描述为“任选取代的”,则该取代基可以:(1)没有被取代,或(2)被取代。可能的取代基包括但不局限于::c1-c6烷基、c2-c6烯基、c2-c6炔基、芳基、环烃基、杂环基、杂芳基、卤素、c1-c6卤代烃基、氧代、cn、no2、orxa、oc(o)rz、oc(o)n(rxa)2、srxa、s(o)2rxa、s(o)2n(rxa)2、c(o)rxa、c(o)orxa、c(o)n(rxa)2、c(o)n(rxa)s(o)2rz、n(rxa)2、n(rxa)c(o)rz、n(rxa)s(o)2rz、n(rxa)c(o)o(rz)、n(rxa)c(o)n(rxa)2、n(rxa)s(o)2n(rxa)2、(c1-c6亚烃基)cn、(c1-c6亚烃基)orxa、(c1-c6亚烃基)oc(o)rz、(c1-c6亚烃基)oc(o)n(rxa)2、(c1-c6亚烃基)srxa、(c1-c6亚烃基)s(o)2rxa、(c1-c6亚烃基)s(o)2n(rxa)2、(c1-c6亚烃基)c(o)rxa、(c1-c6亚烃基)c(o)orxa、(c1-c6亚烃基)c(o)n(rxa)2、(c1-c6亚烃基)c(o)n(rxa)s(o)2rz、(c1-c6亚烃基)n(rxa)2、(c1-c6亚烃基)n(rxa)c(o)rz、(c1-c6亚烃基)n(rxa)s(o)2rz、(c1-c6亚烃基)n(rxa)c(o)o(rz)、(c1-c6亚烃基)n(rxa)c(o)n(rxa)2或(c1-c6亚烃基)n(rxa)s(o)2n(rxa)2;其中,rxa在每次出现时独立地是氢、芳基、环烃基、杂环基、杂芳基、c1-c6烃基或c1-c6卤代烃基;rz在每次出现时独立地是芳基、环烃基、杂环基、杂芳基、c1-c6烃基或c1-c6卤代烃基。
在本文一些实施方案中,参考包括取代基的结构式,例如,取代基ar、z1、z2、r1、r2、r4、r10a、r10b、r10c、r11a、r11b、l、rx、fx、lk、ab、n和/或m,描述了各种adc、合成子和含有adc和/或合成子的bcl-xl抑制剂。可以理解,含有取代基的各个基团可以根据化合价和稳定性来进行组合。本公开所设想的取代基和变量的组合,只是能够形成稳定化合物的那些组合。本文使用的术语“稳定”是指具有足够制备稳定性,并且在足够的时间内保持化合物的完整性,可有效用于本文说明的目的的化合物。
本文使用的下列术语具有下列含义∶
术语“烷氧基”是指式-ora的基团,其中,ra是烃基。代表性的烷氧基包括甲氧基、乙氧基、丙氧基、叔丁氧基,等等。
术语“烷氧基烃基”是指被烷氧基取代的烃基,并且可以由通式-rbora代表,其中,rb是亚烃基,ra是烃基。
单独或作为另一个取代基的一部分的术语“烃基”,是指饱和或不饱和的支链、直链或环状一价烃基,它是从母体烷、烯或炔烃的单个碳原子上除去一个氢原子所产生的。典型的烃基包括但不局限于:甲基;乙基(ethyls),例如,乙烷基(ethanyl)、乙烯基、乙炔基;丙基(propyls),例如,丙-1-基、丙-2-基、环丙-1-基、丙-1-烯-1-基、丙-1-烯-2-基、丙-2-烯-1-基、环丙-1-烯-1-基;环丙-2-烯-1-基、丙-1-炔-1-基、丙-2-炔-1-基、等等;丁基(butanyls),例如丁-1-基、丁-2-基(仲丁基)、2-甲基-丙-1-基(异丁基)、2-甲基-丙-2-基(叔丁基)、环丁烷-1-基、丁-1-烯-1-基、丁-1-烯-2-基、2-甲基-丙-1-烯-1-基、丁-2-烯-1-基、丁-2-烯-2-基、丁-1,3-二烯-1-基、丁-1,3-二烯-2-基、环丁-1-烯-1-基、环丁-1-烯-3-基、环丁-1,3-二烯-1-基、丁-1-炔-1-基、丁-1-炔-3-基、丁-3-炔-1-基,等;等等。如果指定具体的饱和水平,则使用下面定义的“烷基”、“烯基”和/或“炔基”。术语“低级烃基”是指具有1至6个碳的烃基。
单独或作为另一个取代基的一部分的术语“烷基”,是指饱和的支链、直链或环状烃基,它是从母体烷烃的单个碳原子上除去一个氢原子所产生的。典型的烷基包括但不局限于:甲基;乙烷基(ethanyl);丙烷基(propanyls),例如,丙-1-基、丙-2-基(异丙基)、环丙-1-基,等等;丁基,例如,丁-1-基、丁-2-基(仲丁基)、2-甲基-丙-1-基(异丁基)、2-甲基-丙-2-基(叔丁基)、环丁-1-基,等;等等。
单独或作为另一个取代基的一部分的术语“烯基”,是指具有至少一个碳-碳双键的不饱和的支链、直链或环状烃基,它是从母体烯烃的单个碳原子上除去一个氢原子所产生的。典型的烯基包括但不局限于:乙烯基;丙烯基,例如,丙-1-烯-1-基、丙-1-烯-2-基、丙-2-烯-1-基、丙-2-烯-2-基、环丙-1-烯-1-基;环丙-2-烯-1-基;丁烯基例如丁-1-烯-1-基、丁-1-烯-2-基、2-甲基-丙-1-烯-1-基、丁-2-烯-1-基、丁-2-烯-2-基、丁-1,3-二烯-1-基、丁-1,3-二烯-2-基、环丁-1-烯-1-基、环丁-1-烯-3-基、环丁-1,3-二烯-1-基,等;等等。
单独或作为另一个取代基的一部分的术语“炔基”,是指具有至少一个碳-碳三键的不饱和的支链、直链或环状烃基,它是从母体炔烃的单个碳原子上除去一个氢原子所产生的。典型的炔基包括但不局限于:乙炔基;丙炔基,例如,丙-1-炔-1-基、丙-2-炔-1-基、等等;丁炔基,例如,丁-1-炔-1-基、丁-1-炔-3-基、丁-3-炔-1-基,等;等等。
术语“烃基胺”是指式-nhra的基团,“二烃基胺”是指式-nrara的基团,其中,每个ra彼此独立地是烃基。
术语“亚烃基”是指具有两个端部单价基团核心的烷烃、烯烃或炔烃基团,其是从两个端部碳原子的每个碳原子上除去一个氢原子所产生的。典型的亚烃基包括但不局限于:亚甲基;以及饱和或不饱和的亚乙基;亚丙基;亚丁基;等等。术语“低级亚烃基”是指具有1至6个碳的亚烃基。
术语“芳基”是指含有6至14个碳环原子的芳香碳环基。芳基可以是单环或多环的(即,可以含有一个以上的环)。在多环芳环的情况下,只要求多环系统中的一个环是芳香环,而其余的环可以是饱和、部分饱和或不饱和环。芳基的例子包括苯基、萘基、茚基、茚满基和四氢萘基。
前缀“卤代”表示:包括该前缀的取代基被一个或多个独立选择的卤素基团取代。例如,卤代烃基是指至少一个氢被卤素基团代替的烃基取代基。典型的卤素基团包括氯、氟、溴和碘。卤代烃基的例子包括氯甲基、1-溴乙基、氟甲基、二氟甲基、三氟甲基和1,1,1-三氟乙基。应该认识到,如果取代基被一个以上的卤素基团取代,则那些卤素基团可以相同或不同(除非另有说明)。
术语“卤代烷氧基”是指式-orc的基团,其中,rc是卤代烃基。
术语“杂烃基”(heteroalkyl)、“杂烷基”、“杂烯基”、“杂炔基”和“杂亚烃基”(heteroalkylene)分别指的是烃基、烷基、烯基、炔基和亚烃基基团,其中的一个或多个碳原子,例如,1、2或3个碳原子,各自独立地被相同或不同的杂原子或杂原子的基团代替。可以替代碳原子的典型的杂原子和/或杂原子基团包括但不局限于:-o-、-s-、-s-o-、-nrc-、-ph-、-s(o)-、-s(o)2-、-s(o)nrc-、-s(o)2nrc-,等等,包括其组合,其中每个rc独立地是氢或c1-c6烃基。
术语“环烃基”和“杂环基”分别指的是“烃基”和“杂烃基”的环状形式。对于杂环基,杂原子可以占据与分子的其余部分连接的位置。环烃基或杂环基环可以是单个环(单环)或具有两个或多个环(双环或多环)。
单环环烃基和杂环基通常含有3至7个环原子,更通常含有3至6个环原子,且更通常含有5至6个环原子。环烃基的实例包括但不局限于:环丙基;环丁基(cyclobutyls),例如环丁烷基和环丁烯基;环戊基(cyclopentyls),例如环戊烷基和环戊烯基;环己基(cyclohexyls),例如环己烷基和环己烯基;等等。单环杂环基的例子包括但不局限于:氧杂环丁烷、呋喃基、二氢呋喃基、四氢呋喃基、四氢吡喃基、噻吩基(硫呋喃基)、二氢噻吩基、四氢噻吩基、吡咯基、吡咯啉基、吡咯烷基、咪唑基、咪唑啉基、咪唑烷基、吡唑基、吡唑啉基、吡唑烷基、三唑基、四唑基、噁唑基、噁唑烷基、异噁唑啉基、异噁唑基、噻唑基、异噻唑基、噻唑啉基、异噻唑啉基、噻唑烷基、异噻唑烷基、噻二唑基、噁二唑基(包括1,2,3-噁二唑基、1,2,4-噁二唑基、1,2,5-噁二唑基(呋咱基)或1,3,4-噁二唑基)、噁三唑基(包括1,2,3,4-噁三唑基或1,2,3,5-噁三唑基)、二噁唑基(包括1,2,3-二噁唑基、1,2,4-二噁唑基、1,3,2-二噁唑基或1,3,4-二噁唑基)、1,4-二噁烷基、二氧代硫吗啉基、噁噻唑基、氧硫杂环戊烯基、氧杂硫杂环戊烷基、吡喃基、二氢吡喃基、硫吡喃基、四氢硫吡喃基、吡啶基(吖嗪基)、哌啶基、二吖嗪基(包括哒嗪基(1,2-二吖嗪基))、嘧啶基(1,3-二吖嗪基)或吡嗪基(1,4-二吖嗪基))、哌嗪基、三嗪基(包括1,3,5-三嗪基、1,2,4-三嗪基和1,2,3-三嗪基))、噁嗪基(包括1,2-噁嗪基、1,3-噁嗪基或1,4-噁嗪基))、噁噻嗪基(包括1,2,3-噁噻嗪基、1,2,4-噁噻嗪基、1,2,5-噁噻嗪基或1,2,6-噁噻嗪基))、噁二嗪基(包括1,2,3-噁二嗪基、1,2,4-噁二嗪基、1,4,2-噁二嗪基或1,3,5-噁二嗪基))、吗啉基、氮杂卓基、氧杂环庚烯基、硫杂环庚烯基、二氮杂卓基、吡啶酮基(包括吡啶-2(1h)-酮基和吡啶-4(1h)-酮基)、呋喃-2(5h)-酮基、嘧啶酮基(包括嘧啶-2(1h)-酮基和嘧啶-4(3h)-酮基)、噁唑-2(3h)-酮基、1h-咪唑-2(3h)-酮基、哒嗪-3(2h)-酮基和吡嗪-2(1h)-酮基。
多环环烃基和杂环基含有一个以上的环,双环环烃基和杂环基含有两个环。环可以是桥接的、稠合的或螺取向的。多环环烃基和杂环基可以包括桥环、稠合环和/或螺环的组合形式。在螺环环烃基或杂环基中,两个不同的环共用一个原子。螺环烃基的实例是螺[4.5]癸烷,螺杂环基的实例是螺二氢吡唑。
在桥接的环烃基或杂环基中,环共享至少两个常规非相邻的原子。桥接的环烃基的实例包括但不局限于:金刚烷基和降冰片基环。桥接的杂环基的实例包括但不局限于:2-氧杂三环[3.3.1.13,7]癸基。
在稠合的环烃基或杂环基中,两个或多个环稠合在一起,使得两个环共享一个普通键。稠合环环烃基的实例包括十氢化萘、亚萘基、四氢萘和蒽。含有两个或三个环的稠合环杂环基的例子包括:咪唑并吡嗪基(包括咪唑并[1,2-a]吡嗪基)、咪唑并吡啶基(包括咪唑并[1,2-a]吡啶基)、咪唑并哒嗪基(包括咪唑并[1,2-b]哒嗪基)、噻唑并吡啶基(包括噻唑并[5,4-c]吡啶基、噻唑并[5,4-b]吡啶基、噻唑并[4,5-b]吡啶基和噻唑并[4,5-c]吡啶基)、茚嗪基、吡喃并吡咯基、4h-喹嗪基、嘌呤基、萘啶基、吡啶并吡啶基(包括吡啶并[3,4-b]-吡啶基、吡啶并[3,2-b]-吡啶基或吡啶并[4,3-b]-吡啶基)和喋啶基。稠合环杂环基的其它实例包括:苯并稠合的杂环基,例如,二氢色烯基、四氢异喹啉基、吲哚基、异吲哚基(异苯并吖唑基、假异氮茚基)、假吲哚基(异氮杂茚基)、异吲唑基(苯基吡唑基)、苯并吖嗪基(包括喹啉基(1-苯并吖嗪基)或异喹啉基(2-苯并吖嗪基))、酞嗪基、喹喔啉基、喹唑啉基、苯并二吖嗪基(包括噌琳基(1,2-苯并二吖嗪基)或喹唑啉基(1,3-苯并二吖嗪基))、苯并吡喃基(包括苯并二氢吡喃基或异苯并二氢吡喃基)、苯并噁嗪基(包括1,3,2-苯并噁嗪基、1,4,2-苯并噁嗪基、2,3,1-苯并噁嗪基或3,1,4-苯并噁嗪基)、苯并[d]噻唑基和苯并异噁嗪基(包括1,2-苯并异噁嗪基或1,4-苯并异噁嗪基)。
术语“杂芳基”是指含有5至14个环原子的芳香杂环基。杂芳基可以是单环或2或3个稠环。杂芳基的实例包括6元环,例如,吡啶基、吡嗪基、嘧啶基、哒嗪基和1,3,5-、1,2,4-或1,2,3-三嗪基;5元环取代基,例如,三唑基、吡咯基、咪唑基、呋喃基、噻吩基、吡唑基、噁唑基、异噁唑基、噻唑基、1,2,3-、1,2,4-、1,2,5-或1,3,4-噁二唑基和异噻唑基;6/5元稠环取代基,例如,咪唑并吡嗪基(包括咪唑并[1,2-a]吡嗪基)咪唑并吡啶基(包括咪唑并[1,2-a]吡啶基)、咪唑并哒嗪基(包括咪唑并[1,2-b]哒嗪基)、噻唑并吡啶基(包括噻唑并[5,4-c]吡啶基、噻唑并[5,4-b]吡啶基、噻唑并[4,5-b]吡啶基和噻唑并[4,5-c]吡啶基)、苯并[d]噻唑基、苯并硫呋喃基、苯并异噁唑基、苯并噁唑基、嘌呤基和苯邻甲内酰胺基;以及6/6元稠环,例如,苯并吡喃基、喹啉基、异喹啉基、噌琳基、喹唑啉基和苯并噁嗪基。杂芳基也可以是具有芳香(4n+2π电子)共振部分的杂环,例如,吡啶酮基(包括吡啶-2(1h)-酮基和吡啶-4(1h)-酮基)、嘧啶酮基(包括嘧啶-2(1h)-酮基和嘧啶-4(3h)-酮基)、哒嗪-3(2h)-酮基和吡嗪-2(1h)-酮基。
本文使用的术语“磺酸盐或酯(sulfonate)”是指磺酸的盐或酯。
本文使用的术语“磺酸甲酯”是指磺酸的甲基酯。
本文使用的术语“羧酸盐或酯(carboxylate)”是指羧酸的盐或酯。
在连接子的背景下,本文使用的术语“糖”是指单糖类的o-糖苷或n-糖苷碳水化合物衍生物,并且可以来源于天然存在的来源,或可以起源于合成。例如,“糖”包括衍生物,例如但不局限于:衍生自β-葡糖醛酸和β-半乳糖的那些糖。合适的糖取代基包括但不局限于:羟基、胺、羧酸、酯和醚。
术语“nhs酯”是指羧酸的n-羟基琥珀酰亚胺酯衍生物。
当在“或其盐”的背景下使用时,术语“盐”包括通常用于形成碱金属盐的盐、形成游离酸或游离碱的加成盐的盐。通常,这些盐典型地可以利用常规方法制备,例如,通过合适的酸或碱与本发明化合物的反应来制备。
如果预定给予患者盐(例如,与上下文的体外使用相对),优选,盐是可药用和/或生理学相容的盐。在本专利申请中,作形容词用的术语“可药用”是指其所修饰的名词适于用作药物产品或作为药物产品的一部分。术语“可药用盐”包括通常用于形成碱金属盐和形成游离酸或游离碱的加成盐的盐。通常,这些盐典型地可以利用常规方法制备,例如,通过合适的酸或碱与本发明化合物的反应来制备。
5.2示范性的实施方案
正如概述所提到的那样,本公开的一个方面涉及含有bcl-xl抑制剂的adc,其中bcl-xl抑制剂通过连接子与抗体连接。在具体实施方案中,adc是按照下面结构式(i)的化合物,或其盐,其中ab代表抗体,d代表bcl-xl抑制剂(药物),l代表连接子,lk代表在连接子l上的反应性官能团和抗体ab上的互补官能团之间形成的连接基,m代表与抗体连接的d-l-lk单元的数目∶
下面更详细地描述可以包含于本文所描述的adc的各种bcl-xl抑制剂(d)、连接子(l)和抗体(ab)以及与adc连接的bcl-xl抑制剂的数量的具体实施方案。
5.2.1bcl-xl抑制剂
adc包含一或多种bcl-xl抑制剂,它们可以相同或不同,但通常是相同的。在一些实施方案中,包含于adc的bcl-xl抑制剂,和在某些具体实施方案中,上面结构式(i)的d,是按照结构式(iia)的化合物∶
或其盐,其中∶
ar选自
z1选自n、ch和c-cn;
z2选自nh、ch2、o、s、s(o)和s(o2);
r1选自甲基、氯和氰基;
r2选自氢、甲基、氯和氰基;
r4是氢、c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基或c1-4羟烃基,其中r4的c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基和c1-4羟烃基任选被一个或多个独立地选自och3、och2ch2och3和och2ch2nhch3的取代基取代;
r10a、r10b和r10c各自彼此独立地选自氢、卤素、c1-6烷基、c2-6烯基、c2-6炔基和c1-6卤代烃基;
r11a和r11b各自彼此独立地选自氢、甲基、乙基、卤代甲基、羟基、甲氧基、卤素、cn和sch3;
n是0、1、2或3;和
#代表与连接子l的连接点。
在某些实施方案中,式(iia)的ar选自
在某些实施方案中,式(iia)的z1是n。
在某些实施方案中,式(iia)的z1是ch。
在某些实施方案中,式(iia)的z2是o。
在某些实施方案中,式(iia)的r1选自甲基和氯。
在某些实施方案中,式(iia)的r2选自氢和甲基。在具体实施方案中,r2是氢。
在某些实施方案中,式(iia)中的r1是甲基,r2是氢和z1是n。
在某些实施方案中,式(iia)中的r10a是卤素,r10b和r10c各自是氢。在具体实施方案中,r10a是氟。
在某些实施方案中,式(iia)中的r10b是卤素,r10a和r10c各自是氢。在具体实施方案中,r10b是氟。
在某些实施方案中,式(iia)中的r10c是卤素,r10a和r10b各自是氢。在具体实施方案中,r10c是氟。
在某些实施方案中,式(iia)中的r10a、r10b和r10c各自是氢。
在某些实施方案中,式(iia)中的r11a和r11b相同。在具体实施方案中,r11a和r11b各自是甲基。
在某些实施方案中,式(iia)中的n是0或1。
可以包括在本文所描述的adc中的示范性的bcl-xl抑制剂和/或其盐,包括实施例1.1-1.8分别所描述的化合物w1.01-w1.08。
含有adc的bcl-xl抑制剂,当未包括在adc内时,与抗凋亡bcl-xl蛋白结合,并抑制它,诱导细胞程序死亡。按照结构式(iia)的具体bcl-xl抑制剂,当未包括在adc内时(即,按照结构式(iia)的化合物或盐,其中,#代表氢原子),其结合和抑制bcl-xl活性的能力可以在标准结合和活性试验中得到证明,包括,例如,tao等人(2014,acsmed.chem.lett.,5:1088-1093)所描述的tr-fretbcl-xl结合试验。可用于证明bcl-xl结合的具体tr-fretbcl-xl结合试验提供于下面实施例4中。通常,在实施例4的结合试验中,可以在本文描述的adc中使用的bcl-xl抑制剂的ki小于大约10nm,但可以显示出显著更低的ki,例如,小于大约1、0.1nm、或甚至0.01nm的ki。
在标准基于细胞的细胞毒性试验中,例如,tao等人(2014,acsmed.chem.lett.,5:1088-1093)所描述的fl5.12细胞和molt-4细胞毒性试验,也可以证明bcl-xl抑制活性。可以用于证明具体bcl-xl抑制剂的bcl-xl抑制活性的具体molt-4细胞毒性试验提供于下面实施例5中。通常,在实施例5的molt-4细胞毒性试验中,本文描述的adc中所使用的bcl-xl抑制剂表现出小于大约500nm的ec50值,但可以表现出显著更低的ec50值,例如小于大约250、100、50、20、10或甚至5nm的ec50值。
虽然结构式(iia)定义的bcl-xl抑制剂预计是细胞渗透性的抑制剂,并且当未包括在adc内时,可以渗入细胞,但在用渗透细胞的细胞试验中,可以证明不能自由穿过细胞膜的化合物的bcl-xl抑制活性。正如背景部分讨论的那样,线粒体外膜透化(momp)的过程是由bcl-2家族蛋白控制的。具体地说,促凋亡bcl-2家族蛋白bax和bak促进momp,并且它们一旦活化,在线粒体膜外上进行低聚反应,形成孔隙,导致细胞色素c(cytc)的释放。cytc的释放引起凋亡体(apoptosome)的形成,这转而会导致半胱天冬酶活化以及使细胞经受编程性细胞死亡的其它情况(参见,goldstein等人,2005,celldeathanddifferentiation12:453-462)。抗凋亡bcl-2家族成员,包括bcl-2和bcl-xl,拮抗bax和bak的寡聚作用。bcl-xl抑制剂,在存活取决于bcl-xl的细胞中,可以导致bax和/或bak的活化、momp、cytc的释放和导致细胞程序死亡的下游活动。可以通过细胞中的细胞色素c的线粒体和细胞溶质部分的western印迹,确定cytc释放的过程,并且用作细胞中的细胞程序死亡的代表性量度标准。
作为检测低细胞渗透性的分子的bcl-xl抑制活性以及后续的cytc释放的方法,可以用在血浆(但不是线粒体、膜)中形成选择性孔隙的药剂处理细胞。具体地说,与线粒体膜相比,在质膜中,胆固醇/磷脂比例更高。因此,用低浓度的胆固醇引导的净化剂毛地黄皂苷进行短期培养,可选择性地渗透质膜,不会显著地影响线粒体膜。所述药剂与胆固醇形成不溶解的复合物,导致胆固醇从它的正常磷脂结合位点分离出来。随后,这种作用在脂质双层中形成大约40-50å宽的孔。在凋亡细胞中,一旦质膜被透化,可以冲洗掉能够通过毛地黄皂苷形成的孔隙的细胞溶质组分,包括从线粒体释放到细胞溶质中的细胞色素c(campos,2006,cytometrya69(6):515-523)。
通常,在实施例5的molt-4细胞透化cytc试验中,bcl-xl抑制剂的ec50值小于大约10nm,尽管所述化合物还可以表现出显著更低的ec50值,例如,小于大约5、1或甚至0.5nm。
虽然许多结构式(iia)的bcl-xl抑制剂相比于其它抗凋亡bcl-2家族蛋白可选择性或特异性地抑制bcl-xl,但不需要选择性和/或特异性地抑制bcl-xl。除了抑制bcl-xl之外,含有adc的bcl-xl抑制剂还可以抑制一或多种其它抗凋亡bcl-2家族蛋白,例如,比如,bcl-2。在一些实施方案中,含有adc的bcl-xl抑制剂对于bcl-xl具有选择性和/或特异性。特异性或选择性是指,在相同试验条件下,相比于结合或抑制bcl-2,具体bcl-xl抑制剂结合或抑制bcl-xl的程度更大。在具体实施方案中,在bcl-xl结合试验中,相比于bcl-2,含有adc的bcl-xl抑制剂对于bcl-xl显示出10倍、100倍或甚至更大的特异性。
5.2.2连接子
在本文所描述的adc中,bcl-xl抑制剂通过连接子与抗体连接。连接adc的bcl-xl抑制剂与抗体的连接子可以是短的、长的、疏水性的、亲水性的、挠性的或刚性的连接子,或可以由各自独立地具有一或多种上述性能的片段组成,使得连接子可以包括具有不同性能的片段。连接子可以是多价连接子,使得它们将一个以上的bcl-xl抑制剂共价连接到抗体上单一位点,或可以是单价连接子,使得它们将单个bcl-xl抑制剂共价连接到抗体上的单一位点。
正如技术人员所理解的那样,连接子与bcl-xl抑制剂在一个位置形成共价连接基,在另一个位置与抗体形成共价连接基,由此将bcl-xl抑制剂连接到抗体上。在连接子上的官能团和抑制剂与抗体上的官能团之间反应,形成共价连接基。本文使用的术语“连接子”意图包括:(i)连接子的非缀合形式,其包括能够使连接子与bcl-xl抑制剂共价连接的官能团,以及能够使连接子与抗体共价连接的官能团;(ii)连接子的部分缀合形式,其包括能够使连接子与抗体共价连接的官能团,并且共价连接bcl-xl抑制剂,或反之亦然;和(iii)连接子的完全缀合形式,其共价连接bcl-xl抑制剂和抗体。在本文所描述的中间体合成子和adc的一些具体实施方案中,分别用rx和lk具体说明含有连接子上的官能团的部分以及在连接子和抗体之间形成的共价连接基的部分。一个实施方案涉及在合成子共价连接抗体的条件下,通过结合肿瘤细胞上表达的细胞表面受体或肿瘤相关抗原的抗体与本文描述的合成子接触所形成的adc。一个实施方案涉及adc的制备方法,所述adc是在合成子共价连接抗体的条件下,通过接触本文描述的合成子所形成的。一个实施方案涉及在表达bcl-xl的细胞中抑制bcl-xl活性的方法,所述方法包括:在adc结合细胞的条件下,使细胞与能够结合细胞的本文所描述的adc接触。
优选,连接子对于细胞外部的条件是化学上稳定的(但不是必要的),并且可以设计成能在细胞内部断裂、裂解和/或另外特异性地降解。或者,可以使用没有设计成能够在细胞内部特异性裂解或降解的连接子。在adc背景下用于连接药物与抗体的多种连接子在本领域是已知的。这些连接子中的任何连接子以及其它连接子,可以用于连接本文描述的adc中的bcl-xl抑制剂与抗体。
例如,美国专利8,399,512、美国公开申请2010/0152725、美国专利8,524,214、美国专利8,349,308、美国公开申请2013/189218、美国公开申请2014/017265、wo2014/093379、wo2014/093394、wo2014/093640描述了可以将许多bcl-xl抑制剂连接到抗体上的示范性的多价连接子,本文以引证的方式引入它们的全部内容。例如,mersana等人开发的fleximer®连接子技术能够使高dar的adc具有良好的物理化学性能。如下所示,fleximer®连接子技术是基于通过酯键的序列,将药物分子引入到溶解的聚缩醛骨架上。该方法提供高载荷的adc(dar达到20),同时保持良好的物理化学性能。对于bcl-xl抑制剂,可以使用这种方法,如下面反应路线所示。
为了使用上面反应路线描述的fleximer®连接子技术,可以存在脂族醇,或将其引入到bcl-xl抑制剂中。然后,醇部分与丙氨酸部分缀合,然后以合成方式结合到fleximer®连接子上。adc的体外脂质体处理释放含有母体醇的药物。
枝状类型的连接子的其它实例可见于下列文献:us2006/116422;us2005/271615;degroot等人,(2003)angew.chem.int.ed.42:4490-4494;amir等人(2003)angew.chem.int.ed.42:4494-4499;shamis等人(2004)j.am.chem.soc.126:1726-1731;sun等人(2002)bioorganic&medicinalchemistryletters12:2213-2215;sun等人(2003)bioorganic&medicinalchemistry11:1761-1768;king等人(2002)tetrahedronletters43:1987-1990。
下列文献描述了可以使用的示范性的单价连接子:例如,nolting,2013,antibody-drugconjugates,methodsinmolecularbiology1045:71-100;kitson等人,2013,cros/cmos-chemicaoggi-chemistrytoday31(4):30-36;ducry等人,2010,bioconjugatechem.21:5-13;zhao等人,2011,j.med.chem.54:3606-3623;美国专利us7,223,837;美国专利us8,568,728;美国专利us8,535,678;和wo2004010957,本文以引证的方式结合它们各自的全部内容。
例如(但没有限制性),可以包括在本文所描述的adc中的一些可断裂的和不可断裂的连接子如下所述。
5.2.2.1可断裂的连接子
在某些实施方案中,所选择的连接子是体外和体内可断裂的连接子。可断裂的连接子可以包括化学或酶催不稳定或可降解的连接子。可断裂的连接子通常依赖于细胞内部释放药物的过程,例如,在细胞质中还原、在溶酶体中接触酸性条件或通过细胞内的特异性蛋白酶或其它酶来断裂。可断裂的连接子通常包括一个或多个化学或酶催可断裂的化学键,而连接子的其余部分是不可断裂的。
在某些实施方案中,连接子包含化学上不稳定的基团,例如,腙和/或二硫基。包含化学上不稳定基团的连接子利用血浆和一些胞质腔室之间的性能差异。对于含有腙的连接子,促进药物释放的胞内条件是核内体和溶酶体的酸性环境,而含有二硫化物的连接子在含有高浓度的硫醇(例如,谷胱甘肽)的细胞溶质中还原。在某些实施方案中,可以在化学上不稳定的基团附近使用取代基,引入空间位阻,使得包含化学上不稳定基团的连接子的血浆稳定性提高。
对酸不稳定的基团,例如,腙,在血液的中性ph环境(ph7.3-7.5)下的系统循环期间保持完整,并且一旦adc内化到细胞的适度酸性的核内体(ph5.0-6.5)和溶酶体(ph4.5-5.0)的腔室中,将进行水解反应并释放药物。。这种ph依赖性释放机制与药物的非特异性释放有关。为了提高连接子的腙基团的稳定性,可以通过化学改性来改变连接子,例如,取代,使得调整为在溶酶体中更有效地释放,同时使循环过程中的损失最小化。
含有腙的连接子可以包含其它断裂位点,例如,其它对酸不稳定的断裂位点和/或酶催不稳定的断裂位点。包括含有示范性的腙的连接子的adc包括下列结构∶
其中,d和ab分别代表药物和ab,n代表与抗体连接的药物-连接子的数量。在某些连接子中,例如,连接子(id),该连接子包含两个可断裂的基团:二硫化物和腙部分。对于这种连接子,未修饰的游离药物的有效释放需要酸性ph,或二硫化物还原和酸性ph。已经表明,具有单个腙断裂位点的连接子例如(ie)和(if)是有效的连接子。
可以包括在连接子中的其它对酸不稳定的基团包括含有顺式乌头基(aconityl)的连接子。顺式乌头基(aconityl)化学使用与酰胺键并列的羧酸,在酸性条件下促进酰胺水解。
可断裂的连接子还可以包括二硫基。二硫化物在生理ph下保持热力学稳定,并且可以被设计成在细胞内部内化时能够释放药物,与胞外环境相比,在细胞内部,细胞溶质提供显著更好的还原条件。二硫键的断裂通常需要存在胞质的硫醇辅助因子,例如(还原的)谷胱甘肽(gsh),使得含有二硫化物的连接子在循环中保持合理的稳定性,在细胞溶质中选择性地释放药物。胞内酶蛋白二硫键异构酶,或能够断裂二硫键的类似的酶,还可以有助于在细胞内部优先断裂二硫键。与gsh或半胱氨酸(最丰富的低分子量硫醇)在循环中的显著更低的浓度(大约5µm)相比,据报道gsh在细胞中的浓度在0.5-10mm范围。无规律的血流导致含氧量低的状态的肿瘤细胞,导致还原酶的活性提高,并因此导致甚至更高的谷胱甘肽浓度。在某些实施方案中,含有二硫化物的连接子的体内稳定性,可以通过连接子的化学改性得到提高,例如,使用与二硫键相邻的空间位阻。
包括示范性的含有二硫化物的连接子的adc包括下列结构∶
其中,d和ab分别代表药物和抗体,n代表与抗体连接的药物-连接子的数量,r在每次出现时独立地选自氢或烃基。在某些实施方案中,增大与二硫键相邻的空间位阻能够提高连接子的稳定性。结构(例如(ig)和(ii))表明,当一个或多个r基团选自低级烃基(例如,甲基)时,体内稳定性提高。
可以使用的另一个类型的连接子是通过酶特异性断裂的连接子。在一个实施方案中,连接子是溶酶体酶可断裂的连接子。这种连接子通常是基于肽的连接子,或包括起到酶的底物作用的肽域。与化学上不稳定的连接子相比,基于肽的连接子在血浆和胞外环境(milleu)中倾向于更稳定。肽键通常具有良好的血清稳定性,与溶酶体相比,溶酶体的蛋白水解酶在血液中的活性极低,这是由于内源性的抑制剂和不利的血液高ph值。由于溶酶体的蛋白酶的作用,例如,组织蛋白酶和纤溶酶,药物从抗体上特异性地释放。在某些肿瘤组织中,这些蛋白酶的存在水平可以升高。在一个实施方案中,连接子可通过溶酶体酶断裂,溶酶体酶是组织蛋白酶b。在某些实施方案中,可通过溶酶体酶断裂连接子,溶酶体酶是β-葡糖苷酸酶或β-半乳糖苷酶。在某些实施方案中,可通过溶酶体酶断裂连接子,溶酶体酶是β-葡糖苷酸酶。在某些实施方案中,可通过溶酶体酶断裂连接子,溶酶体酶是β-半乳糖苷酶。
在示范性的实施方案中,可断裂的肽选自四肽,例如,gly-phe-leu-gly、ala-leu-ala-leu,或二肽,例如,val-cit、val-ala和phe-lys。在某些实施方案中,由于更长的肽的疏水性,所以,二肽优于更长的多肽。
已经描述了用于连接药物(例如,多柔比星、丝裂霉素、喜树碱、太利苏霉素和阿里他汀(auristatin)/阿里他汀(auristatin)家族成员)与抗体的基于各种二肽的可断裂的连接子(参见,dubowchik等人,1998,j.org.chem.67:1866-1872;dubowchik等人,1998,bioorg.med.chem.lett.8:3341-3346;walker等人,2002,bioorg.med.chem.lett.12:217-219;walker等人,2004,bioorg.med.chem.lett.14:4323-4327;以及francisco等人,2003,blood102:1458-1465,本文以引证的方式结合它们各自的内容)。所有这些二肽连接子,或这些二肽连接子的改进型,可以在本文描述的adc使用。可以使用的其它二肽连接子包括在adc中发现的那些二肽连接子,例如,seattlegenetics的布妥昔单抗(brentuximab)、vendotinsgn-35(adcetris™)、seattlegeneticssgn-75(抗cd-70,mc-一甲基阿里他汀(auristatin)f(mmaf)、celldextherapeuticsglembatumumab(cdx-011)(抗-nmb,val-cit-一甲基阿里他汀(auristatin)e(mmae)和cytogenpsma-adc(psma-adc-1301)(抗psma,val-cit-mmae)。
酶催可断裂的连接子可以包括自降解的间隔基(self-immolativespacer),空间上将药物与酶断裂位点分开。药物与肽连接子的直接连接,可以导致药物的氨基酸加合物的蛋白质水解释放,由此减弱它的活性。使用自降解间隔基,可以允许在酰胺键水解时完全活性的、化学上未修饰的药物的消除。
一种自降解间隔基是双功能的对氨基苄醇基团,它通过氨基与肽连接,形成酰胺键,而含有胺的药物可以通过氨基甲酸酯官能团与连接子的苄基羟基连接(得到对酰胺基苄基氨基甲酸酯,pabc)。一旦出现蛋白酶介导的断裂,所得到的前体药物被活化,引起1,6-消除反应,释放未修饰的药物、二氧化碳和残余的连接子基团。下列反应路线描述了对酰胺基氨基甲酸苄酯的断裂和药物的释放∶
其中,x-d代表未修饰的药物。
还描述了这种自降解基团的杂环变体。参见美国专利us7,989,434。
在某些实施方案中,酶催断裂的连接子是基于ß-葡糖醛酸的连接子。溶酶体酶ß-葡糖苷酸酶将ß-葡糖苷酸糖苷键断裂,可以容易地释放药物。这种酶大量地存在于溶酶体内,并且在一些肿瘤类型中过度表达,同时,细胞外部的酶活性很低。由于ß-葡糖苷酸的亲水性,所以,基于ß-葡糖醛酸的连接子可以用于防止adc聚集的倾向。在某些实施方案中,优选,基于ß-葡糖醛酸的连接子作为与疏水性药物连接的adc的连接子。下列反应路线描述了从含有基于ß-葡糖醛酸的连接子的adc中释放药物∶
已经描述了用于连接药物(例如,阿里他汀(auristatin)、喜树碱(camptothecin)和多柔比星类似物、cbi小沟结合剂(minor-groovebinders)和psymberin)与抗体的各种可断裂的基于ß-葡糖醛酸的连接子(参见,jeffrey等人,2006,bioconjug.chem.17:831-840;jeffrey等人,bioorg.med.chem.lett.17:2278-2280;以及jiang等人,2005,j.am.chem.soc.127:11254-11255,本文以引证的方式结合它们各自的内容)。所有基于这些ß-葡糖醛酸的连接基可以在本文所描述adc中使用。在某些实施方案中,酶催可断裂的连接基是基于ß-半乳糖苷的连接基。ß-半乳糖苷大量地存在于溶酶体内,同时,细胞外部的酶活性很低。另外,含有酚基团的bcl-xl抑制剂可以通过酚上的氧与连接子共价键合。美国公开的app.no.2009/0318668所描述的一种这样的连接子,依赖于使用与基于常规“pabo”的自降解基团结合的二氨基-乙烷“spacelink”来递送酚的方法。下面使用本公开的bcl-xl抑制剂,图解描述连接子的断裂。
可断裂的连接子可以包括不可断裂的部分或片段,和/或,在另外的不可断裂的连接子中可以包括可断裂的片段或部分,以使它可断裂。仅用于举例,聚乙二醇(peg)和相关的聚合物,在聚合物骨架中可以包括可断裂的基团。例如,聚乙二醇或聚合物连接子可以包括一个或多个可断裂的基团,例如二硫化物、腙或二肽。
可以包括在连接子中的其它可降解的连接基,包括由peg羧酸或活化的peg羧酸与生物活性剂上的醇基反应所形成的酯键,其中这种酯基通常在生理条件下水解,释放出生物活性剂。水解可降解的连接基包括但不局限于:碳酸酯连接基;胺和醛反应所产生的亚胺连接基;醇与磷酸基反应所形成的磷酸酯连接基;作为醛和醇的反应产物的缩醛连接基;作为甲酸酯和醇的反应产物的原酸酯连接基;以及由亚磷酰胺基团(包括但不局限于:在聚合物的端部)和寡核苷酸的5'羟基所形成的寡核苷酸连接基。
在某些实施方案中,连接子包含酶催可断裂的肽部分,例如,包含结构式(iva)、(ivb)或(ivc)的连接子∶
或其盐,其中∶
肽代表可通过溶酶体酶断裂的肽(举例说明的n→c,其中,肽包括氨基和羧基“端”);
t代表含有一个或多个乙二醇单元或亚烃基链或其组合的聚合物;
ra选自氢、烃基、磺酸酯和磺酸甲酯;
p是0至5的整数;
q是0或1;
x是0或1;
y是0或1;
*代表与连接子的其余部分的连接点。
在某些实施方案中,连接子包含酶催可断裂的肽部分,例如,包含结构式(iva)、(ivb)或(ivc)或其盐的连接子。
在某些实施方案中,肽选自三肽或二肽。在具体实施方案中,二肽选自∶
或其盐。
可以包括在本文描述的adc中的按照结构式(iva)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如所描述的,连接子包括适合于将连接子与抗体共价连接的基团)∶
可以包括在本文描述的adc中的按照结构式(ivb)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团)∶
在某些实施方案中,连接子包含酶催可断裂的糖部分,例如,包含结构式(va)、(vb)、(vc)或(vd)或其盐的连接子∶
其中∶
q是0或1;
r是0或1;
x1是o或nh;
*代表与连接子的其余部分的连接点。
可以包括在本文描述的adc中的按照结构式(va)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团)∶
可以包括在本文描述的adc中的按照结构式(vb)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团)∶
可以包括在本文描述的adc中的按照结构式(vc)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团)∶
可以包括在本文描述的adc中的按照结构式(vd)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团)∶
5.2.2.2不可断裂的连接子
虽然可断裂的连接子可以提供某些优势,但含有本文所描述的adc的连接子没有必要是可断裂的连接子。对于不可断裂的连接子,药物释放不会依赖于血浆和一些胞质腔室之间的性能差异。adc内化之后,通过抗原介导的胞吞作用,假定出现药物的释放,并且递送到溶酶体的腔室中,在溶酶体的腔室中,通过胞内蛋白质降解,抗体降解至氨基酸的水平。这个过程释放出由药物、连接子以及连接子共价连接的氨基酸残基所形成的药物衍生物。与带有可断裂的连接子的缀合物相比,源自于带有不可断裂的连接子的缀合物的氨基酸药物代谢物更具亲水性,并且通常膜渗透性更小,这导致旁观者效应更小,非特异性的毒性更小。通常,与带有可断裂的连接子的adc相比,带有不可断裂的连接子的adc在循环中的稳定性更大。不可断裂的连接子可以是亚烃基链,或性质上可以是聚合物,例如,基于聚烷撑二醇的聚合物、酰胺聚合物,或可以包括亚烃基链、聚烷撑二醇和/或酰胺聚合物的片段。在某些实施方案中,连接子包含具有1至6个乙二醇单元的聚乙二醇片段。
已经描述了用于连接药物与抗体的各种不可断裂的连接子(参见,jeffrey等人,2006,bioconjug.chem.17;831-840;jeffrey等人,2007,bioorg.med.chem.lett.17:2278-2280;以及jiang等人,2005,j.am.chem.soc.127:11254-11255,本文以引证的方式结合它们的内容)。所有这些连接子可以包括在本文描述的adc中。
在某些实施方案中,连接子是体内不可断裂的连接子,例如,举例说明的按照结构式(via)、(vib)、(vic)或(vid)的连接子,所述连接子包括适合于将连接子与抗体共价连接的基团∶
或其盐,其中∶
ra选自氢、烃基、磺酸酯和磺酸甲酯;
rx是包括能够将连接子与抗体共价连接的官能团的部分;
可以包括在本文描述的adc中的按照结构式(via)-(vid)的连接子的具体示范性实施方案包括下面举例说明的连接子(正如示例所述,连接子包括适合于将连接子与抗体共价连接的基团,和“
5.2.2.3用于连接连接子与抗体的基团
连接基团性质上可以是亲电子的基团,包括∶马来酰亚胺基团、活化的二硫化物、活性酯,例如,nhs酯和hobt酯、卤代甲酸酯,酰卤、烃基和苄基卤,例如,卤代乙酰胺。正如下面讨论的那样,还有与可以按照本公开使用的“自稳定的”马来酰亚胺和“桥接二硫化物”相关的新兴技术。
已经观察到药物-连接子从adc中的缺失,其是用白蛋白、半胱氨酸或谷胱甘肽的马来酰亚胺调换过程的结果(alley等人,2008,bioconjugatechem.19:759-769)。这种情况在缀合物的高度溶剂可及的位点尤其普遍,而部分可及的且具有荷正电环境的位点促进了马来酰亚胺环的水解(junutula等人,2008,nat.biotechnol.26:925-932)。公认的解决方案是使缀合所形成的琥珀酰亚胺水解,这可以抵抗从抗体的脱缀合,由此使得adc在血清中稳定。据以前报道,琥珀酰亚胺环在碱性条件下会进行水解(kalia等人,2007,bioorg.med.chem.lett.17:6286-6289)。下面的示意图描述了在抗体缀合条件下自发水解得到稳定性提高的adc的“自稳定的”马来酰亚胺基团的一个实例。参见美国公开申请2013/0309256。
polytherics已经公开了桥接一对由天然铰链二硫键的还原所获得的巯基的方法。参见badescu等人,2014,bioconjugatechem.25:1124-1136。下面的示意图描述了该反应。这种方法的优越性是,通过iggs的完全还原(得到4对巯基),而后与4当量的烷基化剂反应,能够合成均匀的dar4adc。包含“桥接二硫化物”的adc也使稳定性提高。
类似地,正如下面所描述的那样,已经研发了能够桥接一对巯基的马来酰亚胺衍生物。参见美国公开申请2013/0224228。
5.2.2.4连接子选择因素
正如技术人员所了解的那样,为具体adc所选择的连接子可以受到各种因素的影响,包括但不局限于:与抗体连接的位点(例如,lys、cys或其它氨基酸残基)、药物药效团的结构限制以及药物的亲油性。对于特异性抗体/药物组合,为adc所选择的具体连接子应该设法平衡这些不同因素。对于受到adc中的连接子的选择影响的因素的综述,参见nolting,chapter5“linkertechnologyinantibody-drugconjugates,”in:antibody-drugconjugates:methodsinmolecularbiology,vol.1045,pp.71-100,laurentducry(ed.),springerscience&businessmedica,llc,2013。
例如,已经观察到,adc实施杀伤抗原阳性肿瘤细胞附近的旁观者抗原阴性细胞。adc的旁观者细胞杀伤机理表明,在adc的胞内处理期间形成的代谢产物可以起到作用。adc在抗原阳性细胞中代谢产生的中性细胞毒素代谢物,似乎在旁观者细胞杀伤过程中起一定作用,而带电荷的代谢物则可能被阻止扩散穿过膜进入到培养基中,并由此不能影响旁观者杀伤。在某些实施方案中,为了减弱adc的细胞代谢物所引起的旁观者杀伤效应,对连接子进行选择。在某些实施方案中,为了提高旁观者杀伤效应,对连接子进行选择。
连接子的性能也可以影响adc在使用和/或储存条件下的聚集。通常,据文献报道,每个抗体分子,adc包含至多3-4个药物分子(参见,例如,chari,2008,accchemres41:98-107)。获得更高的药物对抗体比例(“dar”)的企图经常失败,尤其是在药物和连接子都是疏水性的情况下,原因是adc出现聚集(参见,king等人,2002,jmedchem45:4336-4343;hollander等人,2008,bioconjugatechem19:358-361;burke等人,2009bioconjugatechem20:1242-1250)。在许多情况下,作为提高效能的方法,dar高于3-4是有利的。在bcl-xl抑制剂性质上是疏水性的情况下,作为降低adc聚集的方法,合乎需要的是,选择相对亲水性的连接子,特别是在需要dar大于3-4的情况下。由此,在某些实施方案中,连接子引入降低adc在储存和/或使用期间聚集的化学部分。为了降低adc的聚集,连接子可以引入极性或亲水基,例如带电荷的基团、或在生理ph值下变成带电荷的基团。例如,连接子可以引入带电荷的基团,例如盐或去质子化的基团,例如羧酸盐,或在生理ph值下质子化,例如胺。
据报道,能够达到高达20的dar的示范性的多价连接子,可用于将许多bcl-xl抑制剂与抗体连接,如下列文献所述:美国专利us8,399,512;美国公开申请2010/0152725;美国专利us8,524,214;美国专利us8,349,308;美国公开申请2013/189218;美国公开申请2014/017265;wo2014/093379;wo2014/093394;wo2014/093640,本文以引证的方式结合它们的全部内容。
在具体实施方案中,利用分子排阻色谱(sec)测定,adc在储存或使用期间的聚集小于大约40%。在具体实施方案中,利用分子排阻色谱(sec)测定,adc在储存或使用期间的聚集小于35%,例如小于大约30%,例如小于大约25%,例如小于大约20%,例如小于大约15%,例如小于大约10%,例如小于大约5%,例如小于大约4%,或甚至更小。
一个实施方案涉及adc或合成子,其中,连接子l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其盐。
5.3抗体
adc的抗体可以是,通常但不必然是是特异性,与感兴趣的靶细胞的表面上表达的抗原结合的任何抗体。抗原不必将其中结合的adc内化到细胞中,但在一些实施方案中,其能够将其中结合的adc内化到细胞中。感兴趣的靶细胞通常包括:需要抑制抗凋亡bcl-xl蛋白来诱导细胞程序死亡的细胞,包括,例如(没有限制性),表达或过度表达bcl-xl的肿瘤细胞。靶点抗原可以是感兴趣的靶细胞上表达的任何蛋白、糖蛋白、多糖、脂蛋白,等等,但通常是在靶细胞上独特表达并且在正常或健康细胞上没有表达的蛋白、或与正常或健康细胞相比较,在靶细胞上过度表达的蛋白,以便adc选择性地靶向感兴趣的具体细胞,例如,肿瘤细胞。正如技术人员所理解的那样,具体抗原以及由此选择的抗体取决于感兴趣的目标靶细胞的特征。在具体实施方案中,adc的抗体是适合于给予人类的抗体。
抗体(abs)和免疫球蛋白(igs)是具有相同结构特性的糖蛋白。尽管抗体对于具体靶点具有结合特异性,但免疫球蛋白包括抗体及没有靶向特异性的其它抗体类分子。天然的抗体和免疫球蛋白通常是大约150,000道尔顿的异四聚体糖蛋白,由两个相同的轻(l)链和两个相同的重(h)链组成。每个重链在一端具有一个可变域(vh),后面有许多恒定域。每个轻链在一端具有一个可变域(vl),在它的另一端具有一个恒定域。
关于“vh”指的是抗体的免疫球蛋白重链的可变域,包括fv、scfv或fab的重链。关于“vl”指的是免疫球蛋白轻链的可变域,包括fv、scfv、dsfv或fab的轻链。
本文使用的术语“抗体”具有最广义的含义,是指与具体抗原特异性结合或与具体抗原免疫反应的免疫球蛋白分子,并且包括多克隆抗体、单克隆抗体、基因工程抗体以及另外的抗体的修改型,包括但不局限于:鼠抗体、嵌合抗体、人源化的抗体、杂缀合抗体(例如,双特异性抗体、双抗体、三抗体和四抗体)和抗体的抗原结合片段,包括,例如,fab'、f(ab')2、fab、fv、rigg和scfv片段。术语“scfv”是指单链fv抗体,其中,源自于常规抗体的重链和轻链的可变域结合形成一个链。
抗体可以是鼠、人、人源化、嵌合抗体或衍生自其它物种的抗体。抗体是能够辨别具体抗原并与其结合的免疫系统所产生的蛋白(janeway,c.,travers,p.,walport,m.,shlomchik(2001)immunobiology,5thed.,garlandpublishing,newyork)。靶点抗原通常具有许多结合位点,也称为表位,由多个抗体上的cdr所识别。与不同表位特异性结合的每个抗体具有不同的结构。由此,一个抗原可以具有一个以上的对应抗体。抗体包括全长免疫球蛋白分子或全长免疫球蛋白分子的免疫活性部分,即,包含抗原结合位点的分子,所述抗原结合位点能够免疫特异性结合感兴趣的靶点或其一部分,这种靶点包括但不局限于:癌细胞或产生与自身免疫疾病有关的自身免疫抗体的细胞。本文公开的免疫球蛋白可以是免疫球蛋白分子的任何类型(例如,igg、ige、igm、igd和iga)、种类(例如,igg1、igg2、igg3、igg4、iga1和iga2)或亚类。免疫球蛋白可以源自于任何物种。然而,在一方面,免疫球蛋白是人、鼠或兔子源的免疫球蛋白。
术语“抗体片段”是指全长抗体的一部分,通常是靶点结合或可变域。抗体片段的实例包括fab、fab'、f(ab')2和fv片段。“fv”片段是最小的抗体片段,它包含全部靶点识别和结合位点。这个域由一个重链可变域和一个轻链可变域的坚固的、非共价相关的二聚体(vh-vl二聚体)组成。在这个构型中,每个可变域的三个cdr相互作用,限定vh-vl二聚体表面上的靶点结合位点。通常,六个cdr赋予抗体靶点结合特异性。然而,在有些情况下,即使单个可变域(或只包含三个对于靶点特异性的cdr的fv的一半)也可以具有识别和结合靶点的能力。“单链fv”或“scfv”抗体片段在单个多肽链中包含抗体的vh和vl域。通常,fv多肽在vh和vl域之间进一步包含多肽连接子,能够使scfv形成靶向结合的目标结构。“单域抗体”由单一vh或vl域组成,对于靶点表现出足够的亲合性。在一个具体实施方案中,单域抗体是camelized化的抗体(参见,例如,riechmann,1999,journalofimmunologicalmethods231:25-38)。
fab片段包含轻链的恒定域以及重链的第一个恒定域(ch1)。fab'片段由于在重链chl域的羧基端添加几个残基(包括一个或多个抗体绞链区的半胱氨酸),它与fab片段不同。f(ab')片段是由在f(ab')2胃蛋白酶消化产物的铰链半胱氨酸处的二硫键断裂所产生的。本领域普通技术人员了解抗体片段的其它化学缀合。
轻链和重链可变域具有互补性决定区(cdr),亦称高变区。可变域的更加高度保守的部分被称作骨架(fr)。正如本领域已知的那样,根据上下文和本领域已知的各种定义,描绘抗体的高变区的氨基酸位置/边界可以改变。可以把可变域内的一些位置看作混杂型高变位置,原因在于,根据一套标准,可以认为这些位置在高变区内,而根据一套不同的标准,可以认为这些位置在高变区的外面。还可以在延长的高变区中发现这些位置中的一个或多个。每个链中的cdr通过fr区近似紧密地结合在一起,与其它链的cdr一起,有助于形成抗体的靶点结合位点(参见,kabat等人,sequencesofproteinsofimmunologicalinterest(nationalinstituteofhealth,bethesda,md.1987)。本文使用的免疫球蛋白氨基酸残基编号,是按照kabat等人的免疫球蛋白氨基酸残基编号系统进行编号的,除非另外说明。
在某些实施方案中,本公开的adc的抗体是单克隆抗体。术语“单克隆抗体”(mab)是指衍生自单一复制品或克隆体的抗体,包括,例如,任何真核、原核或噬菌体克隆体,并且不是指制备它的方法。优选,本公开的单克隆抗体以均匀或基本上均匀的形式存在。单克隆抗体包括能够与蛋白特异性结合的完整分子以及抗体片段(例如,fab和f(ab')2片段)。与完整抗体相比,fab和f(ab')2片段缺少完整抗体的fc片段,能够更加快速地从动物的循环中清除,并且非特异性的组织结合更少(wahl等人,1983,j.nucl.med.24:316)。可以使用本领域已知的多种技术,制备本公开使用的单克隆抗体,包括使用杂交瘤、重组和噬菌体展示技术,或其组合。本公开的抗体包括嵌合、灵长类化(primatized)、人源化或人类抗体。
尽管在大多数情况下,抗体只由遗传编码的氨基酸组成,但在一些实施方案中,为了控制与抗体连接的bcl-xl抑制剂的数目以及它们的位置,可以在特定位置引入非编码的氨基酸。在下列文献中,讨论了可以引入到抗体中的、用于控制化学计量和连接位置的非编码的氨基酸的实例,以及制备这种修饰的抗体的方法:tian等人,2014,procnat’lacadsciusa111(5):1766-1771,以及axup等人,2012,procnat’lacadsciusa109(40):16101-16106,,本文以引证的方式结合它们的全部内容。在某些实施方案中,非编码的氨基酸将每个抗体的bcl-xl抑制剂的数目限定至大约1-8个或大约2-4个。
在某些实施方案中,本文所描述的adc的抗体是嵌合抗体。本文使用的术语“嵌合”抗体是指具有可变序列的抗体,其衍生自非人免疫球蛋白,例如大鼠或小鼠抗体,以及人免疫球蛋白恒定域,通常选自人免疫球蛋白模板。制备嵌合抗体的方法在本领域是已知的。参见,例如,morrison,1985,science229(4719):1202-7;oi等人,1986,biotechniques4:214-221;gillies等人,1985,j.immunol.methods125:191-202;美国专利us5,807,715;4,816,567;以及4,816397,本文以引证的方式结合它们的全部内容。
在某些实施方案中,本文所描述的adc的抗体是人源化的抗体。非人(例如,鼠)抗体的“人源化”形式是嵌合的免疫球蛋白、免疫球蛋白链或其片段(例如,fv、fab、fab'、f(ab')2或抗体的其它靶点结合子域),其包含衍生自非人免疫球蛋白的最小序列。通常,人源化的抗体包含基本上所有可变域、至少一个可变域,和典型地两个可变域,其中,所有的或基本上所有的cdr区与非人免疫球蛋白的cdr区对应,所有或基本上所有的fr区是人免疫球蛋白序列的fr区。人源化的抗体还可以包含免疫球蛋白恒定区(fc)的至少一部分,典型地是人免疫球蛋白共有序列的恒定区的至少一部分。抗体人源化的方法在本领域是已知的方法。参见,例如,riechmann等人,1988,nature332:323-7;美国专利us5,530,101;5,585,089;5,693,761;5,693,762;和6,180,370(queen等人);ep239400;pct公开出版物wo91/09967;美国专利us5,225,539;ep592106;ep519596;padlan,1991,mol.immunol.,28:489-498;studnicka等人,1994,prot.eng.7:805-814;roguska等人,1994,proc.natl.acad.sci.91:969-973;以及美国专利us5,565,332,本文以引证的方式结合它们的全部内容。
在某些实施方案中,本文所描述的adc的抗体是人类抗体。对于人类患者的治疗处理来说,需要完全“人类”抗体。本文使用的“人类抗体”包括具有人免疫球蛋白的氨基酸序列的抗体,包括从人免疫球蛋白库、或从对于一或多种人免疫球蛋白是转基因的并且不表达内源性免疫球蛋白的动物中分离出来的抗体。可以利用本领域已知的各种方法制备人类抗体,包括使用来源于人免疫球蛋白序列的抗体库的噬菌体展示方法。参见美国专利us4,444,887、4,716,111、6,114,598、6,207,418、6,235,883、7,227,002、8,809,151和美国公开申请2013/189218,本文以引证的方式结合它们的全部内容。还可以使用不能表达功能性的内源性免疫球蛋白但可以表达人免疫球蛋白基因的转基因小鼠制备人类抗体。参见,例如,美国专利us5,413,923;5,625,126;5,633,425;5,569,825;5,661,016;5,545,806;5,814,318;5,885,793;5,916,771;5,939,598;7,723,270;8,809,051,以及美国公开申请2013/117871,本文以引证的方式结合它们的全部内容。另外,使用与上述技术相似的技术,可以使公司(例如,medarex(princeton,nj)、astellaspharma(deerfield,il)和regeneron(tarrytown,ny))提供直接针对所选择的抗原的人类抗体。识别所选择的表位的完全人类抗体,可以使用被称为“导向选择”的技术来产生。在这种方法中,所选择的非人类单克隆抗体,例如,小鼠抗体,用于引导完全人类抗体的选择,所述完全人类抗体识别相同的表位(jespers等人,1988,biotechnology12:899-903)。
在某些实施方案中,本文所描述的adc的抗体是灵长类化(primatized)的抗体。术语“灵长类化(primatized)的抗体”是指包含猴子可变区和人类恒定区的抗体。制备灵长类化(primatized)的抗体的方法在本领域是已知的方法。参见,例如,美国专利us5,658,570、5,681,722和5,693,780,本文以引证的方式结合它们的全部内容。
在某些实施方案中,本文所描述的adc的抗体是双特异性抗体或双可变域抗体(dvd)。双特异性和dvd抗体是单克隆抗体,通常是人类或人源化的抗体,对于至少两种不同的抗原具有结合特异性。例如,美国专利us7,612,181描述了dvd,本文以引证的方式结合其公开内容。
在某些实施方案中,本文所描述的adc的抗体是衍生的抗体。例如(但没有限制性),衍生的抗体通常是利用糖基化、乙酰化、peg化、磷酸化、酰胺化、通过已知的保护/封端基团衍生化、蛋白酶裂解、连接到细胞配体或其它蛋白上等等来进行修饰的。可以利用已知的技术,进行许多化学修饰的任何修饰,包括但不限于:特异性化学裂解、乙酰化、甲酰化、衣霉素的代谢合成,等等。另外,衍生物可以包含一个或多个非天然的氨基酸,例如,使用ambrx技术(参见,例如,wolfson,2006,chem.biol.13(10):1011-2)。
在某些实施方案中,本文描述的adc的抗体具有已被修饰的序列,相对于相应的野生型序列,这种修饰改变至少一个恒定区介导的生物效应子功能。例如,在一些实施方案中,可以修饰抗体,以相对于未修饰的抗体,减少至少一个恒定区介导的生物效应子功能,例如减少与fc受体(fcr)的结合。使抗体的免疫球蛋白恒定区片段在fcr相互作用所必需的特定区域发生突变,可以减少fcr结合(参见,例如,canfϊeldandmorrison,1991,j.exp.med.173:1483-1491;以及lund等人,1991,j.immunol.147:2657-2662)。
在某些实施方案中,修饰本文描述的adc的抗体,相对于未修饰的抗体,获得或增加至少一个恒定区介导的生物效应子功能,例如增强fcγr相互作用(参见,例如,us2006/0134709)。例如,具有结合了fcγriia、fcγriib和/或fcγriiia的恒定区(比相应的野生型恒定区具有更大的亲合性)的抗体,可以按照本文描述的方法制备。
在某些具体实施方案中,本文描述的adc的抗体是结合了肿瘤细胞的抗体,例如针对细胞表面受体或肿瘤相关的抗原(taa)的抗体。为了发现诊断和治疗癌症的有效的细胞靶点,研究人员设法确定横跨膜,或在一或多种具体类型的癌细胞的表面上特异性表达(与一或多种正常的非癌性细胞相比较)的横跨膜或其它肿瘤相关的多肽。通常,与非癌性细胞的表面相比较,这种肿瘤相关的多肽更加充分地在癌细胞的表面上表达。这种细胞表面受体和肿瘤相关的抗原在本领域是已知的,并且可以使用本领域众所周知的方法和信息制备它们,用于产生抗体。
5.3.1示范性的细胞表面受体和taa
本文描述的adc的抗体可以靶向的细胞表面受体和taa的实例包括但不局限于下列各种受体和taa。为方便起见,下面列出了与本领域已知的这些抗原有关的信息,包括名称、别称(alternativenames)、genbank登录号和原始参考文献,按照nationalcenterforbiotechnologyinformation(ncbi)的核酸和蛋白序列鉴定规则。列出的细胞表面受体和taa所对应的核酸和蛋白序列可在公共数据库例如genbank中得到。
4-1bb
5ac
5t4
alpha-胎蛋白
血管生成素(angiopoietin)2
aslg659
tcl1
bmpr1b
brevican(bcan,behab)
c242抗原
c5
ca-125
ca-125(仿制品)
ca-ix(碳酸酐酶9)
ccr4
cd140a
cd152
cd19
cd20
cd200
cd21(c3dr)1)
cd22(b细胞受体cd22-b异构型)
cd221
cd23(ge受体)
cd28
cd30(tnfrsf8)
cd33
cd37
cd38(环状adp核糖水解酶)
cd4
cd40
cd44v6
cd51
cd52
cd56
cd70
cd72(lyb-2,b细胞分化抗原cd72)
cd74
cd79a(cd79a,cd79α,免疫球蛋白相关的alpha)genbank登录号(accessionno.):np__001774.10)
cd79b(cd79b,cd79β,b29)
cd80
cea
cea-相关抗原
ch4d5
cldn18.2
cripto(cr,cr1,crgf,tdgf1畸胎癌衍生的生长因子)
ctla-4
cxcr5
dll4
dr5
e16(lat1,slc7a5)egfl7
egfr
epcam
ephb2r(drt,erk,hek5,epht3,tyro5)
episialin
erbb3
etbr(内皮素b型受体)
fcrh1(fc受体类蛋白1)
fcrh2(包含磷酸酶锚定蛋白的ifgp4、irta4、spap1、spap1b、spap1c、sh2域)
纤连蛋白额外域-b
叶酸受体1
突变型受体
gd2
gd3神经节苷脂
geda
gpnmb
her1
her2(erbb2)
her2/neu
her3
hgf
hla-dob
hla-dr
人类扩散因子受体激酶
igf-1受体
igg4
il-13
il20rα(il20ra,zcytor7)
il-6
ilgf2
ilfr1r
整联蛋白α
整联蛋白α5β1
整联蛋白αvβ3
irta2(免疫球蛋白超家族受体迁移相关的2,基因染色体1q21)
lewis-y抗原
ly64(rp105)
mcp-1
mdp(dpep1)
mpf(msln、smr、间皮素(mesothelin)、巨核细胞强化因子)
ms4a1
msg783(rnf124、假定蛋白flj20315)
muc1
粘蛋白(mucin)canag
napi3(napi-3b、nptiib、slc34a2、ii型钠依赖性磷酸转运蛋白3b)
nca(ceacam6)
p2x5(嘌呤能受体p2x配体门控的离子通道5)
pd-1
pdcd1
pdgf-rα
前列腺特异性膜抗原
psca(前列腺干细胞抗原前体物)
pscahlg
rankl
ron
sdc1
sema5b
slamf7(cs-1)
steap1
steap2(hgnc__8639、pcanap1、stamp1、steap2、stmp、前列腺癌相关的基因1)
tag-72
tem1
结合腕蛋白(tenascin)c
tenb2(tmeff2,tomoregulin,tpef,hpp1,tr)
tgf-β
trail-e2
trail-r1
trail-r2
trpm4(br22450、flj20041、trpm4、trpm4b,瞬时型受体电位阳离子通道子族m,成员4)
tactaa16.88
tweak-r
tyrp1(糖蛋白75)
vegf
vegf-a
egfr-1
vegfr-2
波形蛋白。
5.3.2示范性的抗体
本公开的adc所使用的示范性的抗体包括但不局限于:3f8(gd2)、阿巴伏单抗(abagovomab)(ca-125(仿制品))、adecatumumab(epcam)、阿夫土珠(afutuzumab)(cd20)、alacizumabpegol(vegfr2)、ald518(il-6)、阿仑单抗(alemtuzumab)(cd52)、阿替木单抗五钠(altumomabpentetate)(cea)、amatuximab(间皮素(mesothelin))、anatumomabmafenatox(tag-72)、apolizumab(hla-dr)、arcitumomab(cea)、bavituximab(磷脂酰丝氨酸)、bectumomab(cd22)、贝利木单抗(belimumab)(baff)、besilesomab(cea-相关抗原)、贝伐单抗(bevacizumab)(vegf-a)、bivatuzumabmertansine(cd44v6)、blinatumomab(cd19)、brentuximabvedotin((cd30(tnfrsf8))、cantuzumabmertansine(mucincanag)、cantuzumabravtansine(muc1)、capromabpendetide(前列腺癌细胞)、carlumab(mcp-1)、卡曲单抗(catumaxomab)(epcam,cd3)、cc49(tag-72)、cbr96-doxadc(lewis-y抗原)、西妥昔单抗(cetuximab)(egfr)、citatuzumabbogatox(epcam)、cixutumumab(igf-1受体)、氯沙坦四氮四烯(clivatuzumabtetraxetan)(muc1)、conatumumab(trail-e2)、dacetuzumab(cd40)、dalotuzumab(胰岛素样生长因子i受体)、daratumumab((cd38(环状adp核糖水解酶))、demcizumab(dll4)、狄诺塞麦(denosumab)(rankl)、detumomab(b-淋巴瘤细胞)、drozitumab(dr5)、dusigitumab(ilgf2)、ecromeximab(gd3神经节糖苷)、依库珠单抗(eculizumab)(c5)、依决洛单抗(edrecolomab)(epcam)、elotuzumab(slamf7)、elsilimomab(il-6)、enavatuzumab(tweak受体)、enoticumab(dll4)、ensituximab(5ac)、依托米单抗(epitumomabcituxetan)(episialin)、依帕珠单抗(epratuzumab)(cd22)、ertumaxomab((her2/neu,cd3))、etaracizumab(整联蛋白αvβ3)、farletuzumab(叶酸受体1)、fbta05(cd20)、ficlatuzumab(hgf)、figitumumab(igf-1受体)、flanvotumab((tyrp1(糖蛋白75))、fresolimumab(tgf-β)、galiximab(cd80)、ganitumab(igf-i)、吉妥珠单抗ozogamicin(cd33)、girentuximab((碳酸酐酶9(ca-ix))、glembatumumabvedotin(gpnmb)、ibritumomabtiuxetan(cd20)、icrucumab(vegfr-1)、igovomab(ca-125)、imab362(cldn18.2)、imgatuzumab(egfr)、indatuximabravtansine(sdc1)、intetumumab(cd51)、依托珠单抗ozogamicin(cd22)、易普利姆玛(ipilimumab)(cd152)、iratumumab((cd30(tnfrsf8))、labetuzumab(cea)、lambrolizumab(pdcd1)、来沙木单抗(lexatumumab)(trail-r2)、林妥珠单抗(lintuzumab)(cd33)、洛伐单抗米黄丹(lorvotuzumabmertansine)(cd56)、lucatumumab(cd40)、lumiliximab((cd23(ige受体))、马帕木单抗(mapatumumab)(trail-r1)、margetuximab(ch4d5)、马妥珠单抗(matuzumab)(egfr)、milatuzumab(cd74)、mitumomab(gd3神经节苷脂)、莫加木珠单抗(mogamulizumab)(ccr4)、moxetumomabpasudotox(cd22)、nacolomabtafenatox(c242抗原)、naptumomabestafenatox(5t4)、narnatumab(ron)、那他珠单抗(natalizumab)(整联蛋白α4)、necitumumab(egfr)、nesvacumab(血管生成素(angiopoietin)2)、尼妥珠单抗(nimotuzumab)(egfr)、nivolumab(igg4)、ocaratuzumab(cd20)、奥法木单抗(ofatumumab)(cd20)、olaratumab(pdgf-rα)、onartuzumab(人类扩散因子受体激醇)、ontuxizumab(tem1)、oportuzumabmonato(epcam)、oregovomab(ca-125)、otlertuzumab(cd37)、panitumumab(egfr)、pankomab(muc1的肿瘤特异性糖基化)、parsatuzumab(egfl7)、patritumab(her3)、pemtumomab(muc1)、培妥珠单抗(pertuzumab)(her2/neu)、pidilizumab(pd-1)、pinatuzumabvedotin(cd22)、pritumumab(波形蛋白)、racotumomab(n-羟乙酰基神经氨酸)、radretumab(纤连蛋白额外域-b)、ramucirumab(vegfr2)、利妥木单抗(rilotumumab)(hgf)、利妥昔单抗(rituximab)(cd20)、robatumumab(igf-1受体)、samalizumab(cd200)、satumomabpendetide(tag-72)、seribantumab(erbb3)、sibrotuzumab(fap)、sgn-cd19a(cd19)、sgn-cd33a(cd33)、siltuximab(il-6)、solitomab(epcam)、sonepcizumab(鞘氨醇-1-磷酸)、tabalumb(baff)、塔卡单抗四氮芥(tacatuzumabtetraxetan)(alpha-胎蛋白)、塔利珠单抗paptox(taplitumomabpaptox)(cd19)、tenatumomab(结合腕蛋白(tenascin)c)、teprotumumab(cd221)、tgn1412(cd28)、替尼米特单抗(ticilimumab)(ctla-4)、tigatuzumab(trail-r2)、tnx-650(il-13)、tovetumab(cd140a)、曲妥单抗(trastuzumab)(her2/neu)、trbs07(gd2)、tremelimumab(ctla-4)、tucotuzumabcelmoleukin(epcam)、ublituximab(ms4a1)、urelumab(4-1bb)、凡德他尼(vandetanib)(vegf)、vantictumab(突变型受体)、volociximab(整联蛋白α5β1)、vorsetuzumabmafodotin(cd70)、votumumab(肿瘤抗原ctaa16.88)、扎鲁木单抗(zalutumumab)(egfr)、扎木单抗(zanolimumab)(cd4)和zatuximab(her1)。
在某些实施方案中,adc的抗体结合egfr、ncam1或epcam。
5.4制备抗体的方法
可以通过免疫球蛋白轻链和重链基因在宿主细胞中的重组表达,制备adc的抗体。例如,为了重组地表达抗体,用一或多种携带编码抗体的免疫球蛋白轻链和重链的dna片段的重组表达载体转染宿主细胞,使得轻链和重链在宿主细胞中表达,并且任选,分泌到培养宿主细胞的培养基中,可以从所述培养基中回收抗体。标准的重组dna方法用于获得抗体重链和轻链基因,将这些基因结合到重组表达载体中,并且将载体引入到宿主细胞中,例如,下列文献所描述的那些:molecularcloning;alaboratorymanual,secondedition(sambrook,fritschandmaniatis(eds),coldspringharbor,n.y.,1989),currentprotocolsinmolecularbiology(ausubel,f.m.等人,eds.,greenepublishingassociates,1989)以及美国专利us4,816,397。
在一个实施方案中,fc变体抗体与它们的野生型同等物相似,但在它们的fc域方面有变化。为了产生编码这种fc变体抗体的核苷酸,可以合成编码野生型抗体的fc域或一部分fc域(称为“野生型fc域”)的dna片段,并且用作使用常规诱变技术诱变产生本文所描述的抗体的模板;或者,可以直接合成编码所述抗体的dna片段。
一旦获得编码野生型fc域的dna片段,可以利用标准重组dna技术,进一步操作这些dna片段,例如,将恒定区基因转变为全长抗体链基因。在这些操作中,将ch-编码的dna片段操作性地连接到编码另一个蛋白的另一个dna片段上,例如,抗体可变区或挠性连接子。在这方面使用的术语“操作性地连接”是指连接两个dna片段,使得这两个dna片段编码的氨基酸序列保留在框内。
为了表达fc变体抗体,将上述获得的编码部分或全长轻和重链的dna插入到表达载体中,使得基因操作性地连接到转录和转译控制序列上。在这种情况下,术语“操作性地连接”是指抗体基因连接到载体上,使得在载体内的转录和转译控制序列提供它们调节抗体基因的转录和转译的预期功能。选择表达载体和表达控制序列,使它们与所使用的表达宿主细胞相容。可以将变体抗体轻链基因和抗体重链基因插入到单独的载体中,或更典型地是,将两种基因插入到相同的表达载体中。
利用标准方法(例如,连接抗体基因片段和载体上的互补限制位点,或如果不存在限制位点,则进行平端连接),将抗体基因插入表达载体中。在插入变体fc域序列之前,表达载体可以已经携带抗体可变域序列。另外或者可选地,重组表达载体可以编码促进宿主细胞分泌抗体链的信号肽。可以将抗体链基因克隆到载体上,使得信号肽在框内与所述抗体链基因的氨基端连接。信号肽可以是免疫球蛋白信号肽或异源信号肽(即,源自于非免疫球蛋白的信号肽)。
除了抗体链基因之外,重组表达载体还携带控制抗体链基因在宿主细胞中表达的调节序列。术语“调节序列”意指包括启动子、增强子及控制抗体链基因的转录或转译的其它表达控制要素(例如,聚腺苷酸化信号)。例如,这种调节序列描述在下列文献中:goeddel,geneexpressiontechnology:methodsinenzymology185(academicpress,sandiego,ca,1990)。本领域技术人员能够理解,可以根据下列因素来设计表达载体,包括调节序列的选择:例如所转染的宿主细胞的选择、目标蛋白的表达水平,等等。哺乳动物宿主细胞表达的合适的调节序列包括:在哺乳动物细胞中引导蛋白高水平表达的病毒要素,例如启动子和/或增强子,它们来源于巨细胞病毒(cmv)(例如,cmv启动子/增强子)、猿猴病毒40(sv40)(例如,sv40启动子/增强子)、腺病毒(例如,腺病毒主要晚期启动子(admlp))和多瘤病毒。为了进一步说明病毒调控要素和其序列,参见,例如,美国专利us5,168,062(stinski)、美国专利us4,510,245(bell等人)和美国专利us4,968,615(schaffner等人)。
除了抗体链基因和调节序列之外,重组表达载体还可以携带其它序列,例如,调节载体在宿主细胞中复制(例如,复制起点)和选择性标记基因的序列。选择性标记基因便于选择已经引入了载体的宿主细胞(参见,例如,美国专利us4,399,216、4,634,665和5,179,017(所述专利都属于axel等人))。例如,通常,选择性标记基因在已经引入载体的宿主细胞上会带来抗药性,例如,对于g418、嘌呤霉素、杀稻瘟菌素、潮霉素或氨甲喋呤的抗药性。合适的选择性标记基因包括二氢叶酸还原酶(dhfr)基因(用于带有氨甲喋呤选择/扩增的dhfr宿主细胞)和新基因(用于g418选择)。为了表达轻和重链,利用标准技术,将编码重和轻链的表达载体转染到宿主细胞中。术语“转染”的各种形式意在包括多种通常用于将外源性dna引入到原核或真核宿主细胞中的技术,例如电穿孔、脂转染、磷酸钙沉淀法、deae-葡聚糖转染等等。
可能在原核或真核宿主细胞中表达抗体。在某些实施方案中,为了适当折叠的和免疫活性的抗体的最佳分泌,在真核细胞中表达抗体,例如,在哺乳动物宿主细胞中表达。表达重组抗体的示范性的哺乳动物宿主细胞包括中国仓鼠卵巢(cho细胞)(包括dhfr-cho细胞,描述在下列文献中:urlaubandchasin,1980,proc.natl.acad.sci.usa77:4216-4220,与dhfr选择性标记一起使用,例如,如kaufmanandsharp,1982,mol.biol.159:601-621所述)、ns0骨髓瘤细胞、cos细胞、293细胞和sp2/0细胞。当编码抗体基因的重组表达载体被引入到哺乳动物宿主细胞中时,将宿主细胞在足以使抗体在宿主细胞中表达或使抗体分泌到宿主细胞所生长的培养基中的一段时间内培养,由此制备抗体。可以使用标准蛋白纯化方法,从培养基中回收抗体。宿主细胞还可以用于制备完整抗体的一部分,例如,fab片段或scfv分子。
在一些实施方案中,adc的抗体可以是双功能抗体。其中一个重链和一个轻链对于一个抗原具有特异性、另一个重链和轻链对于第二个抗原具有特异性的这种抗体,可以利用标准化学交联方法,通过一个抗体与第二个抗体交联来制备。双功能抗体还可以通过表达设计为编码双功能抗体的核酸来制备。
在某些实施方案中,双特异性抗体,即,使用相同结合位点,结合一个抗原与第二个无关的抗原的抗体,可以通过使轻链和/或重链cdr中的氨基酸残基突变来制备。示范性的第二个抗原包括促炎细胞因子(例如,淋巴细胞毒素、干扰素-γ或白介素-1)。例如,通过使抗原结合位点周围的氨基酸残基突变,可以制备双特异性抗体(参见,例如,bostrom等人,2009,science323:1610-1614)。双功能抗体可以通过表达设计为编码双特异性抗体的核酸来制备。
还可以通过化学合成来制备抗体(例如,利用solidphasepeptidesynthesis,第二版,1984,thepiercechemicalco.,rockford,ill.所描述的方法)。还可以使用无细胞的平台产生抗体(参见,例如,chu等人,biochemiano.2,2001(rochemolecularbiologicals))。
flanagan等人(methodsinmolecularbiology,vol.378∶monoclonalantibodies∶methodsandprotocols)描述了fc融合蛋白的重组表达的方法。
一旦已经通过重组表达制备了抗体,则可以利用本领域已知的纯化免疫球蛋白分子的任何方法,将抗体纯化,例如,利用色谱(例如,离子交换色谱、亲合色谱,尤其是通过a蛋白或g蛋白选择之后对于抗原的亲合性和sizing柱色谱)、离心、差异溶解度或纯化蛋白的任何其它标准技术。
分离之后,如果需要的话,可以进一步纯化抗体,例如,利用高效液相色谱(参见,例如,fisher,laboratorytechniquesinbiochemistryandmolecularbiology(workandburdon,eds.,elsevier,1980)),或凝胶过滤色谱(在superdextm75柱(pharmaciabiotechab,uppsala,sweden)上)。
5.5抗体-药物缀合物合成子
抗体-药物缀合物合成子是用于形成adc的合成中间体。所述合成子通常是按照结构式(iii)的化合物,
或其盐,其中,d是如先前描述的bcl-xl抑制剂,l是如先前描述的连接子,rx是包含适合于共价连接合成子与抗体的官能团的部分。在具体实施方案中,合成子是按照结构式(iiia)的化合物或其盐,其中,ar、r1、r2、r4、r10a、r10b、r10c、r11a、r11b、z1、z2和n如先前结构式(iia)所定义,l和rx如结构式(iii)所定义∶
为了合成adc,在官能团rx与抗体上的“互补”官能团fx反应的条件下,使按照结构式(iii)的中间体合成子或其盐与感兴趣的抗体接触,形成共价连接基。
基团rx和fx的特征取决于连接合成子与抗体所使用的化学过程。通常,使用的化学过程不应该改变抗体的完整性,例如,它结合它的靶点的能力。优选,缀合抗体的结合性能与非缀合抗体的结合性能极其类似。分子与生物分子(例如,抗体)缀合的各种化学过程和技术在本领域是已知的,尤其是与抗体缀合的各种化学过程和技术是众所周知的。参见,例如,amon等人,“monoclonalantibodiesforimmunotargetingofdrugsincancertherapy,”inmonoclonalantibodiesandcancertherapy,reisfeld等人eds.,alanr.liss,inc.,1985;hellstrom等人,“antibodiesfordrugdelivery,”incontrolleddrugdelivery(robinson等人eds.,marceldekker,inc.,2nded.1987;thorpe,“antibodycarriersofcytotoxicagentsincancertherapy:areview,”inmonoclonalantibodies'84:biologicalandclinicalapplications,pinchera等人eds.,1985;“analysis,results,andfutureprospectiveofthetherapeuticuseofradiolabeledantibodyincancertherapy,”inmonoclonalantibodiesforcancerdetectionandtherapy,baldwin等人eds.,academicpress,1985;以及thorpe等人,1982,immunol.rev.62:119-58;和wo89/12624。任何这些化学过程可以用于连接合成子与抗体。
在一个实施方案中,rx包含能够连接合成子与抗体上的氨基的官能团。在另一个实施方案中,rx包含nhs-酯或异硫氰酸酯。在另一个实施方案中,rx包含能够连接合成子与抗体上的巯基的官能团。在另一个实施方案中,rx包含卤代乙酰基或马来酰亚胺。
通常,合成子连接抗体的氨基酸残基的侧链,包括,例如,可及的赖氨酸残基的伯氨基,或可及的半胱氨酸残基的巯基。通过还原链间二硫键,可以获得游离巯基。
在一个实施方案中,lk是与抗体ab上的氨基形成的连接基。在另一个实施方案中,lk是酰胺或硫脲。在另一个实施方案中,lk是与抗体ab上的巯基形成的连接基。在另一个实施方案中,lk是硫醚。
在一个实施方案中,d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08和其盐;l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其盐;lk选自酰胺、硫脲和硫醚;和m是1至8的整数。
很多官能团rx和用于连接合成子与可及的赖氨酸残基的化学过程是已知的,并且包括,例如(没有限制性),nhs-酯和异硫氰酸酯。
很多官能团rx和用于连接合成子与可及的半胱氨酸残基的游离巯基的化学过程是已知的,并且包括,例如(没有限制性),卤代乙酰基和马来酰亚胺。
在一个实施方案中,d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08和其盐;l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其盐;rx包含选自nhs-酯、异硫氰酸酯、卤代乙酰基和马来酰亚胺的官能团。
然而,缀合化学不局限于合适的侧链基团。合适的小分子与胺连接,侧链(例如,胺)可以转变为其它有用的基团,例如,羟基。通过多功能小分子与抗体可及的氨基酸残基的侧链的缀合,这个策略可用于增加抗体中的合适连接位点的数目。
也可以设计抗体,使其包括用于缀合的氨基酸残基。下列文献描述了设计抗体的方法,使抗体包括在adc环境下用于缀合药物的非遗传编码的氨基酸残基:axup等人,2003,procnatlacadsci109:16101-16106,以及tian等人,2014,procnatlacadsci111:1776-1771。
用于制备本文所描述的adc的示范性合成子包括但不局限于下列合成子∶
5.6抗体药物缀合物
在合适的靶细胞的细胞试验和/或体内试验中,可以证明本文所描述的adc的bcl-xl抑制活性。可以用于证明靶向egfr、epcam或ncam1的adc的活性的具体试验提供于实施例7和8中。通常,在这种细胞试验中,adc的ec50值小于大约5000nm,不过,adc可以显示出显著更低的ec50值,例如,小于大约500、300或甚至小于100nm。使用表达特定靶点抗原的细胞所进行的类似的细胞试验,可以用于证明靶向其它抗原的adc的bcl-xl抑制活性。
5.7合成方法
本文所描述的bcl-xl抑制剂以及合成子,可以使用已知的有机化学的标准技术来合成。下面提供了合成bcl-xl抑制剂以及合成子的常规反应路线,它们可以原样使用,或进行修改,用于合成全部范围的本文所描述的bcl-xl抑制剂以及合成子。合成示范性的bcl-xl抑制剂与合成子的可以用于指导的具体方法提供于实施例部分中。
也可以利用标准方法制备adc,例如,与下列文献所描述的方法类似的方法:hamblett等人,2004,“effectsofdrugloadingontheantitumoractivityofamonoclonalantibodydrugconjugate,”clin.cancerres.10:7063-7070;doronina等人,2003,“developmentofpotentandhighlyefficaciousmonoclonalantibodyauristatinconjugatesforcancertherapy,”nat.biotechnol.21(7):778-784;以及francisco等人,2003,“caclo-vcmmae,ananti-cd30-monomethylauristatineconjugatewithpotentandselectiveantitumoractivity,”blood102:1458-1465。例如,每个抗体带有四个药物的adc,可以如下制备:在37℃下,使用过量的还原剂,例如,dtt或tcep,部分还原抗体30分钟,然后,使用1mmdtpa/dpbs,通过sephadex®g-25树脂洗脱,进行缓冲液交换。用dpbs进一步稀释洗脱液,可以使用5,5'-二硫代双(2-硝基苯甲酸)[ellman's试剂],测定抗体的硫醇浓度。在4℃下,加入过量(例如,5倍)的连接子-药物合成子(经过1小时),并通过加入实质上过量(例如,20倍)的半胱氨酸,淬灭该缀合反应。可以在sephadexg-25(在pbs中平衡)上纯化所得到的adc混合物,除去未反应的合成子,如果需要的话,进行脱盐,并且通过分子排阻色谱纯化。然后,可以通过0.2μm过滤器,将得到的adc无菌过滤,如果需要的话,冷冻干燥,储存。在某些实施方案中,所有链间的半胱氨酸二硫键被连接子-药物缀合物替代。
合成示范性的adc的具体方法提供于实施例部分中,可以用于合成全部范围的本文所描述的adc。
5.7.1合成bcl-xl抑制剂的一般方法
5.7.1.1合成化合物(9)
反应路线1
反应路线1描述了吡唑中间体式(9)的合成方法。可以用bh3·thf处理3-溴-5,7-二甲基金刚烷甲酸(1),提供3-溴-5,7-二甲基金刚烷甲醇(2)。该反应通常在环境温度下、在溶剂(例如但不局限于:四氢呋喃)中进行。在氰基亚甲基三丁基正膦的存在下,用1h-吡唑处理3-溴-5,7-二甲基金刚烷甲醇(2),可以制备1-((3-溴-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基)-1h-吡唑(3)。该反应通常在升高的温度下、在溶剂(例如但不局限于:甲苯)中进行。在碱(例如但不局限于:三乙胺)的存在下,可以用乙烷-1,2-二醇处理1-((3-溴-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基)-1h-吡唑(3),提供2-{[3,5-二甲基-7-(1h-吡唑-1-基甲基)三环[3.3.1.13,7]癸-1-基]氧基}乙醇(4)。该反应通常在升高的温度下进行,并且该反应可以在微波条件下进行。可以用强碱(例如但不局限于:正丁基锂)处理2-{[3,5-二甲基-7-(1h-吡唑-1-基甲基)三环[3.3.1.13,7]癸-1-基]氧基}乙醇(4),而后加入碘甲烷,提供2-({3,5-二甲基-7-[(5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)乙醇(5)。加入和反应通常在溶剂(例如但不局限于:四氢呋喃)中、在降低的温度下进行,而后升温至环境温度进行后处理。可以用n-碘代琥珀酰亚胺处理2-({3,5-二甲基-7-[(5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)乙醇(5),提供1-({3,5-二甲基-7-[2-(羟基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-4-碘代-5-甲基-1h-吡唑(6)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:三乙胺)的存在下,1-({3,5-二甲基-7-[2-(羟基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-4-碘代-5-甲基-1h-吡唑(6)与甲磺酰氯反应,而后加入胺h2nr4,可以制备式(7)的化合物。与甲磺酰氯的反应通常在低温下进行,而后升高温度用于与胺的反应,且该反应通常在溶剂(例如但不局限于:四氢呋喃)中进行。在4-二甲基氨基吡啶的存在下,可以使式(7)的化合物与二碳酸二叔丁基酯反应,提供式(8)的化合物。该反应通常在环境温度下、在溶剂(例如但不局限于:四氢呋喃)中进行。式(8)的化合物的硼化以提供式(9)的化合物,可以在本文描述的条件下和容易在文献中得到的条件下进行。
5.7.1.2合成化合物(14)
反应路线2
反应路线2描述了式(14)的中间体的合成方法。在碱(例如但不局限于:n,n-二异丙基乙胺或三乙胺)的存在下,式(10)的化合物与3-溴-6-氟吡啶甲酸叔丁基酯(11)反应,可以制备式(12)的化合物。该反应通常在惰性气氛中、在升高的温度下、在溶剂(例如但不局限于:二甲亚砜)中进行。在本文或在文献中所描述的硼化条件下,可以使式(12)的化合物与4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(13)反应,提供式(14)的化合物。
5.7.1.3合成化合物(24)
反应路线3
反应路线3描述了制备中间体的方法,所述中间体包含与金刚烷连接的-nu(亲核试剂)和以叔丁基酯形式保护的吡啶甲酸酯。在本文或文献所描述的suzuki偶合条件下,可以使2-(6-(叔丁氧羰基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯(14)与1-({3,5-二甲基-7-[2-(羟基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-4-碘代-5-甲基-1h-吡唑(6)反应,提供2-(6-(叔丁氧羰基)-5-(1-((3-(2-羟乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯(17)。可以用碱(例如但不局限于:三乙胺)处理2-(6-(叔丁氧羰基)-5-(1-((3-(2-羟乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯(17),而后用甲磺酰氯处理,提供2-(6-(叔丁氧羰基)-5-(1-((3,5-二甲基-7-(2-((甲磺酰基)氧基)乙氧基)金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯(18)。加入通常在溶剂(例如但不局限于:二氯甲烷)中在低温下进行,而后升温至环境温度。可以使2-(6-(叔丁氧羰基)-5-(1-((3,5-二甲基-7-(2-((甲磺酰基)氧基)乙氧基)金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯(18)与式(19)的亲核试剂(nu)反应,提供式(20)的化合物。亲核试剂的实例包括但不局限于:叠氮化钠、甲胺、氨和亚氨基二碳酸二叔丁基酯。可以使式(20)的化合物与氢氧化锂反应,提供式(21)的化合物。该反应通常在环境温度下、在溶剂(例如但不局限于:四氢呋喃、甲醇、水或其混合物)中进行。在本文所描述或容易在文献中获得的酰胺化条件下,可以使式(21)的化合物与式(22)的化合物反应,其中,ar如本文所述,提供式(23)的化合物。在溶剂(例如但不局限于:二氯甲烷或二噁烷)中,可以用酸(例如,三氟乙酸或hcl)处理式(23)的化合物,提供式(24)的化合物。
5.7.1.4合成化合物(34)
反应路线4
反应路线4描述了化合物(34)的合成方法。在本文所描述或容易在文献中获得的酰胺化条件下,可以使式(25)的化合物与式(26)的化合物反应,其中,ar如本文所述,提供式(27)的化合物。在碱(例如但不局限于:碳酸铯)的存在下,可以使式(27)的化合物与3-溴-6-氟吡啶甲酸叔丁基酯(11)反应,提供式(28)的化合物。该反应通常在升高的温度下、在溶剂(例如但不局限于:n,n-二甲基乙酰胺)中进行。在本文或文献所描述的suzuki偶合条件下,式(28)的化合物与式(29)的硼酸酯(或等效的硼酸)反应,可以制备式(30)的化合物。用三氟乙酸处理式(30)的化合物,可以制备式(31)的化合物。该反应通常在环境温度下、在溶剂(例如但不局限于:二氯甲烷)中进行。可以使式(31)的化合物与2-甲氧基乙醛(32)反应,而后使用还原剂,例如但不局限于:硼氢化钠,提供式(33)的化合物。该反应通常在环境温度下、在溶剂(例如但不局限于:二氯甲烷、甲醇或其混合物)中进行。在溶剂(例如但不局限于:二氯甲烷或二噁烷)中,可以用酸(例如,三氟乙酸或hcl)处理式(33)的化合物,提供式(34)的化合物。
5.7.2合成合成子的一般方法
在下面的反应路线中,变量ar2在式(iia)的化合物中代表
5.7.2.1合成化合物(89)
反应路线5
如反应路线5所示,在本文所描述或容易在文献中得到的酰胺化条件下,式(77)的化合物,其中,pg是合适的对碱不稳定的保护基,和aa(2)是cit、ala或lys,可以与4-(氨基苯基)甲醇(78)反应,提供化合物(79)。化合物(79)与碱(例如但不局限于:二乙胺)反应,可以制备化合物(80)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在本文所描述或在文献中容易得到的酰胺化条件下,化合物(81),其中,pg是合适的对碱或酸敏感的保护基,和aa(1)是val或phe,可以与化合物(80)反应,提供化合物(82)。视情况而定,用二乙胺或三氟乙酸处理化合物(82),可以制备化合物(83)。该反应通常在环境温度下、在溶剂(例如但不局限于:二氯甲烷)中进行。化合物(84),其中,sp是间隔基,可以与化合物(83)反应,提供化合物(85)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使化合物(85)与双(4-硝基苯基)碳酸酯(86)反应,提供化合物(87)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使化合物(87)与化合物(88)反应,提供化合物(89)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.7.2.2合成化合物(94)和(96)
反应路线6
反应路线6描述了另一个mab-连接子与二肽合成子连接的方法。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使化合物(88)与化合物(90)反应,提供化合物(91)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。化合物(91)与二乙胺反应,可以制备化合物(92)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在本文所描述或容易在文献中得到的酰胺化条件下,化合物(93),其中,x1是cl、br或i,可以与化合物(92)反应,提供化合物(94)。在本文所描述或容易在文献中获得的酰胺化条件下,可以使化合物(92)与式(95)的化合物反应,提供化合物(96)。
5.7.2.3合成化合物(106)
反应路线7
反应路线7描述了乙烯基葡糖苷酸连接子中间体与合成子的合成方法。可以用氧化银处理(2r,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(97),而后使用4-溴-2-硝基苯酚(98),提供(2s,3r,4s,5s,6s)-2-(4-溴-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(99)。该反应通常在环境温度下、在溶剂(例如但不局限于:乙腈)中进行。在碱(例如但不局限于:碳酸钠)和催化剂(例如但不局限于:三(二亚苄基丙酮)二钯(pd2(dba)3))的存在下,可以使(2s,3r,4s,5s,6s)-2-(4-溴-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(99)与(e)-叔丁基二甲基((3-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)烯丙基)氧基)硅烷(100)反应,提供(2s,3r,4s,5s,6s)-2-(4-((e)-3-((叔丁基二甲基甲硅烷基)氧基)丙-1-烯-1-基)-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(101)。该反应通常在升高的温度下、在溶剂(例如但不局限于:四氢呋喃)中进行。在酸(例如但不局限于:盐酸)的存在下,(2s,3r,4s,5s,6s)-2-(4-((e)-3-((叔丁基二甲基甲硅烷基)氧基)丙-1-烯-1-基)-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(101)与锌反应,可以制备(2s,3r,4s,5s,6s)-2-(2-氨基-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(102)。加入通常在低温下在溶剂(例如但不局限于:四氢呋喃、水或其混合物)中进行,而后升温至环境温度。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使(2s,3r,4s,5s,6s)-2-(2-氨基-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(102)与(9h-芴-9-基)甲基(3-氯-3-氧代丙基)氨基甲酸酯(103)反应,提供(2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(104)。加入通常在低温下在溶剂(例如但不局限于:二氯甲烷)中进行,而后升温至环境温度。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使化合物(88)与(2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(104)反应,而后进行后处理,并在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下与式(105)的化合物反应,提供化合物(106)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.7.2.4合成化合物(115)
反应路线8
反应路线8描述了代表性的2-醚葡糖苷酸连接子中间体与合成子的合成方法。在碳酸银的存在下,可以使(2s,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(97)与2,4-二羟基苯甲醛(107)反应,提供(2s,3r,4s,5s,6s)-2-(4-甲酰基-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(108)。该反应通常在升高的温度下、在溶剂(例如但不局限于:乙腈)中进行。可以用硼氢化钠处理(2s,3r,4s,5s,6s)-2-(4-甲酰基-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(108),提供(2s,3r,4s,5s,6s)-2-(3-羟基-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(109)。加入通常在低温下在溶剂(例如但不局限于:四氢呋喃、甲醇或其混合物)中进行,而后升温至环境温度。在咪唑的存在下,(2s,3r,4s,5s,6s)-2-(3-羟基-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(109)与叔丁基二甲基甲硅烷基氯反应,可以制备(2s,3r,4s,5s,6s)-2-(4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(110)。该反应通常在低温下、在溶剂(例如但不局限于:二氯甲烷)中进行。在三苯基膦和偶氮二甲酸酯(例如但不局限于:二氮烯-1,2-二甲酸二叔丁基酯)的存在下,(2s,3r,4s,5s,6s)-2-(4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(110)与(9h-芴-9-基)甲基(2-(2-羟乙氧基)乙基)氨基甲酸酯反应,可以制备(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(111)。该反应通常在环境温度下、在溶剂中进行,例如但不局限于:甲苯。可以用乙酸处理(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(111),提供(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(112)。该反应通常在环境温度下、在溶剂(例如但不局限于:水、四氢呋喃或其混合物)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(112)与双(4-硝基苯基)碳酸酯反应,可以制备(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(113)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以用化合物(88)处理(2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(113),而后用氢氧化锂处理,提供化合物(114)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺、四氢呋喃、甲醇或其混合物)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,化合物(114)与化合物(84)反应,可以制备化合物(115)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.7.2.5合成化合物(119)
反应路线9
反应路线9描述了将第二个增溶基团引入到糖连接子上的方法。在本文描述的或容易在文献中得到的酰胺化条件下,可以使化合物(116)与(r)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-磺丙酸(117)反应,而后用碱(例如但不局限于:二乙胺)处理,提供化合物(118)。在本文所描述或容易在文献中获得的酰胺化条件下,可以使化合物(118)与化合物(84)反应,其中,sp是间隔基,提供化合物(119)。
5.7.2.6合成化合物(129)
反应路线10
反应路线10描述了4-醚葡糖苷酸连接子中间体与合成子的合成方法。在碱(例如但不局限于:碳酸钾)的存在下,2,4-二羟基苯甲醛(120)与1-溴-2-(2-溴乙氧基)乙烷(121)反应,可以制备4-(2-(2-溴乙氧基)乙氧基)-2-羟基苯甲醛(122)。该反应通常在升高的温度下、在溶剂(例如但不局限于:乙腈)中进行。可以用叠氮化钠处理4-(2-(2-溴乙氧基)乙氧基)-2-羟基苯甲醛(122),提供4-(2-(2-叠氮基乙氧基)乙氧基)-2-羟基苯甲醛(123)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在氧化银的存在下,4-(2-(2-叠氮基乙氧基)乙氧基)-2-羟基苯甲醛(123)与(3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(124)反应,可以制备(2s,3r,4s,5s,6s)-2-(5-(2-(2-叠氮基乙氧基)乙氧基)-2-甲酰基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(125)。该反应通常在环境温度下、在溶剂(例如但不局限于:乙腈)中进行。在pd/c的存在下,(2s,3r,4s,5s,6s)-2-(5-(2-(2-叠氮基乙氧基)乙氧基)-2-甲酰基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(125)的氢化,提供(2s,3r,4s,5s,6s)-2-(5-(2-(2-氨基乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(126)。该反应通常在环境温度下、在溶剂中进行,例如但不局限于:四氢呋喃。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,用(9h-芴-9-基)甲基氯甲酸酯处理(2s,3r,4s,5s,6s)-2-(5-(2-(2-氨基乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(126),可以制备(2s,3r,4s,5s,6s)-2-(5-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(127)。该反应通常在低温下、在溶剂(例如但不局限于:二氯甲烷)中进行。在碱(例如但不局限于:n,n二异丙基乙胺)的存在下,可以使化合物(88)与(2s,3r,4s,5s,6s)-2-(5-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(127)反应,而后用氢氧化锂处理,提供化合物(128)。该反应通常在低温下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,化合物(128)与化合物(84)反应,可以制备化合物(129)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.7.2.7合成化合物(139)
反应路线11
反应路线11描述了氨基甲酸酯葡糖苷酸中间体与合成子的合成方法。可以用氢化钠处理2-氨基-5-(羟甲基)苯酚(130),而后与2-(2-叠氮基乙氧基)乙基4-甲基苯磺酸酯(131)反应,提供(4-氨基-3-(2-(2-叠氮基乙氧基)乙氧基)苯基)甲醇(132)。该反应通常在升高的温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在咪唑的存在下,(4-氨基-3-(2-(2-叠氮基乙氧基)乙氧基)苯基)甲醇(132)与叔丁基二甲基氯硅烷反应,可以制备2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯胺(133)。该反应通常在环境温度下、在溶剂中进行,例如但不局限于:四氢呋喃。在碱(例如但不局限于:三乙胺)的存在下,可以用光气处理2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯胺(133),而后在碱(例如但不局限于:三乙胺)的存在下,与(3r,4s,5s,6s)-2-羟基-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(134)反应,提供(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(135)。该反应通常在溶剂(例如但不局限于:甲苯)中进行,加入通常在低温下进行,加入光气之后,升温至环境温度,并在加入(3r,4s,5s,6s)-2-羟基-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(134)之后,在升高的温度下加热。(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(135)与对甲苯磺酸一水合物反应,可以制备(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(羟甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(136)。该反应通常在环境温度下、在溶剂中进行,例如但不局限于:甲醇。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(羟甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(136)与双(4-硝基苯基)碳酸酯反应,提供(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(137)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使(2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(137)与化合物反应,而后用氢氧化锂水溶液处理,提供化合物(138)。第一步通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行,第二步通常在低温下、在溶剂(例如但不局限于:甲醇)中进行。可以用三(2-羧乙基))膦盐酸盐处理化合物(138),而后在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,与化合物(84)反应,提供化合物(139)。与三(2-羧乙基)膦盐酸盐的反应通常在环境温度下、在溶剂(例如但不局限于:四氢呋喃、水或其混合物)中进行,与n-琥珀酰亚胺基6-马来酰亚胺基(maleimido)己酸盐的反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.7.2.8合成化合物(149)
反应路线12
反应路线20描述了半乳糖苷连接子中间体与合成子的合成方法。可以用乙酸中的hbr处理(2s,3r,4s,5s,6r)-6-(乙酰氧基甲基)四氢-2h-吡喃-2,3,4,5-四基四乙酸酯(140),提供(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-溴四氢-2h-吡喃-3,4,5-三基三乙酸酯(141)。该反应通常在环境温度下、在氮气氛围中进行。在4-羟基-3-硝基苯甲醛(142)的存在下,用氧化银(i)处理(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-溴四氢-2h-吡喃-3,4,5-三基三乙酸酯(141),可以制备(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-甲酰基-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(143)。该反应通常在环境温度下、在溶剂(例如但不局限于:乙腈)中进行。可以用硼氢化钠处理(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-甲酰基-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(143),提供(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-(羟甲基)-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(144)。该反应通常在低温下、在溶剂(例如但不局限于:四氢呋喃、甲醇或其混合物)中进行。在盐酸的存在下,用锌处理(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-(羟甲基)-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(144),可以制备(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(2-氨基-4-(羟甲基)苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(145)。该反应通常在低温下、在氮气氛围中、在溶剂(例如但不局限于:四氢呋喃)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,(2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(2-氨基-4-(羟甲基)苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(145)与(9h-芴-9-基)甲基(3-氯-3-氧代丙基)氨基甲酸酯(103)反应,可以制备(2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(羟甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(146)。该反应通常在低温下、在溶剂(例如但不局限于:二氯甲烷)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使(2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(羟甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(146)与双(4-硝基苯基)碳酸酯反应,提供(2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(147)。该反应通常在低温下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以使(2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(147)与化合物(88)反应,而后用氢氧化锂处理,提供化合物(148)。第一步通常在低温下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行,第二步通常在环境温度下、在溶剂(例如但不局限于:甲醇)中进行。在碱(例如但不局限于:n,n-二异丙基乙胺)的存在下,可以用化合物(84)处理化合物(148),其中,sp是间隔基,提供化合物(149)。该反应通常在环境温度下、在溶剂(例如但不局限于:n,n-二甲基甲酰胺)中进行。
5.8组合物
本文描述的bcl-xl抑制剂和/或adc可以是包含所述抑制剂或adc以及一或多种载体、赋形剂和/或稀释剂的组合物形式。可以将所述组合物配制用于具体使用,例如兽用或在人类中的药物用途。组合物的形式(例如,干粉、液体制剂,等等)和所使用的赋形剂、稀释剂和/或载体取决于所述抑制剂和/或adc的目标用途,对于治疗用途,取决于给药模式。
对于治疗用途,bcl-xl抑制剂和/或adc组合物可以作为包括药用载体的无菌药物组合物的一部分来提供。这种组合物可以是任何合适的形式(取决于给予患者的目标方法)。可以通过各种途径给予患者所述药物组合物,例如口服、透皮、皮下、鼻内、静脉内、肌肉内、鞘内、局部地(topically)或局部给药(locally)。在任何给定的情况中,最合适的给药途径取决于具体bcl-xl抑制剂或adc、患者、疾病的性质和严重程度以及患者的实际条件。通常,口服或胃肠外给予bcl-xl抑制剂,静脉内或皮下给予adc药物组合物。
药物组合物可以方便地存在于单位剂型中,每个剂量包含预定数量的本文所描述的bcl-xl抑制剂或adc。单位剂量所包括的抑制剂或adc的数量取决于所治疗的疾病,以及本领域众所周知的其它因素。对于bcl-xl抑制剂,这种单位剂量可以是片剂、胶囊剂、锭剂等等形式,其含有适合于单一给药数量的bcl-xl抑制剂。对于adc,这种单位剂量可以是含有适合于单一给药数量的adc的冷冻干燥的干粉形式,或液体形式。干粉单位剂型可以包装在具有注射器、合适数量的稀释剂和/或用于给药的其它组分的试剂盒中。液体形式的单位剂量,可以方便地以预先填充了适合于单一给药数量的adc的注射器形式提供。
也可以提供散装形式的、含有适合于多次给药数量的adc的药物组合物。
通过将具有目标纯度的adc与任选的本领域通常使用的可药用载体、赋形剂或稳定剂(所有这些在本文中被称为“载体”)混合,即,缓冲剂、稳定剂、防腐剂、等渗剂(isotonifiers)、非离子型清洗剂、抗氧化剂及其它各种添加剂,可以将adc的药物组合物制备为用于储存的冷冻干燥制剂或水溶液形式。参见remington’spharmaceuticalsciences,16thedition(osol,ed.1980)。这种添加剂在使用剂量和浓度下对于接受者应该是无毒的。
缓冲剂有助于保持ph值在接近生理条件的范围内。它们的浓度范围可以为大约2mm至大约50mm。本公开使用的合适的缓冲剂包括有机和无机酸以及其盐,例如,柠檬酸盐缓冲剂(例如,柠檬酸一钠-柠檬酸二钠混合物、柠檬酸-柠檬酸三钠混合物、柠檬酸-柠檬酸一钠混合物,等等)、琥珀酸盐缓冲剂(例如,琥珀酸-琥珀酸一钠混合物、琥珀酸-氢氧化钠混合物、琥珀酸-琥珀酸二钠混合物,等等)、酒石酸盐缓冲剂(例如,酒石酸-酒石酸钠混合物、酒石酸-酒石酸钾混合物、酒石酸-氢氧化钠混合物,等等)、富马酸盐缓冲剂(例如,富马酸-富马酸一钠混合物、富马酸-富马酸二钠混合物、富马酸一钠-富马酸二钠混合物,等等)、葡糖酸盐缓冲剂(例如,葡糖酸-葡糖酸钠混合物、葡糖酸-氢氧化钠混合物、葡糖酸-葡糖酸钾混合物,等等)、草酸盐缓冲剂(例如,草酸-草酸钠混合物、草酸-氢氧化钠混合物、草酸-草酸钾混合物,等等)、乳酸盐缓冲剂(例如,乳酸-乳酸钠混合物、乳酸-氢氧化钠混合物、乳酸-乳酸钾混合物,等等)和乙酸盐缓冲剂(例如,乙酸-乙酸钠混合物、乙酸-氢氧化钠混合物,等等)。另外,可以使用磷酸盐缓冲剂、组氨酸缓冲剂和三甲胺盐,例如tris。
为了抑制微生物生长,可以加入防腐剂,加入数量可以为大约0.2%-1%(w/v)。本公开使用的合适的防腐剂包括:苯酚、苯甲醇、间甲酚、对羟基苯甲酸甲酯、对羟基苯甲酸丙酯、十八烷基二甲基苄基氯化铵、苯扎卤铵(benzalconiumhalides)(例如,苯扎氯铵、苯扎溴铵和苯扎碘铵)、氯化六烃季铵和对羟基苯甲酸烷基酯,例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯、儿茶酚、间苯二酚、环己醇和3-戊醇。为了保证本公开的液体组合物的等渗性,可以加入等渗剂(isotonicifiers)(有时称为“稳定剂”),包括多羟基糖醇,例如,三元醇或高级糖醇,例如,丙三醇、赤藓醇、阿糖醇、木糖醇、山梨糖醇和甘露糖醇。稳定剂指的是广泛类型的赋形剂,其功能范围可以从填充剂至添加剂,其增溶治疗剂,或有助于防止变性,或防止与容器壁粘附。典型的稳定剂可以是多羟基的糖醇(上面列举的);氨基酸,例如精氨酸、赖氨酸、甘氨酸、谷氨酰胺、天冬酰胺、组氨酸、丙氨酸、鸟氨酸、l-亮氨酸、2-苯丙氨酸、谷氨酸、苏氨酸,等等,有机糖或糖醇,例如乳糖、海藻糖、水苏糖、甘露糖醇、山梨糖醇、木糖醇、核糖醇、肌醇(myoinisitol)、卫矛醇、丙三醇等等,包括环醇,例如肌醇;聚乙二醇;氨基酸聚合物;含硫还原剂,例如脲、谷胱甘肽、硫辛酸、硫代乙酸钠、硫代甘油、α-单硫代甘油和硫代硫酸钠;低分子量多肽(例如,10个残基或残基更少的肽);蛋白,例如,人血清白蛋白、牛血清白蛋白、明胶或免疫球蛋白;亲水性的聚合物,例如聚乙烯吡咯烷酮,单糖,例如木糖、甘露糖、果糖、葡萄糖;二糖,例如乳糖、麦芽糖、蔗糖,以及三糖(trisaccacharides),例如棉子糖;和多糖,例如葡聚糖。
可以加入非离子型表面活性剂或清洗剂(亦称“润湿剂”),帮助溶解糖蛋白,以及保护糖蛋白避免搅拌引起的聚集,也可以使制剂受到剪切表面应力的影响,不会引起蛋白的变性。合适的非离子型表面活性剂包括聚山梨醇酯(20、80,等等)、泊洛沙姆(polyoxamers)(184、188,等等)、复合多元醇、聚氧乙烯脱水山梨糖醇单醚(tween®-20、tween®-80,等等)。可以存在大约0.05mg/ml至大约1.0mg/ml的非离子型表面活性剂,例如大约0.07mg/ml至大约0.2mg/ml。
其它各种赋形剂包括:填充剂(例如,淀粉)、螯合剂(例如,edta)、抗氧化剂(例如,抗坏血酸、甲硫氨酸、维生素e)和共溶剂。
5.9使用方法
adc所包括的bcl-xl抑制剂,以及adc所提供的合成子,抑制bcl-xl活性,并且在表达bcl-xl的细胞中诱导细胞程序死亡。相应地,可以在抑制bcl-xl活性和/或在细胞中诱导细胞程序死亡的方法中,使用bcl-xl抑制剂和/或adc。
对于bcl-xl抑制剂,所述方法通常包括:使存活至少部分地取决于bcl-xl表达的细胞,与足以抑制bcl-xl活性和/或诱导细胞程序死亡数量的bcl-xl抑制剂接触。对于adc,所述方法通常包括:在adc能结合抗原的条件下,使存活至少部分地取决于bcl-xl表达,且表达adc的抗体的细胞表面抗原的细胞与adc接触。
在某些实施方案中,尤其是包含adc的bcl-xl抑制剂具有低或极低细胞渗透性的那些实施方案,adc的抗体结合能够将adc内化到细胞中的靶点,在细胞中,它可以递送它的bcl-xl抑制合成子。可以在抑制bcl-xl活性和/或抑制细胞程序死亡的细胞试验中体外进行所述方法,或作为需要抑制细胞程序死亡和/或诱导细胞程序死亡的疾病的治疗途径来体内进行。
细胞程序死亡失调涉及各种疾病,包括,例如,自身免疫病症(例如,系统性红斑狼疮、类风湿性关节炎、移植物抗宿主病、重症肌无力或舍格伦氏综合征)、慢性炎性病症(例如,牛皮癣、哮喘或克罗恩氏病)、高增殖病症(例如,乳腺癌、肺癌)、病毒感染(例如,疱疹、乳头状瘤或hiv),及其它病症,例如,骨关节炎和动脉粥样硬化。本文所描述的bcl-xl抑制剂或adc可以用于治疗或改善这些疾病中的任何疾病。这种治疗通常包括:给予患有所述疾病的患者足以提供治疗益处数量的本文所描述的bcl-xl抑制剂或adc。对于adc,所给予的adc的抗体的特征取决于所治疗的疾病,由此,抗体应该结合在受益于抑制bcl-xl活性的细胞类型中表达的细胞表面抗原。获得的治疗益处也取决于所治疗的具体疾病。在某些情况下,当以单一疗法给予时,bcl-xl抑制剂或adc可以治疗或改善疾病本身,或疾病的症状。在其它情况下,bcl-xl抑制剂或adc可以是整个治疗方案的一部分,所述治疗方案包括其它药剂,所述其它药剂和抑制剂或adc一起,用于治疗或改善所治疗的疾病,或所述疾病的症状。可以辅助本文所描述的bcl-xl抑制剂和/或adc给予或与本文所描述的bcl-xl抑制剂和/或adc一起给予的、用于治疗或改善具体疾病的药剂,对于本领域技术人员来说是显而易见的。
虽然绝对治愈始终是任何治疗方案的目标,但提供治疗益处不要求达到治愈效果。治疗益处可以包括:终止或减缓疾病的进展、使没有治愈的疾病衰退,和/或,改善或减缓疾病症状的发展。延长存活时间(与统计平均数相比较)和/或改善生活品质也可以认为是治疗益处。
涉及细胞程序死亡失调并且在世界范围内是显著的健康负担的一个具体类别的疾病是癌症。在一个具体实施方案中,本文所描述的bcl-xl抑制剂和/或adc可以用于治疗癌症。癌症可以是,例如,实体肿瘤或血液肿瘤。可以用本文所描述的adc治疗的癌症包括但不局限于:膀胱癌、脑癌、乳腺癌、骨髓癌、子宫颈癌、慢性淋巴细胞性白血病、结肠直肠癌、食道癌、肝细胞癌、淋巴细胞性白血病、滤泡性淋巴瘤、t细胞或b细胞的淋巴恶性肿瘤、黑素瘤、粒细胞性白血病、骨髓瘤、口腔癌、卵巢癌、非小细胞肺癌、慢性淋巴细胞性白血病、骨髓瘤、前列腺癌、小细胞肺癌或脾癌。adc尤其有益于治疗癌症,这是由于,抗体可用于靶向对于肿瘤细胞特异性的bcl-xl抑制合成子,由此潜在地避免或改善不合乎需要的、与全身性给予非缀合抑制剂相关的副作用和/或毒性。一个实施方案涉及治疗涉及内在的细胞程序死亡失调的疾病的方法,所述方法包括:给予患有涉及细胞程序死亡失调的疾病的患者有效提供治疗益处数量的本文所描述的adc,其中,adc的抗体结合内在的细胞程序死亡失调的细胞上的细胞表面受体。一个实施方案涉及治疗癌症的方法,所述方法包括:给予患有癌症的患者有效提供治疗益处数量的本文所描述的adc,所述adc能够结合在癌细胞的表面上表达的细胞表面受体或肿瘤相关抗原。
在致瘤性癌症的环境中,治疗益处,除了包括上述效果之外,还可以具体包括:终止或减缓肿瘤生长的进展、使肿瘤生长衰退、根除一或多种肿瘤和/或增加患者存活时间(与所治疗癌症的类型和阶段的统计平均数相比较)。在一个实施方案中,所治疗的癌症是致瘤性癌症。
为了提供治疗益处,可以以单一疗法给予bcl-xl抑制剂和/或adc,或辅助其它化学治疗剂和/或放射治疗给予,或与它们一起给予。本文所描述的抑制剂和/或adc可以作为辅助治疗的化学治疗剂可以是靶向化学治疗剂(例如,其它bcl-xl抑制剂或adc、蛋白激酶抑制剂,等等)或非靶向化学治疗剂(例如,非特异性的细胞毒素药剂,例如,放射性核苷(radionucleotides)、烷基化剂和嵌合剂)。可以辅助给予的本文所描述的抑制剂和/或adc的非靶向化学治疗剂包括但不局限于:氨甲喋呤、紫杉酚、左旋天冬酰胺酶、巯基嘌呤、硫鸟嘌呤、羟基脲、阿糖胞苷、环磷酰胺、异环磷酰胺、亚硝基脲、顺铂、卡铂、丝裂霉素、达卡巴嗪、甲基苄肼(procarbizine)、托泊替康、氮芥、环磷酰胺、依托泊苷、5-氟尿嘧啶、bcnu、依立替康、喜树碱、博来霉素、多柔比星、伊达比星、柔红霉素、放线菌素、普卡霉素、米托蒽醌、门冬酰胺酶、长春碱、长春花新碱、长春瑞滨、太平洋紫杉醇、刺孢霉素和多西他赛。
已经表明,升高的bcl-xl表达与耐受化学治疗和放射治疗有关。本文的数据证明,不能有效作为治疗癌症的单一疗法的bcl-xl抑制剂和/或adc,可以辅助其它化学治疗剂或放射治疗给予,或与它们一起给予,从而提供治疗益处。尽管不受任何手术疗法束缚,但人们相信,给予对于标准化学治疗剂和/或放射治疗变得耐受性的肿瘤本文所描述的bcl-xl抑制剂和/或adc,可以使肿瘤敏感,这样就可以使它们再次对化学治疗剂和/或放射治疗作出响应。一个实施方案涉及给予有效使肿瘤细胞对于标准化学治疗和/或放射治疗敏感数量的本文所描述的adc的方法。相应地,在治疗癌症的背景下,“治疗益处”包括给予还没有开始化学和/或放射治疗的患者、或对于已经开始化学和/或放射治疗但还没有显示出耐受性迹象的患者、或对于化学和/或放射治疗已经开始显示出耐受性的患者本文所描述的抑制剂和/或adc,所述抑制剂和/或adc可以作为化学治疗剂和/或放射治疗的辅助治疗,或与它们一起给予所述患者,作为使肿瘤对于化学和/或放射治疗敏感的方法。
5.10剂量和给药方案
所给予的adc的数量将取决于各种因素,包括但不局限于:所治疗的具体疾病、给药模式、目标治疗益处、疾病的阶段或严重程度、患者的年龄、体重及其它特征,等等。有效剂量的确定在本领域技术人员的能力范围之内。
有效剂量可以最初用细胞试验来评估。例如,可以配制人类使用的初剂量,达到adc的循环血液或血清浓度,希望这个浓度能够使bcl-xl抑制剂的细胞浓度处于或高于特定抑制分子的ic50值或ed50值(在细胞试验中测定)。
还可以用体内动物模型来评估人类使用的初始剂量。各种疾病的合适的动物模型在本领域是已知的。
当辅助其它药剂(例如,其它化学治疗剂)给予或与它们一起给予时,可以以与其它药剂相同的时间表或不同的时间表,给予adc。当以相同时间表给予时,可以在给予其它药剂之前、之后给予adc,或与其它药剂同时给予。在一些实施方案中,如果adc作为标准化学和/或放射疗法的辅助治疗给予,或与它们一起给予,可以在标准治疗之前开始给予adc,例如,在标准化学和/或放射治疗开始之前一天、几天、一周、几周、一个月或甚至几个月之前开始给予adc。
当辅助其它药剂给予或与其它药剂(例如,标准化学治疗剂)一起给予时,其它药剂通常是根据它的标准给药时间表(途径、剂量和频率)给予的。然而,在有些情况下,当作为adc治疗的辅助治疗来给予时,达到效力所需要的数量可能需要少于标准数量。
6.实施例
实施例1.合成示范性的bcl-xl抑制剂
该实施例提供了示范性的bcl-xl抑制化合物w1.01-w1.08的合成方法。使用acd/name(2012年发行)(build56084,2012年4月5日,advancedchemistrydevelopmentinc.,toronto,ontario),将bcl-xl抑制剂(w1.01-w1.08)和合成子(实施例2.1-2.53)命名。使用acd/name(2012年发行)(build56084,2012年4月5日,advancedchemistrydevelopmentinc.,toronto,ontario),或chemdraw®ver.9.0.7(cambridgesoft,cambridge,ma),或chemdraw®ultraver.12.0(cambridgesoft,cambridge,ma),将bcl-xl抑制剂和合成子中间体命名。
1.1. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-[1-({3,5-二甲基-7-[2-(甲基氨基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-5-甲基-1h-吡唑-4-基]吡啶-2-甲酸(化合物w1.01)
1.1.1. 3-溴-5,7-二甲基金刚烷甲酸
在50ml圆底烧瓶中,在0℃,加入溴(16ml)。然后加入铁粉(7g),并将该反应在0℃下搅拌30分钟。然后加入3,5-二甲基金刚烷-1-甲酸(12g)。将该混合物升温至室温,并搅拌3天。将冰和浓hcl的混合物倒入该反应混合物中。将得到的悬浮液用na2so3(50g,在200ml水中)处理两次,破坏溴,并用二氯甲烷提取三次。将合并的有机物用1nhcl水溶液洗涤,用na2so4干燥,过滤,并浓缩,得到粗品标题化合物。
1.1.2. 3-溴-5,7-二甲基金刚烷甲醇
向实施例1.1.1(15.4g)的四氢呋喃(200ml)溶液中加入bh3(1m,在四氢呋喃中,150ml)。将该混合物在室温下搅拌过夜。然后逐滴加入甲醇,小心地淬灭该反应混合物。然后,真空浓缩该混合物,并将残余物在乙酸乙酯(500ml)和2nhcl水溶液(100ml)之间平衡。用乙酸乙酯进一步提取水层两次,并将合并的有机提取物用水和盐水洗涤,用na2so4干燥,并过滤。蒸发溶剂,得到标题化合物。
1.1.3. 1-((3-溴-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基)-1h-吡唑
向实施例1.1.2(8.0g)的甲苯(60ml)溶液中加入1h-吡唑(1.55g)和氰基亚甲基三丁基正膦(2.0g)。将该混合物在90℃下搅拌过夜。然后,浓缩该反应混合物,并将残余物用硅胶柱色谱纯化(10∶1的庚烷∶乙酸乙酯),得到标题化合物。ms(esi)m/e324.2(m+h)+。
1.1.4. 2-{[3,5-二甲基-7-(1h-吡唑-1-基甲基)三环[3.3.1.13,7]癸-1-基]氧基}乙醇
向实施例1.1.3(4.0g)的乙烷-1,2-二醇(12ml)溶液中加入三乙胺(3ml)。将该混合物在150℃下、在微波条件(biotageinitiator)下搅拌45分钟。将该混合物倒入水(100ml)中,并用乙酸乙酯提取三次。用水和盐水洗涤合并的有机提取物,用na2so4干燥,并过滤。蒸发溶剂,得到粗品,用硅胶色谱纯化粗品,用20%的乙酸乙酯/庚烷洗脱,而后用5%的甲醇/二氯甲烷洗脱,得到标题化合物。ms(esi)m/e305.2(m+h)+。
1.1.5. 2-({3,5-二甲基-7-[(5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)乙醇
向冷却(-78℃)的实施例1.1.4(6.05g)的四氢呋喃(100ml)溶液中加入n-buli(40ml,2.5m,在己烷中)。将该混合物在-78℃下搅拌1.5小时。通过注射器加入碘甲烷(10ml),并将该混合物在-78℃下搅拌3小时。然后,用nh4cl水溶液淬灭该反应混合物,用乙酸乙酯提取两次,并将合并的有机提取物用水和盐水洗涤。用na2so4干燥之后,过滤该溶液,浓缩,并将残余物用硅胶柱色谱纯化,用5%的甲醇/二氯甲烷洗脱,得到标题化合物。ms(esi)m/e319.5(m+h)+。
1.1.6. 1-({3,5-二甲基-7-[2-(羟基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-4-碘代-5-甲基-1h-吡唑
向实施例1.1.5(3.5g)的n,n-二甲基甲酰胺(30ml)溶液中加入n-碘代琥珀酰亚胺(3.2g)。将该混合物在室温下搅拌1.5小时。然后,用乙酸乙酯(600ml)稀释该反应混合物,并用nahso3水溶液、水和盐水洗涤。用na2so4干燥之后,过滤该溶液,浓缩,并将残余物用硅胶色谱纯化(20%的乙酸乙酯/二氯甲烷),得到标题化合物。ms(esi)m/e445.3(m+h)+。
1.1.7. 2-({3-[(4-碘代-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基甲磺酸酯
向冷却的实施例1.1.6(6.16g)的二氯甲烷(100ml)溶液中加入三乙胺(4.21g),而后加入甲磺酰氯(1.6g)。将该混合物在室温下搅拌1.5小时。然后,用乙酸乙酯(600ml)稀释该反应混合物,并用水和盐水洗涤。用na2so4干燥之后,过滤该溶液,并浓缩,不用进一步纯化残余物,直接在下一个反应中使用。ms(esi)m/e523.4(m+h)+。
1.1.8. 1-({3,5-二甲基-7-[2-(甲基氨基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-4-碘代-5-甲基-1h-吡唑
在微波条件(biotageinitiator)下,将实施例1.1.7(2.5g)的2m甲胺/甲醇(15ml)溶液在100℃下搅拌20分钟。真空浓缩该反应混合物。然后,用乙酸乙酯(400ml)稀释残余物,并用nahco3水溶液、水和盐水洗涤。用na2so4干燥之后,过滤该溶液,并浓缩,不用进一步纯化残余物,直接在下一个反应中使用。ms(esi)m/e458.4(m+h)+。
1.1.9. [2-({3-[(4-碘代-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]甲基氨基甲酸叔丁基酯
向实施例1.1.8(2.2g)的四氢呋喃(30ml)溶液中加入二碳酸二叔丁基酯(1.26g)和催化数量的4-二甲基氨基吡啶。将该混合物在室温下搅拌1.5小时,并用乙酸乙酯(300ml)稀释。用饱和nahco3水溶液、水(60ml)和盐水(60ml)洗涤该溶液。用na2so4干燥有机层,过滤,并浓缩。将残余物用硅胶色谱纯化,用20%的乙酸乙酯/二氯甲烷洗脱,得到标题化合物。ms(esi)m/e558.5(m+h)+。
1.1.10. 6-氟-3-溴吡啶甲酸
在5℃,用1小时将6-氨基-3-溴吡啶甲酸(25g)的400ml二氯甲烷/氯仿(1:1)浆液加入到二氯甲烷(100ml)中的四氟硼酸亚硝(18.2g)中,并将得到的混合物再搅拌30分钟,然后升温至35℃,并搅拌过夜。将该反应冷却至室温,而后用nah2po4水溶液调节至ph4。将得到的溶液用二氯甲烷提取三次,用盐水洗涤合并的提取物,用硫酸钠干燥,过滤,并浓缩,提供标题化合物。
1.1.11. 3-溴-6-氟吡啶甲酸叔丁基酯
在0℃,将对甲苯磺酰氯(27.6g)加入到实施例1.1.10(14.5g)和吡啶(26.7ml)的二氯甲烷(100ml)和叔丁醇(80ml)溶液中。将该反应搅拌15分钟,升温至室温,并搅拌过夜。浓缩该溶液,并在乙酸乙酯和na2co3水溶液之间分配。分离各层,并用乙酸乙酯提取水层。将有机层合并,用na2co3水溶液和盐水冲洗,用硫酸钠干燥,过滤,并浓缩,提供标题化合物。
1.1.12. 2-(5-溴-6-(叔丁氧羰基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
向1,2,3,4-四氢异喹啉-8-甲酸甲酯盐酸盐(12.37g)和实施例1.1.11(15g)的二甲亚砜(100ml)溶液中加入n,n-二异丙基乙胺(12ml)。将该混合物在50℃下搅拌24小时。然后,用乙酸乙酯(500ml)稀释该混合物,用水和盐水洗涤,并用na2so4干燥。过滤,蒸发溶剂,得到残余物,用硅胶色谱纯化残余物,用20%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e448.4(m+h)+。
1.1.13. 2-(6-(叔丁氧羰基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
向实施例1.1.12(2.25g)和[1,1'-双(二苯基膦基)二茂铁]二氯钯(ii)(205mg)的乙腈(30ml)溶液中加入三乙胺(3ml)和频哪醇硼烷(2ml)。在回流下,将该混合物搅拌3小时。将该混合物用乙酸乙酯(200ml)稀释,用水和盐水洗涤,并用na2so4干燥。过滤,蒸发溶剂,硅胶色谱(用20%的乙酸乙酯/庚烷洗脱),得到标题化合物。ms(esi)m/e495.4(m+h)+。
1.1.14. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-((叔丁氧羰基)(甲基)氨基)乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
向实施例1.1.13(4.94g)的四氢呋喃(60ml)和水(20ml)溶液中加入实施例1.1.9(5.57g)、1,3,5,7-四甲基-8-十四烷基-2,4,6-三氧杂-8-磷杂金刚烷(412mg)、三(二亚苄基丙酮)二钯(0)(457mg)和k3po4(11g)。将该混合物在回流下搅拌24小时。冷却该反应混合物,用乙酸乙酯(500ml)稀释,用水和盐水洗涤,并用na2so4干燥。过滤,蒸发溶剂,得到残余物,用硅胶色谱纯化残余物,用20%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e799.1(m+h)+。
1.1.15. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-((叔丁氧羰基)(甲基)氨基)乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸
向实施例1.1.14(10g)的四氢呋喃(60ml)、甲醇(30ml)和水(30ml)溶液中加入氢氧化锂一水合物(1.2g)。将该混合物在室温下搅拌24小时。用2%hcl水溶液中和该反应混合物,并真空浓缩。将残余物用乙酸乙酯(800ml)稀释,用水和盐水洗涤,并用na2so4干燥。过滤,蒸发溶剂,得到标题化合物。ms(esi)m/e785.1(m+h)+。
1.1.16. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[(叔丁氧羰基)(甲基)氨基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸叔丁基酯
向实施例1.1.15(10g)的n,n-二甲基甲酰胺(20ml)溶液中加入苯并[d]噻唑-2-胺(3.24g)、氟-n,n,n',n'-四甲基甲脒(tetramethylformamidinium)六氟磷酸盐(5.69g)和n,n-二异丙基乙胺(5.57g)。将该混合物在60℃下搅拌3小时。将该反应混合物用乙酸乙酯(800ml)稀释,用水和盐水洗涤,并用na2so4干燥。过滤,蒸发溶剂,得到残余物,用硅胶色谱纯化残余物,用20%的乙酸乙酯/二氯甲烷洗脱,得到标题化合物。ms(esi)m/e915.5(m+h)+。
1.1.17. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-[1-({3,5-二甲基-7-[2-(甲基氨基)乙氧基]三环[3.3.1.13,7]癸-1-基}甲基)-5-甲基-1h-吡唑-4-基]吡啶-2-甲酸
向实施例1.1.16(5g)的二氯甲烷(20ml)溶液中加入三氟乙酸(10ml)。将该混合物搅拌过夜。真空蒸发溶剂,并将残余物溶于二甲亚砜/甲醇(1:1,10ml)中,通过反相色谱分离,使用analogix系统和c18柱(300g),用10-85%的乙腈和0.1%三氟乙酸/水洗脱,得到标题化合物的tfa盐。1hnmr(300mhz,二甲亚砜d6)δppm12.85(s,1h),8.13-8.30(m,2h),8.03(d,1h),7.79(d,1h),7.62(d,1h),7.32-7.54(m,3h),7.28(d,1h),6.96(d,1h),4.96(dd,1h),3.80-3.92(m,4h),3.48-3.59(m,1h),2.91-3.11(m,2h),2.51-2.59(m,4h),2.03-2.16(m,2h),1.21-1.49(m,6h),0.97-1.20(m,4h),0.87(s,6h)。ms(esi)m/e760.4(m+h)+。
1.2. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[(1r,3r,5s,7s)-3,5-二甲基-7-(2-{2-[2-(甲基氨基)乙氧基]乙氧基}乙氧基)三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸(化合物w1.02)
1.2.1. 2-(2-(2-(((3-((1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
向实施例1.1.3(2.65g)的2,2'-(乙烷-1,2-二基双(氧基))二乙醇(15g)溶液中加入三乙胺(3ml)。将该混合物在180℃下、在微波条件(biotageinitiator)下搅拌120分钟。将该混合物用水和乙腈(1:1,40ml)稀释。将粗品加入到反相柱(c18,sf65-800g)上,用10-100%的乙腈/水(含有0.1%三氟乙酸)洗脱,得到标题化合物。ms(esi)m/e393.0(m+h)+。
1.2.2. 2-(2-(2-((3,5-二甲基-7-((5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
向冷却(0℃)的实施例1.2.1(2.69g)的四氢呋喃(20ml)溶液中加入n-buli(10ml,2.5m,在己烷中)。将该混合物在0℃下搅拌1.5小时。通过注射器加入碘甲烷(1ml),并将该混合物在0℃下搅拌1.5小时。将该反应混合物用三氟乙酸(1ml)淬灭。蒸发溶剂之后,残余物直接用于下一步。ms(esi)m/e407.5(m+h)+。
1.2.3. 2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
向冷却(0℃)的实施例1.2.2(2.78g)的n,n-二甲基甲酰胺(30ml)溶液中加入n-碘代琥珀酰亚胺(1.65g)。将该混合物在室温下搅拌2小时。将粗品加入到反相柱(c-18,sf65-800g)上,用10-100%的乙腈/水(含有0.1%三氟乙酸)洗脱,得到标题化合物。ms(esi)m/e533.0(m+h)+。
1.2.4. 2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙氧基)-n-甲基乙胺
向冷却(0℃)的实施例1.2.3(2.45g)的四氢呋喃(10ml)溶液中加入三乙胺(1ml),而后加入甲磺酰氯(0.588g)。将该混合物在室温下搅拌2小时。将甲胺(10ml,2m,在甲醇中)加入到该反应混合物中,并转入到20ml微波管中。将该混合物在微波条件(biotageinitiator)下、在100℃下加热60分钟。冷却至室温之后,真空除去溶剂。将残余物加入到反相柱(c18,sf40-300g)上,用40-100%的乙腈/水(含有0.1%三氟乙酸)洗脱,得到标题化合物。ms(esi)m/e546.0(m+h)+。
1.2.5. (2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙氧基)乙基)(甲基)氨基甲酸叔丁基酯
向实施例1.2.4(1.41g)的四氢呋喃(20ml)溶液中加入二碳酸二叔丁基酯(1g)和4-二甲基氨基吡啶(0.6g)。将该混合物在室温下搅拌3小时,并真空除去溶剂。将残余物用硅胶色谱纯化,用10-100%的乙酸乙酯/己烷洗脱,得到标题化合物。ms(esi)m/e645.8(m+h)+。
1.2.6. (2-(2-(2-((3,5-二甲基-7-((5-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙基)(甲基)氨基甲酸叔丁基酯
向实施例1.2.5(1.25g)、二环己基膦基-2',6'-二甲氧基联苯(0.09g)、频哪醇硼烷(1.5ml)和三乙胺(1.5ml)的二噁烷(20ml)溶液中加入双(苄腈)氯化钯(ii)(0.042g)。脱气之后,将该混合物在90℃下搅拌过夜。蒸发溶剂,硅胶柱纯化(用20-100%的乙酸乙酯/己烷洗脱),得到标题化合物。ms(esi)m/e646.1(m+h)+。
1.2.7. 8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-甲酸叔丁基酯
向2-(叔丁氧羰基)-1,2,3,4-四氢异喹啉-8-甲酸(6.8g)和苯并[d]噻唑-2-胺(5.52g)的二氯甲烷(80ml)溶液中加入1-乙基-3-[3-(二甲基氨基)丙基]-碳二亚胺盐酸盐(9.4g)和4-二甲基氨基吡啶(6g)。将该混合物在室温下搅拌过夜。将该反应混合物用二氯甲烷(400ml)稀释,用5%hcl水溶液、水和盐水洗涤,并用na2so4干燥。过滤该混合物,并将滤液减压浓缩,提供标题化合物。
1.2.8. n-(苯并[d]噻唑-2-基)-1,2,3,4-四氢异喹啉-8-甲酰胺二盐酸盐
向实施例1.2.7(8.5g)的二氯甲烷(80ml)溶液中加入2nhcl/乙醚(80ml)。将该反应混合物在室温下搅拌过夜,并减压浓缩,提供标题化合物。
1.2.9. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-溴吡啶甲酸叔丁基酯
将实施例1.1.11(0.736g)、实施例1.2.8(1.62g)和cs2co3(4.1g)在12ml无水n,n-二甲基乙酰胺中、在120℃下搅拌12小时。然后,将冷却的反应混合物用乙酸乙酯和10%柠檬酸稀释。用柠檬酸洗涤有机相三次,用水和盐水洗涤一次,并用na2so4干燥。过滤,浓缩,得到粗品,在硅胶上色谱分离,使用0-40%的乙酸乙酯/己烷,提供标题化合物。
1.2.10. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(1-(((1s,7s)-3,5-二甲基-7-((2,2,5-三甲基-4-氧代-3,8,11-三氧杂-5-氮杂十三烷-13-基)氧基)金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸叔丁基酯
向实施例1.2.6(0.135g)的四氢呋喃(1ml)和水(1ml)溶液中加入实施例1.2.9(0.12g)、1,3,5,7-四甲基-8-十四烷基-2,4,6-三氧杂-8-磷杂金刚烷(0.023g)、三(二亚苄基丙酮)二钯(0)(0.015g)和k3po4(0.2g)。将该混合物在140℃下、在微波条件(biotageinitiator)下搅拌5分钟。将该反应混合物用甲苯(5ml)稀释,并过滤。蒸发溶剂,硅胶纯化(20-100%的乙酸乙酯/庚烷),得到标题化合物。ms(esi)m/e1004.8(m+h)+。
1.2.11. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[3,5-二甲基-7-(2-{2-[2-(甲基氨基)乙氧基]乙氧基}乙氧基)三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸
将二氯甲烷(10ml)中的实施例1.2.10(1.42g)用三氟乙酸(6ml)处理,并将该反应在室温下搅拌24小时。减压除去挥发物。用反相色谱纯化残余物,使用gilson系统(c18,sf40-300g),用30-100%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物的tfa盐。1hnmr(300mhz,二甲亚砜-d6)δppm12.85(br.s,1h),8.33(br.s,2h),8.03(d,1h),7.79(d,1h),7.62(d,1h),7.41-7.54(m,3h),7.32-7.40(m,2h),7.28(s,1h),6.95(d,1h),4.95(s,2h),3.85-3.93(m,2h),3.81(s,2h),3.60-3.66(m,2h),3.52-3.58(m,4h),3.45(s,3h),2.97-3.12(m,4h),2.56(t,2h),2.10(s,3h),1.34-1.41(m,2h),1.18-1.31(m,4h),0.95-1.18(m,6h),0.85(s,6h)。ms(esi)m/e848.2(m+h)+。
1.3. 合成3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸(化合物w1.03)
1.3.1. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-羟乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
向实施例1.1.13(2.25g)的四氢呋喃(30ml)和水(10ml)溶液中加入实施例1.1.6(2.0g)、1,3,5,7-四甲基-6-苯基-2,4,8-三氧杂-6-磷杂金刚烷(329mg)、三(二亚苄基丙酮)二钯(0)(206mg)和磷酸三钾(4.78g)。将该混合物回流过夜,冷却,并用乙酸乙酯(500ml)稀释。将得到的混合物用水和盐水洗涤,用na2so4干燥有机层,过滤,并浓缩。将残余物用快速色谱纯化,用20%的乙酸乙酯/庚烷洗脱,而后用5%的甲醇/二氯甲烷洗脱,提供标题化合物。
1.3.2. 2-(6-(叔丁氧羰基)-5-(1-((3,5-二甲基-7-(2-((甲磺酰基)氧基)乙氧基)金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
在冰浴中,向冷的实施例1.3.1(3.32g)的二氯甲烷(100ml)溶液中顺序加入三乙胺(3ml)和甲磺酰氯(1.1g)。在室温下搅拌该反应混合物1.5小时,用乙酸乙酯稀释,并用水和盐水洗涤。用na2so4干燥有机层,过滤,浓缩,提供标题化合物。
1.3.3. 2-(5-(1-((3-(2-叠氮基乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(叔丁氧羰基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
向实施例1.3.2(16.5g)的n,n-二甲基甲酰胺(120ml)溶液中加入叠氮化钠(4.22g)。将该混合物在80℃下加热3小时,冷却,用乙酸乙酯稀释,并用水和盐水洗涤。用na2so4干燥有机层,过滤,浓缩。用快速色谱纯化残余物,用20%的乙酸乙酯/庚烷洗脱,提供标题化合物。
1.3.4. 2-(5-(1-((3-(2-叠氮基乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(叔丁氧羰基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸
向在四氢呋喃(60ml)、甲醇(30ml)和水(30ml)的混合物中的实施例1.3.3(10g)的溶液中加入氢氧化锂一水合物(1.2g)。将该混合物在室温下搅拌过夜,并用2%hcl水溶液中和。将得到的混合物浓缩,将残余物溶于乙酸乙酯(800ml)中,并用水和盐水洗涤。用na2so4干燥有机层,过滤,浓缩,提供标题化合物。
1.3.5. 3-(1-((3-(2-叠氮基乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸叔丁基酯
按照1.1.16所描述的方法,用实施例1.3.4替代实施例1.1.15,制备标题化合物。
1.3.6. 3-(1-((3-(2-氨基乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸叔丁基酯
向实施例1.3.5(2.0g)的四氢呋喃(30ml)溶液中加入pd/c(10%,200mg)。将该混合物在氢气氛围中搅拌过夜。过滤该反应,并将滤液浓缩,提供标题化合物。
1.3.7. 3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
将二氯甲烷(3ml)中的实施例1.3.6(300mg)用三氟乙酸(3ml)处理过夜。浓缩该反应混合物,用反相色谱纯化残余物,使用gilson系统(300gc18柱),用10-70%的乙腈/0.1%三氟乙酸水溶液洗脱,提供标题化合物的三氟乙酸盐。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h)8.03(d,1h)7.79(d,1h)7.59-7.73(m,4h)7.41-7.53(m,3h)7.32-7.40(m,2h)7.29(s,1h)6.96(d,1h)4.96(s,2h)3.89(t,2h)3.83(s,2h)3.50(t,2h)3.02(t,2h)2.84-2.94(m,2h)2.11(s,3h)1.41(s,2h)1.21-1.36(m,4h)1.08-1.19(m,4h)0.96-1.09(m,2h)0.87(s,6h)。ms(esi)m/e744.3(m-h)-。
1.4. 合成3-[1-({3-[2-(2-氨基乙氧基)乙氧基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}甲基)-5-甲基-1h-吡唑-4-基]-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸(化合物w1.04)
1.4.1. 2-(2-((3-((1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙醇
如实施例1.1.4所述,用2,2'-氧基二乙醇替代乙烷-1,2-二醇,制备标题化合物。ms(esi)m/e349.2(m+h)+。
1.4.2. 2-(2-((3,5-二甲基-7-((5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙醇
如实施例1.1.5所述,制备标题化合物,用实施例1.4.1替代实施例1.1.4。ms(esi)m/e363.3(m+h)+。
1.4.3. 2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙醇
如实施例1.1.6所述,制备标题化合物,用实施例1.4.2替代实施例1.1.5。ms(esi)m/e489.2(m+h)+。
1.4.4. 2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙基甲磺酸酯
如实施例1.1.7所述,制备标题化合物,用实施例1.4.3替代实施例1.1.6。ms(esi)m/e567.2(m+h)+。
1.4.5. 2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙胺
如实施例1.1.8所述,制备标题化合物,用实施例1.4.4替代实施例1.1.7,用7n氨/甲醇替代2n甲胺/甲醇。ms(esi)m/e488.2(m+h)+。
1.4.6. (2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙氧基)乙基)氨基甲酸叔丁基酯
如实施例1.1.9所述,制备标题化合物,用实施例1.4.5替代实施例1.1.8。ms(esi)m/e588.2(m+h)+。
1.4.7. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(2-((叔丁氧羰基)氨基)乙氧基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸甲酯
如实施例1.1.14所述,制备标题化合物,用实施例1.4.6替代实施例1.1.9。ms(esi)m/e828.5(m+h)+。
1.4.8. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(2-((叔丁氧羰基)氨基)乙氧基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-1,2,3,4-四氢异喹啉-8-甲酸
如实施例1.1.15所述,制备标题化合物,用实施例1.4.7替代实施例1.1.14。ms(esi)m/e814.5(m+h)+。
1.4.9. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(1-((3-(2-(2-((叔丁氧羰基)氨基)乙氧基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸叔丁基酯
如实施例1.1.16所述,制备标题化合物,用实施例1.4.8替代实施例1.1.15。ms(esi)m/e946.2(m+h)+。
1.4.10. 3-[1-({3-[2-(2-氨基乙氧基)乙氧基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}甲基)-5-甲基-1h-吡唑-4-基]-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
如实施例1.1.17所述,制备标题化合物,用实施例1.4.9替代实施例1.1.16。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h),7.99-8.08(m,1h),7.60-7.82(m,4h),7.20-7.52(m,5h),6.93-6.99(m,1h),4.96(s,2h),3.45-3.60(m,6h),2.09-2.14(m,4h),0.95-1.47(m,19h),0.81-0.91(m,6h)。ms(esi)m/e790.2(m+h)+。
1.5. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[(2-甲氧基乙基)氨基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(化合物w1.05)
1.5.1. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(1-(((1r,3r)-3-(2-((2-甲氧基乙基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸叔丁基酯
将实施例1.3.6(0.050g)和2-甲氧基乙醛(6.93mg)的溶液在二氯甲烷(0.5ml)中在室温下一起搅拌1小时。向该反应中加入硼氢化钠(2mg)的甲醇(0.2ml)悬浮液。搅拌30分钟之后,用二氯甲烷(2ml)稀释该反应,并用饱和碳酸氢钠水溶液(1ml)淬灭。分离有机层,用硫酸镁干燥,过滤,并浓缩,得到标题化合物。ms(elsd)m/e860.5(m+h)+。
1.5.2. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[(2-甲氧基乙基)氨基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸
将实施例1.5.1的二氯甲烷(1ml)溶液用三氟乙酸(0.5ml)处理。搅拌过夜之后,浓缩该反应,溶于n,n-二甲基甲酰胺(1.5ml)和水(0.5ml)中,用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物的tfa盐。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,2h),8.39(s,2h),8.03(d,1h),7.79(d,1h),7.62(d,1h),7.53-7.42(m,3h),7.40-7.33(m,2h),7.29(s,1h),6.96(d,1h),4.96(s,2h),3.89(t,2h),3.83(s,2h),3.61-3.53(m,10h),3.29(s,3h),3.17-3.09(m,2h),3.09-2.97(m,4h),2.10(s,3h),1.41(s,2h),1.35-1.23(m,4h),1.20-1.10(m,4h),1.10-0.98(m,2h)。ms(esi)m/e804.3(m+h)+。
1.6. 合成3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸(化合物w1.06)
1.6.1. 3-氰基甲基-4-氟苯甲酸甲基酯
用20分钟向三甲硅烷甲腈(1.49ml)的四氢呋喃(2.5ml)溶液中逐滴加入1m四丁基氟化铵(11.13ml)。然后,将该溶液在室温下搅拌30分钟。将4-氟-3-(溴甲基)苯甲酸甲酯(2.50g)溶于乙腈(12ml)中,并用10分钟逐滴加入到第一个溶液中。然后,将该溶液加热到80℃,保持60分钟,并冷却。减压浓缩该溶液,用硅胶快速柱色谱纯化,用20-30%的乙酸乙酯/庚烷洗脱。减压蒸发溶剂,提供标题化合物。
1.6.2. 3-(2-氨乙基)-4-氟苯甲酸甲基酯
将实施例1.6.1(1.84g)溶于四氢呋喃(50ml)中,并加入1m硼烷(在四氢呋喃中,11.9ml)。将该溶液在室温下搅拌16小时,并慢慢地用甲醇淬灭。加入4m盐酸水溶液(35ml),并将该溶液在室温下搅拌16小时。减压浓缩该混合物,并使用固体碳酸钾,将ph值调节至11和12之间。然后,用二氯甲烷(3x100ml)提取该溶液。将有机提取物合并,并用无水硫酸钠干燥。过滤该溶液,减压浓缩,并将物质用硅胶快速柱色谱纯化,用10-20%的甲醇/二氯甲烷洗脱。减压蒸发溶剂,提供标题化合物。ms(esi)m/e198(m+h)+。
1.6.3. 4-氟-3-[2-(2,2,2-三氟乙酰氨基)乙基]苯甲酸甲基酯
将实施例1.6.2(1.207g)溶于二氯甲烷(40ml)中,并加入n,n-二异丙基乙胺(1.3ml)。然后,逐滴加入三氟乙酸酐(1.0ml)。将该溶液搅拌15分钟。加入水(40ml),并将该溶液用乙酸乙酯(100ml)稀释。加入1m盐酸水溶液(50ml),分离有机层,用1m盐酸水溶液洗涤,而后用盐水洗涤。用无水硫酸钠干燥该溶液。过滤后,减压蒸发溶剂,提供标题化合物。
1.6.4. 5-氟-2-(2,2,2-三氟乙酰基)-1,2,3,4-四氢异喹啉-8-甲酸甲基酯
将实施例1.6.3(1.795g)和多聚甲醛(0.919g)放在烧瓶中,并加入浓硫酸(15ml)。将该溶液在室温下搅拌一个小时。加入冷水(60ml),并将该溶液用乙酸乙酯(2x100ml)提取。将提取物合并,用饱和碳酸氢钠水溶液(100ml)和水(100ml)洗涤,并用无水硫酸钠干燥。过滤该溶液,减压浓缩,并将物质用硅胶快速柱色谱纯化,用10-20%的乙酸乙酯/庚烷洗脱。减压蒸发溶剂,提供标题化合物。ms(esi)m/e323(m+nh4)+。
1.6.5. 5-氟-1,2,3,4-四氢异喹啉-8-甲酸甲基酯
将实施例1.6.4(685mg)溶于甲醇(6ml)和四氢呋喃(6ml)中。加入水(3ml),而后加入碳酸钾(372mg)。将该反应在室温下搅拌三小时,而后用乙酸乙酯(100ml)稀释。用饱和碳酸氢钠水溶液洗涤该溶液,并用无水硫酸钠干燥。滤出溶剂,减压蒸发,提供标题化合物。ms(esi)m/e210(m+h)+。
1.6.6. 2-(5-溴-6-叔丁氧羰基吡啶-2-基)-5-氟-1,2,3,4-四氢异喹啉-8-甲酸甲基酯
在1.1.12中,用实施例1.6.5替代1,2,3,4-四氢异喹啉-8-甲酸甲酯盐酸盐,制备标题化合物。ms(esi)m/e465,467(m+h)+。
1.6.7. 2-[6-叔丁氧羰基-5-(4,4,5,5-四甲基-[1,3,2]二氧杂硼杂环戊烷-2-基)-吡啶-2-基]-5-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.13中,用实施例1.6.6替代实施例1.1.12,制备标题化合物。ms(esi)m/e513(m+h)+。
1.6.8. 2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙胺
在微波条件(biotageinitiator)下,将实施例1.1.7(4.5g)的7n铵/甲醇(15ml)溶液在100℃下搅拌20分钟。将该反应混合物真空浓缩,用乙酸乙酯(400ml)稀释残余物,并用nahco3水溶液、水(60ml)和盐水(60ml)洗涤。用无水na2so4干燥有机层,过滤,并浓缩。残余物不用进一步纯化,直接在下一个反应中使用。ms(esi)m/e444.2(m+h)+。
1.6.9. (2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)-5,7-二甲基金刚烷-1-基)氧基)乙基)氨基甲酸叔丁基酯
向实施例1.6.8(4.4g)的四氢呋喃(100ml)溶液中加入二碳酸二叔丁基酯(2.6g)和n,n-二甲基-4-氨基吡啶(100mg)。将该混合物搅拌1.5小时。然后,用乙酸乙酯(300ml)稀释该反应混合物,并用nahco3水溶液、水(60ml)和盐水(60ml)洗涤。干燥之后(无水na2so4),过滤该溶液,浓缩,并将残余物用硅胶柱色谱纯化(20%的乙酸乙酯/二氯甲烷),得到标题化合物。ms(esi)m/e544.2(m+h)+。
1.6.10. 2-(6-叔丁氧羰基-5-{1-[5-(2-叔丁氧羰基氨基-乙氧基)-3,7-二甲基-金刚烷-1-基甲基]-5-甲基-1h-吡唑-4-基}-吡啶-2-基)-5-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.14中,用实施例1.6.7替代实施例1.1.13,实施例1.6.9替代实施例1.1.9,制备标题化合物。ms(esi)m/e802(m+h)+。
1.6.11. 2-(6-叔丁氧羰基-5-{1-[5-(2-叔丁氧羰基氨基-乙氧基)-3,7-二甲基-金刚烷-1-基甲基]-5-甲基-1h-吡唑-4-基}-吡啶-2-基)-5-氟-1,2,3,4-四氢-异喹啉-8-甲酸
在实施例1.1.15中,用实施例1.6.10替代实施例1.1.14,制备标题化合物。ms(esi)m/e788(m+h)+。
1.6.12. 6-[8-(苯并噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢-1h-异喹啉-2-基]-3-{1-[5-(2-叔丁氧羰基氨基-乙氧基)-3,7-二甲基-金刚烷-1-基甲基]-5-甲基-1h-吡唑-4-基}-吡啶-2-甲酸叔丁基酯
在实施例1.1.16中,用实施例1.6.11替代实施例1.1.15,制备标题化合物。ms(esi)m/e920(m+h)+。
1.6.13. 3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
在实施例1.1.17中,用实施例1.6.12替代实施例1.1.16,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.88(bs,1h),8.03(d,1h),7.79(d,1h),7.73(m,1h),7.63(m,2h),7.52(d,1h),7.48(t,1h),7.36(t,1h),7.28(dd,2h),7.04(d,1h),5.02(s,2h),3.95(t,2h),3.83(s,2h),3.49(t,2h),2.90(m,4h),2.11(s,3h),1.41(s,2h),1.35-1.23(m,4h),1.19-0.99(m,6h),0.87(bs,6h)。ms(esi)m/e764(m+h)+。
1.7 合成3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-6-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
1.7.1 (3-溴-5-氟-苯基)-乙腈
在实施例1.6.1中,用1-溴-3-(溴甲基)-5-氟苯替代4-氟-3-(溴甲基)苯甲酸甲酯,制备标题化合物。
1.7.2 2-(3-溴-5-氟-苯基)-乙胺
在实施例1.6.2中,用实施例1.7.1替代实施例1.6.1,制备标题化合物。
1.7.3 [2-(3-溴-5-氟-苯基)-乙基]-氨基甲酸叔丁基酯
将实施例1.7.2(1.40g)和n,n-二甲基吡啶-4-胺(0.078g)溶于乙腈(50ml)中。加入二碳酸二叔丁基酯(1.54g)。将该溶液在室温下搅拌30分钟。将该溶液用乙醚(150ml)稀释,用0.1mhcl水溶液(25ml)洗涤两次,用盐水(50ml)洗涤,并用无水硫酸钠干燥。过滤该溶液,减压浓缩,并将粗品用硅胶快速柱色谱纯化,用5-10%的乙酸乙酯/庚烷洗脱。减压蒸发溶剂,提供标题化合物。
1.7.4 3-(2-叔丁氧羰基氨基-乙基)-5-氟-苯甲酸甲基酯
将实施例1.7.3(775mg)和二氯[1,1'-双(二苯基膦基)二茂铁]钯(ii)(36mg)加入到50ml耐压瓶中。加入甲醇(10ml)和三甲胺(493mg)。将该溶液脱气,并用氩气吹扫三次,而后脱气,用一氧化碳吹扫。在60psi一氧化碳条件下,将该反应加热至100℃,保持16小时。再加入二氯[1,1'-双(二苯基膦基)二茂铁]钯(ii)(36mg),并重复脱气和吹扫过程。在60psi一氧化碳条件下,将该反应加热至100℃,再保持16小时。减压除去溶剂,并将残余物用硅胶快速柱色谱纯化,用20-30%的乙酸乙酯/庚烷洗脱。减压蒸发溶剂,提供标题化合物。
1.7.5 3-(2-氨基-乙基)-5-氟-苯甲酸甲基酯
将实施例1.7.4(292mg)溶于二氯甲烷(3ml)中。加入2,2,2-三氟乙酸(1680mg),并将该溶液在室温下搅拌两个小时。减压除去溶剂,提供标题化合物,其不用进一步纯化,在下一步中直接使用。
1.7.6 3-氟-5-[2-(2,2,2-三氟-乙酰氨基)-乙基]-苯甲酸甲基酯
在实施例1.6.3中,用实施例1.7.5替代实施例1.6.2,制备标题化合物。
1.7.7 6-氟-2-(2,2,2-三氟-乙酰基)-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.6.4中,用实施例1.7.6替代实施例1.6.3,制备标题化合物。
1.7.8 6-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.6.5中,用实施例1.7.7替代实施例1.6.4,制备标题化合物。
1.7.9 2-(5-溴-6-叔丁氧羰基-吡啶-2-基)-6-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.12中,用实施例1.7.8替代1,2,3,4-四氢异喹啉-8-甲酸甲酯盐酸盐,制备标题化合物。ms(esi)m/e464,466(m+h)+。
1.7.10. 2-[6-叔丁氧羰基-5-(4,4,5,5-四甲基-[1,3,2]二氧杂硼杂环戊烷-2-基)-吡啶-2-基]-6-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.13中,用实施例1.7.9替代实施例1.1.12,制备标题化合物。ms(esi)m/e513(m+h)+,543(m+meoh-h)-。
1.7.11. 亚胺二甲酸{2-[5-(4-碘代-5-甲基-吡唑-1-基甲基)-3,7-二甲基-金刚烷-1-基氧基]-乙基}-二叔丁基酯
将实施例1.1.6(5.000g)溶于二氯甲烷(50ml)中。加入三乙胺(1.543g),并将该溶液在冰浴中冷却。逐滴加入甲磺酰氯(1.691g)。将该溶液升温至室温,并搅拌30分钟。加入饱和碳酸氢钠水溶液(50ml)。分离各层,并将有机层用盐水(50ml)洗涤。然后,将水溶液部分合并,并用二氯甲烷(50ml)反萃取。将有机部分合并,用无水硫酸钠干燥,过滤,并浓缩。将残余物溶于乙腈(50ml)中。加入亚胺二甲酸二叔丁基酯(2.689g)和碳酸铯(7.332g),并将该溶液回流16小时。冷却该溶液,并加入到乙醚(100ml)和水(100ml)中。分离各层。将有机部分用盐水(50ml)洗涤。然后,将水溶液部分合并,并用乙醚(100ml)反萃取。将有机部分合并,用无水硫酸钠干燥,过滤,并减压浓缩。将物质用硅胶快速柱色谱纯化,用20%的乙酸乙酯/庚烷洗脱。减压蒸发溶剂,提供标题化合物。ms(esi)m/e666(m+na)+。
1.7.12. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-6-氟-1,2,3,4-四氢异喹啉-8-甲酸甲酯
在实施例1.1.14中,用实施例1.7.10替代实施例1.1.13,实施例1.7.11替代实施例1.1.9,制备标题化合物。ms(esi)m/e902(m+h)+。
1.7.13. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-6-氟-1,2,3,4-四氢异喹啉-8-甲酸
在实施例1.1.15中,用实施例1.7.12替代实施例1.1.14,制备标题化合物。ms(esi)m/e888(m+h)+,886(m-h)-。
1.7.14. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-6-氟-3,4-二氢异喹啉-2(1h)-基)-3-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸叔丁基酯
在实施例1.1.16中,用实施例1.7.13替代实施例1.1.15,制备标题化合物。
1.7.15. 3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-6-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
在实施例1.1.17中,用实施例1.7.14替代实施例1.1.16,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm8.04(d,1h),7.79(d,1h),7.65(bs,3h),7.50(m,2h),7.40-7.29(m,3h),6.98(d,1h),4.91(d,2h),3.88(t,2h),3.83(s,2h),3.02(t,2h),2.89(t,4h),2.10(s,3h),1.44-1.20(m,6h),1.19-1.00(m,6h),0.86(bs,6h)。ms(esi)m/e764(m+h)+,762(m-h)-。
1.8 合成3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-7-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
1.8.1 [2-(3-溴-4-氟-苯基)-乙基]-氨基甲酸叔丁基酯
在实施例1.7.3中,用2-(3-溴-4-氟苯基)乙胺盐酸盐替代实施例1.7.2,制备标题化合物。
1.8.2 5-(2-叔丁氧羰基氨基-乙基)-2-氟-苯甲酸甲基酯
在实施例1.7.4中,用实施例1.8.1替代实施例1.7.3,制备标题化合物。ms(esi)m/e315(m+nh4)+。
1.8.3 5-(2-氨基-乙基)-2-氟-苯甲酸甲基酯
在实施例1.7.5中,用实施例1.8.2替代实施例1.7.4,制备标题化合物。
1.8.4 2-氟-5-[2-(2,2,2-三氟-乙酰氨基)-乙基]-苯甲酸甲基酯
在实施例1.6.3中,用实施例1.8.3替代实施例1.6.2,制备标题化合物。
1.8.5 7-氟-2-(2,2,2-三氟-乙酰基)-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.6.4中,用实施例1.8.4替代实施例1.6.3,制备标题化合物。ms(esi)m/e323(m+nh4)+。
1.8.6 7-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.6.5中,用实施例1.8.5替代实施例1.6.4,制备标题化合物。ms(esi)m/e210(m+h)+,208(m-h)-。
1.8.7 2-(5-溴-6-叔丁氧羰基-吡啶-2-基)-7-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.12中,用实施例1.8.6替代1,2,3,4-四氢异喹啉-8-甲酸甲酯盐酸盐,制备标题化合物。ms(esi)m/e465,467(m+h)+。
1.8.8 2-[6-叔丁氧羰基-5-(4,4,5,5-四甲基-[1,3,2]二氧杂硼杂环戊烷-2-基)-吡啶-2-基]-7-氟-1,2,3,4-四氢-异喹啉-8-甲酸甲基酯
在实施例1.1.13中,用实施例1.8.7替代实施例1.1.12,制备标题化合物。ms(esi)m/e513(m+h)+,543(m+meoh-h)-。
1.8.9 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-7-氟-1,2,3,4-四氢异喹啉-8-甲酸甲酯
在实施例1.1.14中,用实施例1.8.8替代实施例1.1.13,实施例1.7.11替代实施例1.1.9,制备标题化合物。ms(esi)m/e902(m+h)+,900(m-h)-。
1.8.10. 2-(6-(叔丁氧羰基)-5-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶-2-基)-7-氟-1,2,3,4-四氢异喹啉-8-甲酸
在实施例1.1.15中,用实施例1.8.9替代实施例1.1.14,制备标题化合物。ms(esi)m/e788(m+h)+,786(m-h)-。
1.8.11. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-7-氟-3,4-二氢异喹啉-2(1h)-基)-3-(1-((3-(2-(二-(叔丁氧羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸叔丁基酯
在实施例1.1.16中,用实施例1.8.10替代实施例1.1.15,制备标题化合物。
1.8.12. 3-(1-{[3-(2-氨基乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)-6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-7-氟-3,4-二氢异喹啉-2(1h)-基]吡啶-2-甲酸
在实施例1.1.17中,用实施例1.8.11替代实施例1.1.16,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm13.08(bs,1h),11.41(bs,1h),8.05(d,1h),7.81(d,1h),7.63(m,4h),7.55-7.22(m,6h),6.95(d,1h),4.78(s,2h),3.86(m,4h),3.50(m,2h),2.97(m,2h),2.90(m,2h),2.09(s,3h),1.48-1.40(m,2h),1.38-1.23(m,4h),1.20-1.01(m,6h),0.88(bs,6h)。ms(esi)m/e764(m+h)+,762(m-h)。
实施例2.合成示范性的合成子
该实施例提供了可以用于制备adc的示范性的合成子的合成方法。
2.1. 合成n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子e)
2.1.1. (s)-(9h-芴-9-基)甲基(1-((4-(羟甲基)苯基)氨基)-1-氧代-5-脲基戊-2-基)氨基甲酸酯
将(s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-5-脲基戊酸(40g)溶于二氯甲烷(1.3l)中。将(4-氨基苯基)甲醇(13.01g)、2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(42.1g)和n,n-二异丙基乙胺(0.035l)加入到该溶液中,并将得到的混合物在室温下搅拌16小时。过滤收集产物,并用二氯甲烷冲洗。将合并的固体真空干燥,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e503.3(m+h)+。
2.1.2. (s)-2-氨基-n-(4-(羟甲基)苯基)-5-脲基戊酰胺
将实施例2.1.1(44g)溶于n,n-二甲基甲酰胺(300ml)中。将该溶液用二乙胺(37.2ml)处理,并在室温下搅拌一个小时。过滤该反应混合物,并将溶剂减压浓缩。用碱性氧化铝色谱纯化粗品,用0-30%的甲醇/乙酸乙酯的梯度进行洗脱,得到标题化合物。ms(esi)m/e281.2(m+h)+。
2.1.3. ((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代-5-脲基戊-2-基)氨基)-3-甲基-1-氧代丁-2-基)氨基甲酸叔丁基酯
将(s)-2-(叔丁氧羰基氨基)-3-甲基丁酸(9.69g)溶于n,n-二甲基甲酰胺(200ml)中。向该溶液中加入2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(18.65g),并将该反应在室温下搅拌一个小时。加入实施例2.1.2(12.5g)和n,n-二异丙基乙胺(15.58ml),并将该反应混合物在室温下搅拌16小时。减压浓缩溶剂,用硅胶色谱纯化残余物,用10%的甲醇/二氯甲烷洗脱,得到标题化合物。ms(esi)m/e480.2(m+h)+。
2.1.4. (s)-2-((s)-2-氨基-3-甲基丁酰胺基)-n-(4-(羟甲基)苯基)-5-脲基戊酰胺
将实施例2.1.4(31.8g)溶于二氯甲烷(650ml)中,并向该溶液中加入三氟乙酸(4.85ml)。将该反应混合物在室温下搅拌三小时。减压浓缩溶剂,得到粗品标题化合物和4-((s)-2-((s)-2-氨基-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基2,2,2-三氟乙酸酯的混合物。将粗品溶于1:1的二噁烷/水溶液(300ml)中,并向该溶液中加入氢氧化钠(5.55g)。将该混合物在室温下搅拌三小时。真空浓缩溶剂,用反相hplc纯化粗品,使用combiflash系统,用5-60%的乙腈/水(含有0.05%v/v氢氧化铵)的梯度进行洗脱,得到标题化合物。ms(esi)m/e380.2(m+h)+。
2.1.5. 1-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)-n-((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代-5-脲基戊-2-基)氨基)-3-甲基-1-氧代丁-2-基)-3,6,9,12-四氧杂十五烷-15-酰胺
向实施例2.1.4(1.5g)的n,n-二甲基甲酰胺(50ml)溶液中加入2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯(2.03g)。将该混合物在室温下搅拌三天。将粗品加入到反相柱(c18,sf65-800g)上,用20-100%的乙腈/水(含有0.1%三氟乙酸)洗脱,得到标题化合物。ms(esi)m/e778.3(m+1)+。
2.1.6. 4-((2s,5s)-25-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-5-异丙基-4,7,23-三氧代-2-(3-脲基丙基)-10,13,16,19-四氧杂-3,6,22-三氮杂二十五烷酰胺基)苄基(4-硝基苯基)碳酸酯
向实施例2.1.5(2.605g)和n,n-二异丙胺(1.8ml)的n,n-二甲基甲酰胺(20ml)溶液中加入双(4-硝基苯基)碳酸酯(1.23g)。将该混合物在室温下搅拌16小时。将粗品加入到反相柱(c18,sf65-800g)上,用20-100%的乙腈/水(含有0.1%三氟乙酸)洗脱,得到标题化合物。ms(esi)m/e943.2(m+1)+。
2.1.7. n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺
在0℃,向实施例2.1.6(49.6mg)和实施例1.1.17(30mg)的n,n-二甲基甲酰胺(2ml)混合物中加入n,n-二异丙基乙胺(0.018ml)。将该反应混合物在室温下搅拌过夜,用二甲亚砜稀释,用rp-hplc纯化,使用gilson系统,用20-70%的乙腈/0.1%三氟乙酸水溶液洗脱,提供标题化合物。ms(esi)m/e1563.4(m+h)+。
2.2. 合成n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子d)
向4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺基)-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基4-硝基苯基碳酸酯(购买于synchem,57mg)和实施例1.1.17(57mg)的n,n-二甲基甲酰胺(6ml)溶液中加入n,n-二异丙基乙胺(0.5ml)。将该混合物搅拌过夜,而后真空浓缩。用甲醇(3ml)和乙酸(0.3ml)稀释残余物,用rp-hplc(gilson系统,c18柱)纯化,用30-70%的乙腈/水(含有0.1%三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。1hnmr(300mhz,二甲亚砜-d6)δppm12.86(d,1h),9.98(s,1h),7.96-8.10(m,2h),7.74-7.83(m,2h),7.54-7.64(m,3h),7.31-7.52(m,6h),7.24-7.29(m,3h),6.99(s,2h),6.94(d,1h),4.96(d,4h),4.33-4.43(m,2h),4.12-4.24(m,2h),3.22-3.42(m,7h),2.77-3.07(m,7h),1.86-2.32(m,7h),0.92-1.70(m,22h),0.72-0.89(m,13h)。ms(esi)m/e1358.2(m+h)+。
2.3. 合成n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺(合成子j)
2.3.1. (s)-2-((s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)丙酸
将(s)-2,5-二氧代吡咯烷-1-基2-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酸酯(5g)的40ml二甲氧基乙烷溶液加入到l-丙氨酸(1.145g)和碳酸氢钠(1.08g)水(40ml)溶液中。将该反应混合物在室温下搅拌16小时。将柠檬酸水溶液(15%v/v,75ml)加入到该反应中。滤出沉淀,用水(2x250ml)洗涤,并真空干燥。将固体进一步与乙醚(100ml)一起研磨,过滤,并用硫酸钠干燥,得到产物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e383.0(m+h)+。
2.3.2. (9h-芴-9-基)甲基((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-1-氧代丙-2-基)氨基甲酸酯
将n-乙氧羰基-2-乙氧基-1,2-二氢喹啉(eedq)(6.21g)加入到实施例2.3.1(3.2g)和4-氨基苄醇(1.546g)的50ml二氯甲烷∶甲醇(2∶1)溶液中。将该反应在室温下搅拌2天。真空浓缩溶剂。将残余物与75ml乙酸乙酯一起研磨,过滤收集固体,真空干燥,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e488.0(m+h)+。
2.3.3. (s)-2-氨基-n-((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)丙酰胺
将二乙胺(11.75ml)加入到实施例2.3.2(1.58g)的n,n二甲基甲酰胺(50ml)溶液中,并将该反应在室温下静置16小时。真空蒸发溶剂。将残余物与乙酸乙酯(100ml)一起研磨,过滤收集产物,真空干燥,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e266.0(m+h)+。
2.3.4. 1-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)-n-((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-1-氧代丙-2-基)-3,6,9,12-四氧杂十五烷-15-酰胺
将实施例2.3.3(1.033g)与2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯(2g)在含有1%n,n-二异丙基乙胺的n,n-二甲基甲酰胺(19.5ml)中混合16小时。用反相hplc纯化粗品反应物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(esi)m/e664.0(m+h)+。
2.3.5. 4-((2s,5s)-25-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2,5-二甲基-4,7,23-三氧代-10,13,16,19-四氧杂-3,6,22-三氮杂二十五烷酰胺基)苄基(4-硝基苯基)碳酸酯
将实施例2.3.4(1.5g)与双(4-硝基苯基)碳酸酯(1.38g)在含有1%n,n-二异丙基乙胺的n,n-二甲基甲酰胺(11.3ml)中混合。将该反应在室温下搅拌16小时。用反相hplc纯化粗品反应物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(esi)m/e829.0(m+h)+。
2.3.6. n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
将实施例1.1.17的三氟乙酸盐(15mg)与实施例2.3.5(21.3mg)在n,n-二甲基甲酰胺(1ml)和n,n-二异丙基乙胺(0.006ml)中混合。将该反应混合物在室温下搅拌一个小时。用反相hplc纯化粗品反应物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(esi)m/e1450.7(m+h)+。
2.4. 合成n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺(合成子k)
2.4.1. 6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-1-氧代丙-2-基)己酰胺
在实施例2.3.4中,用n-琥珀酰亚胺基6-马来酰亚胺基(maleimido)己酸酯替代2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯,制备标题化合物。ms(esi)m/e640.8(m+nh4)+。
2.4.2. 4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺基)丙酰胺基)丙酰胺基)苄基(4-硝基苯基)碳酸酯
在实施例2.3.5中,用实施例2.4.1替代实施例2.3.4,制备标题化合物。
2.4.3. n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
在实施例2.3.6中,用实施例2.4.2替代实施例2.3.5,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm9.56(s,1h),7.98(d,1h),7.76(d,1h),7.71-7.52(m,3h),7.51-7.21(m,4h),6.97-6.84(m,1h),4.98(d,2h),4.42(p,1h),4.27(p,1h),3.89(t,1h),3.80(s,2h),3.43(d,19h),3.03(t,7h),2.87(s,2h),2.32(s,1h),2.11(d,3h),1.52(h,2h),1.41-0.94(m,12h),0.84(s,3h)。ms(esi)m/e1244.2(m+h)+。
2.5. 合成n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-缬氨酰-n-{4-[12-({(1s,3s)-3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子l)
2.5.1. (3-溴金刚烷-1-基)甲醇
在实施例1.1.2中,用3-溴金刚烷-1-甲酸替代实施例1.1.1,制备标题化合物。
2.5.2. 1-((3-溴金刚烷-1-基)甲基)-1h-吡唑
在实施例1.1.3中,用实施例2.5.1替代实施例1.1.2,制备标题化合物。ms(esi)m/e295.2(m+h)+。
2.5.3. 2-(2-(2-((3-((1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
在实施例1.2.1中,用实施例2.5.2替代实施例1.1.3,用硫酸银替代三乙胺,制备标题化合物。ms(esi)m/e365.1(m+h)+。
2.5.4. 2-(2-(2-((3-((5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
在实施例1.2.2中,用实施例2.5.3替代实施例1.2.1,制备标题化合物。ms(esi)m/e379.1(m+h)+。
2.5.5. 2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙醇
在实施例1.2.3中,用实施例2.5.4替代实施例1.2.2,制备标题化合物。ms(esi)m/e504.9(m+h)+。
2.5.6. 2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)-n-甲基乙胺
在实施例1.2.4中,用实施例2.5.5替代实施例1.2.3,制备标题化合物。ms(esi)m/e518.4(m+h)+。
2.5.7. (2-(2-(2-((3-((4-碘代-5-甲基-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙基)(甲基)氨基甲酸叔丁基酯
在实施例1.2.5中,用实施例2.5.6替代实施例1.2.4,制备标题化合物。ms(esi)m/e617.9(m+h)+。
2.5.8. 甲基(2-(2-(2-((3-((5-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1h-吡唑-1-基)甲基)金刚烷-1-基)氧基)乙氧基)乙氧基)乙基)氨基甲酸叔丁基酯
在实施例1.2.6中,用实施例2.5.7替代实施例1.2.5,制备标题化合物。ms(esi)m/e618.2(m+h)+。
2.5.9. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(5-甲基-1-((3-((2,2,5-三甲基-4-氧代-3,8,11-三氧杂-5-氮杂十三烷-13-基)氧基)金刚烷-1-基)甲基)-1h-吡唑-4-基)吡啶甲酸叔丁基酯
在实施例1.2.10中,用实施例2.5.8替代实施例1.2.6,制备标题化合物。ms(esi)m/e976.1(m+h)+。
2.5.10. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(5-甲基-1-(((1s,3s)-3-(2-(2-(2-(甲基氨基)乙氧基)乙氧基)乙氧基)金刚烷-1-基)甲基)-1h-吡唑-4-基)吡啶甲酸
在实施例1.2.11中,用实施例2.5.9替代实施例1.2.10,制备标题化合物。ms(esi)m/e820.3(m+h)+。
2.5.11. n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-缬氨酰-n-{4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺
在实施例2.2中,用实施例2.5.10替代实施例1.1.17,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm9.96(br.s,1h),7.96-8.12(m,2h),7.73-7.83(m,2h),7.29-7.66(m,9h),7.17-7.30(m,3h),6.89-7.01(m,2h),4.86-5.01(m,4h),4.28-4.45(m,1h),4.12-4.21(m,1h),3.69-3.92(m,3h),3.27-3.62(m,9h),2.78-3.06(m,7h),2.01-2.23(m,7h),1.87-2.01(m,1h),1.54-1.72(m,4h),1.01-1.54(m,22h),0.72-0.89(m,6h)。ms(esi)m/e1418.4(m+h)+。
2.6. 合成n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-l-缬氨酰-n-{4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子m)
在实施例2.1.7中,用实施例2.5.10替代实施例1.1.17,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm9.97(s,1h),8.07-8.13(m,1h),7.97-8.05(m,2h),7.86(d,1h),7.78(d,1h),7.55-7.63(m,3h),7.40-7.51(m,3h),7.32-7.38(m,2h),7.25-7.30(m,2h),6.98(s,1h),6.93(d,1h),4.91-5.01(m,4h),4.31-4.41(m,1h),4.17-4.24(m,1h),3.83-3.91(m,2h),3.76(s,2h),3.30-3.62(m,21h),3.10-3.17(m,1h),2.89-3.05(m,4h),2.81-2.88(m,3h),2.42-2.47(m,1h),2.27-2.40(m,3h),2.04-2.15(m,5h),1.91-2.00(m,1h),1.30-1.72(m,16h),0.76-0.88(m,6h)。ms(esi)m/e1623.3(m+h)+。
2.7. 合成n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-l-缬氨酰-n-{4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子v)
在实施例2.2中,用实施例1.2.11替代实施例1.1.17,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm9.61(s,1h),7.97(d,1h),7.76(d,1h),7.67(d,1h),7.61(d,1h),7.51-7.57(m,2h),7.38-7.48(m,4h),7.29-7.36(m,2h),7.23-7.28(m,3h),6.86-6.94(m,2h),4.97(d,4h),4.38-4.45(m,1h),4.12-4.19(m,1h),3.89(t,2h),3.80(s,2h),3.47-3.54(m,5h),3.44(s,3h),3.33-3.41(m,6h),2.93-3.06(m,6h),2.87(s,2h),2.11-2.22(m,2h),2.08(s,3h),1.97-2.05(m,1h),1.70-1.81(m,2h),1.33-1.68(m,10h),0.95-1.32(m,14h),0.80-0.91(m,13h)。ms(+esi)m/e1446.3(m+h)+。
2.8. 合成n-({2-[2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基]乙氧基}乙酰基)-l-缬氨酰-n-{4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子ds)
2.8.1. (s)-2-((s)-2-(2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙酰胺基)-3-甲基丁酰胺基)-n-(4-(羟甲基)苯基)-5-脲基戊酰胺
在实施例2.1.5中,用2,5-二氧代吡咯烷-1-基2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙酸酯替代2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯,制备标题化合物。
2.8.2. 4-((2s,5s)-14-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-5-异丙基-4,7-二氧代-2-(3-脲基丙基)-9,12-二氧杂-3,6-二氮杂十四烷酰胺基)苄基(4-硝基苯基)碳酸酯
在实施例2.3.5中,用实施例2.8.1替代实施例2.3.4,制备标题化合物。
2.8.3. n-({2-[2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基]乙氧基}乙酰基)-l-缬氨酰-n-{4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺
在实施例2.2中,用实施例1.2.11替代实施例1.1.17,用实施例2.8.2替代4-((s)-2-((s)-2-(6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰胺基)-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基4-硝基苯基碳酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm9.64(s,1h),7.97(d,1h),7.92(d,1h),7.75(d,1h),7.60(d,1h),7.54(d,2h),7.45(d,2h),7.38-7.43(m,1h),7.29-7.36(m,2h),7.22-7.28(m,4h),6.88-6.93(m,2h),4.98(d,4h),4.39-4.46(m,1h),4.24-4.31(m,1h),3.86-3.93(m,4h),3.80(s,2h),3.46-3.61(m,15h),3.43-3.45(m,5h),3.33-3.38(m,4h),2.87(s,3h),1.99-2.11(m,4h),1.56-1.80(m,2h),1.34-1.50(m,4h),0.94-1.32(m,11h),0.80-0.91(m,13h)。ms(+esi)m/e1478.3(m+h)。
2.9. 该段落特意留白。
2.10. 合成n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子bg)
2.10.1. (s)-2-((s)-2-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)-3-甲基丁酰胺基)-n-(4-(羟甲基)苯基)-5-脲基戊酰胺
将实施例2.1.4(3g)和2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯(1.789g)溶于甲醇(30ml)中,并在室温下搅拌三小时。减压浓缩溶剂,并将残余物用硅胶色谱纯化,用5-30%的甲醇/二氯甲烷的梯度进行洗脱,得到标题化合物。ms(esi)m/e531.0(m+h)+。
2.10.2. 4-((s)-2-((s)-2-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基(4-硝基苯基)碳酸酯
将双(4-硝基苯基)碳酸酯(2.293g)、n,n-二异丙基乙胺(1.317ml)和实施例2.10.1(2g)溶于n,n-二甲基甲酰胺(30ml)中,并在室温下搅拌16小时。减压浓缩溶剂,并将残余物用硅胶色谱纯化,用0-10%的甲醇/二氯甲烷的梯度进行洗脱,得到标题化合物。ms(esi)m/e696.9(m+h)+。
2.10.3. n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺
在实施例2.9.5中,用实施例2.10.2替代实施例2.9.4,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.86(bs,1h),9.95(s,1h),8.10(d,1h),8.01(dd,2h),7.79(d,1h),7.65-7.56(m,3h),7.55-7.40(m,3h),7.40-7.33(m,2h),7.35-7.24(m,3h),6.99(s,2h),6.95(d,1h),4.42-4.28(m,1h),4.15(dd,1h),3.92-3.85(m,2h),3.83-3.77(m,2h),3.77-3.52(m,2h),3.45-3.38(m,2h),3.30-3.23(m,2h),3.08-2.90(m,4h),2.90-2.81(m,3h),2.09(s,3h),2.02-1.86(m,1h),1.79-1.52(m,2h),1.52-0.92(m,15h),0.91-0.75(m,13h)。ms(esi)m/e1316.1(m+h)+。
2.11. 该段落特意留白。
2.12. 合成n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺(合成子bi)
2.12.1. 3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-n-((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-1-氧代丙-2-基)丙酰胺
将实施例2.3.3(9g)和2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯(9.03g)在n,n-二甲基甲酰胺(50ml)中的混合物、在室温下搅拌16小时。用水稀释该反应混合物。将水层用二氯甲烷(3x100ml)反萃取。真空浓缩有机溶剂。将得到的粗品吸收到硅胶上,用硅胶色谱纯化,用50:1的二氯甲烷/甲醇洗脱,得到标题化合物。ms(esi)m/e439.1(m+na)+。
2.12.2. 4-((s)-2-((s)-2-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)丙酰胺基)丙酰胺基)苄基(4-硝基苯基)碳酸酯
在实施例2.10.2中,用实施例2.12.1替代实施例2.10.1,制备标题化合物。用硅胶色谱纯化产物,用25%的四氢呋喃/二氯甲烷洗脱。ms(esi)m/e604.0(m+h)+。
2.12.3. n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-l-丙氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
在实施例2.9.5中,用实施例2.12.2替代实施例2.9.4,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm9.51(s,1h),7.97(dd,1h),7.90-7.83(m,1h),7.76(d,1h),7.72-7.66(m,1h),7.64-7.57(m,1h),7.60-7.55(m,1h),7.55(s,1h),7.48-7.37(m,3h),7.37-7.29(m,2h),7.29-7.22(m,3h),6.91(d,1h),6.88(s,1h),4.98(s,2h),4.96(bs,2h),4.40(p,1h),4.24(p,1h),3.89(t,2h),3.79(s,2h),3.64(t,2h),3.44(t,2h),3.29-3.14(m,2h),3.02(t,2h),2.86(s,3h),2.08(s,3h),1.36(bs,2h),1.31(d,3h),1.29-0.94(m,14h),0.83(s,6h)。ms(esi)m/e1202.1(m+h)+。
2.13. 该段落特意留白。
2.14. 该段落特意留白。
2.15. 该段落特意留白。
2.16. 该段落特意留白。
2.17. 合成n-[(2r)-4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2-磺基丁酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子bo)
2.17.1. 3-(1-((3-(2-((((4-((s)-2-((s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.3.6中,用(9h-芴-9-基)甲基((s)-3-甲基-1-(((s)-1-((4-((((4-硝基苯氧基)羰基)氧基)甲基)苯基)氨基)-1-氧代-5-脲基戊-2-基)氨基)-1-氧代丁-2-基)氨基甲酸酯替代实施例2.3.5,制备标题化合物。ms(esi)m/e1387.3(m+h)+。
2.17.2. 3-(1-((3-(2-((((4-((s)-2-((s)-2-氨基-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例2.17.1(15mg)与30%的二乙胺/n,n-二甲基甲酰胺溶液(0.5ml)混合,并将该反应混合物在室温下搅拌过夜。用反相hplc直接纯化粗品反应混合物,使用c18柱和10-100%的乙腈/水(含有0.1%三氟乙酸)的梯度。将含有产物的级分冷冻干燥,得到标题化合物的三氟乙酸盐。ms(esi)m/e1165.5(m+h)+。
2.17.3. 4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-1-((2,5-二氧代吡咯烷-1-基)氧基)-1-氧代丁烷-2-磺酸酯
在用氮气吹扫的100ml烧瓶中,将1-羧基-3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙烷-1-磺酸酯溶于二甲基乙酰胺(20ml)中。向此溶液中加入n-羟基琥珀酰亚胺(440mg,)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(1000mg),并将该反应在氮气氛围中、在室温下搅拌16小时。减压浓缩溶剂,并将残余物用硅胶色谱纯化,用1-2%的甲醇/二氯甲烷(含有0.1%v/v乙酸)的梯度进行洗脱,得到标题化合物的~80%活化酯和20%酸的混合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e360.1(m+h)+。
2.17.4. n-[(2r)-4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2-磺基丁酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺
将实施例2.17.2的三氟乙酸盐(6mg)与实施例2.17.3(16.85mg)和n,n-二异丙基乙胺(0.025ml)在n,n-二甲基甲酰胺(0.500ml)中混合,并将该反应混合物在室温下搅拌过夜。用反相hplc纯化该粗品反应混合物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到在新增加的位置上立体化学构型不同的两个非对映异构体(衍生自外消旋的实施例2.17.3)。在该中心处的两个产物的立体化学构型是随机指定的。ms(esi)m/e1408.5(m-h)-。
2.18. 合成n-[(2s)-4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2-磺基丁酰基]-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]苯基}-n5-氨基甲酰基-l-鸟氨酰胺(合成子bp)
如实施例2.17.4所述,标题化合物是在制备实施例2.17.4期间分离的第二个非对映异构体。ms(esi)m/e1408.4(m-h)-。
2.19. 该段落特意留白。
2.20. 该段落特意留白。
2.21. 合成n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺(合成子iq)
2.21.1. (s)-(9h-芴-9-基)甲基(1-((4-(羟甲基)苯基)氨基-1-氧代丙-2-基)氨基甲酸酯
向(s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酸(50g)的甲醇(400ml)和二氯甲烷(400ml)溶液中加入(4-氨基苯基)甲醇(23.73g)和2-乙氧基喹啉-1(2h)-甲酸乙酯(79g),并将该反应在室温下搅拌过夜。蒸发溶剂,并将残余物用二氯甲烷洗涤,得到标题化合物。
2.21.2. (s)-2-氨基-n-(4-(羟甲基)苯基)丙酰胺
向实施例2.21.1(10g)的n,n-二甲基甲酰胺(100ml)溶液中加入哌啶(40ml),并将该反应搅拌2小时。蒸发溶剂,并将残余物溶于甲醇中。滤出固体,并浓缩滤液,得到粗品。
2.21.3. (9h-芴-9-基)甲基((s)-1-(((s)-1-((4-(羟甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-3-甲基-1-氧代丁-2-基)氨基甲酸酯
向实施例2.21.2(5g)的n,n-二甲基甲酰胺(100ml)溶液中加入(s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-甲基丁酸(10.48g)和2-(1h-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(14.64g),并将该反应搅拌过夜。蒸发溶剂,用二氯甲烷洗涤残余物,滤出固体,得到粗品。
2.21.4. (9h-芴-9-基)甲基((s)-3-甲基-1-(((s)-1-((4-((((4-硝基苯氧基)羰基)氧基)甲基)苯基)氨基)-1-氧代丙-2-基)氨基)-1-氧代丁-2-基)氨基甲酸酯
在实施例2.10.2中,用实施例2.21.3替代实施例2.10.1,制备标题化合物。
2.21.5. 3-(1-((3-(2-((((4-((s)-2-((s)-2-氨基-3-甲基丁酰胺基)丙酰胺基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例1.3.7(0.102g)、实施例2.21.4(0.089g)和n,n-二异丙基乙胺(0.104ml)的n,n-二甲基甲酰胺(1ml)溶液在室温下搅拌。搅拌过夜之后,加入二乙胺(0.062ml),并将该反应再搅拌2小时。用水(1ml)稀释该反应,用三氟乙酸淬灭,并用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。
2.21.6. 3-(1-((3-(2-((((4-((s)-2-((s)-2-((r)-2-氨基-3-磺基丙酰胺基)-3-甲基丁酰胺基)丙酰胺基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向(r)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-磺基丙酸(0.028g)和2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(0.027g)的n,n-二甲基甲酰胺(1ml)溶液中加入n,n-二异丙基乙胺(0.042ml),并将该反应搅拌5分钟。将该混合物加入到实施例2.21.5(0.050g)中,并将该混合物搅拌1小时。然后,将二乙胺(0.049ml)加入到该反应中,并继续搅拌1小时。将该反应用n,n-二甲基甲酰胺(1ml)和水(0.5ml)稀释,用三氟乙酸淬灭,用反相hplc纯化,使用gilson系统,用10-88%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。ms(esi)m/e1214.4(m-h)-。
2.21.7. n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
向实施例2.21.6(0.030g)和2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(8.34mg)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙基乙胺(0.020ml),并将该反应搅拌1小时。将该反应用n,n-二甲基甲酰胺(1ml)和水(0.5ml)稀释,用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.84(s,1h),9.41(s,1h),8.26(d,1h),8.11-7.95(m,3h),7.79(d,1h),7.68(d,2h),7.61(d,1h),7.57-7.27(m,6h),7.24(d,2h),7.12(t,1h),7.02-6.90(m,3h),4.94(d,4h),4.67(td,2h),4.34-4.22(m,2h),4.04-3.94(m,2h),3.88(t,2h),3.82(s,2h),3.42-3.27(m,4h),3.11-2.96(m,5h),2.84(dd,1h),2.30-1.98(m,6h),1.56-1.41(m,4h),1.41-0.79(m,28h)。ms(esi)m/e1409.1(m+h)+。
2.22. 合成4-[(1e)-3-({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)丙-1-烯-1-基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子db)
2.22.1. (e)-叔丁基二甲基((3-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)烯丙基)氧基)硅烷
在氮气氛围中,向装有叔丁基二甲基(丙-2-炔-1-基氧基)硅烷(5g)和二氯甲烷(14.7ml)的烧瓶中逐滴加入4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷(3.94g)。将该混合物在室温下搅拌一分钟,然后通过小管转移到氮气吹扫的、包含cp2zrclh(氯化双(η5-环戊二烯基)氢化锆,schwartz's试剂)(379mg)的烧瓶中。将得到的反应混合物在室温下搅拌16小时。将该混合物小心地用水(15ml)淬灭,而后用乙醚(3x30ml)提取。用水(15ml)洗涤合并的有机相,用mgso4干燥,过滤,浓缩,用硅胶色谱纯化,用0-8%的乙酸乙酯/庚烷的梯度进行洗脱,得到标题化合物。ms(esi)m/z316.0(m+nh4)+。
2.22.2. (2s,3r,4s,5s,6s)-2-(4-溴-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将(2r,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(5g)溶于乙腈(100ml)中。将ag2o(2.92g)加入到该溶液中,并将该反应在室温下搅拌5分钟。加入4-溴-2-硝基苯酚(2.74g),并将该反应混合物在室温下搅拌4小时。通过硅藻土过滤银盐残余物,并将滤液减压浓缩。用硅胶色谱纯化残余物,用梯度10-70%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi+)m/z550.9(m+nh4)+。
2.22.3. (2s,3r,4s,5s,6s)-2-(4-((e)-3-((叔丁基二甲基甲硅烷基)氧基)丙-1-烯-1-基)-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.22.2(1g)、碳酸钠(0.595g)、三(二亚苄基丙酮)二钯(0.086g)和1,3,5,7-四甲基-6-苯基-2,4,8-三氧杂-6-磷杂金刚烷(0.055g)在配备有回流冷凝器的50ml三颈圆底烧瓶中混合,并将该系统用氮气脱气。将实施例2.22.1(0.726g)的四氢呋喃(15ml)溶液用氮气单独脱气30分钟。将后面的溶液通过小管转移到包含固体试剂的烧瓶中,而后通过注射器加入脱气的水(3ml)。将该反应加热至60℃,保持两小时。将该反应混合物在乙酸乙酯(3x30ml)和水(30ml)之间分配。将合并的有机相干燥(na2so4),过滤,并浓缩。用硅胶色谱纯化残余物,用梯度0-35%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(esi+)m/z643.1(m+nh4)+。
2.22.4. (2s,3r,4s,5s,6s)-2-(2-氨基-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向配备有压力平衡加入漏斗的氮气吹扫的500ml三颈烧瓶中加入锌粉(8.77g)。通过小管,加入脱气的实施例2.22.3(8.39g)的四氢呋喃(67ml)溶液。将得到的悬浮液在冰浴中冷却,并通过加入漏斗逐滴加入6nhcl(22.3ml),加入速率应该使反应的内部温度不超过35℃。加入完成之后,将该反应在室温下搅拌两个小时,通过硅藻土的垫过滤,用水和乙酸乙酯冲洗。用饱和nahco3水溶液处理滤液,直到水层不再是酸性为止,并过滤该混合物,取出所得到的固体。将滤液转入分液漏斗中,并分离各层。用乙酸乙酯(3x75ml)提取水层,并将合并的有机层用水(100ml)洗涤,用na2so4干燥,过滤,并浓缩。将残余物与乙醚一起研磨,并过滤收集固体,提供标题化合物。ms(esi+)m/z482.0(m+h)+。
2.22.5. (9h-芴-9-基)甲基(3-氯-3-氧代丙基)氨基甲酸酯
向3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酸(5.0g)的二氯甲烷(53.5ml)溶液中加入亚硫酰氯(0.703ml)。将该混合物在60℃下搅拌一小时。冷却该混合物,并浓缩,得到标题化合物,其不用进一步纯化,在下一步中直接使用。
2.22.6. (2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((e)-3-羟基丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.22.4(6.78g)溶于二氯甲烷(50ml)中,并将该溶液在冰浴中冷却至0℃。加入n,n-二异丙基乙胺(3.64g),而后逐滴加入实施例2.22.5(4.88g)的二氯甲烷(50ml)溶液。将该反应搅拌16小时,并使冰浴达到室温。加入饱和nahco3水溶液(100ml),并分离各层。将水层进一步用二氯甲烷(2x50ml)提取。用na2so4干燥提取物,过滤,浓缩,用硅胶色谱纯化,用梯度5-95%的乙酸乙酯/庚烷洗脱,得到起始苯胺和目标产物的不可分离的混合物。将该混合物在1nhcl水溶液(40ml)和乙醚与乙酸乙酯的1:1混合物(40ml)之间分配,而后用乙酸乙酯(2x25ml)进一步提取水相。将有机相合并,用水(2x25ml)洗涤,用na2so4干燥,过滤,并浓缩,得到标题化合物。ms(esi+)m/z774.9(m+h)+。
2.22.7. (2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((e)-3-(((4-硝基苯氧基)羰基)氧基)丙-1-烯-1-基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.22.6(3.57g)溶于二氯甲烷(45ml)中,并加入双(4-硝基苯基)碳酸酯(2.80g),而后逐滴加入n,n-二异丙基乙胺(0.896g)。将该反应混合物在室温下搅拌两个小时。将硅胶(20g)加入到该反应溶液中,并将该混合物减压浓缩至干,保持浴温等于或低于25℃。将硅渣加载到柱顶上,并用硅胶色谱纯化产物,用梯度0-100%的乙酸乙酯-庚烷洗脱,提供部分纯化的产物,它被硝基苯酚污染。将该物质与甲基叔丁基醚(250ml)一起研磨,并将得到的浆液静置1小时。过滤收集产物。以类似的方式,收集连续三批产物,得到标题化合物。ms(esi+)m/z939.8(m+h)+。
2.22.8. 3-(1-((3-(2-(((((e)-3-(3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苯基)烯丙基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例1.1.17的三氟乙酸盐(77mg)和实施例2.22.7(83mg)的n,n-二甲基甲酰胺(3.5ml)溶液中加入n,n-二异丙基乙胺(0.074ml)。将该反应慢慢地升温至室温,并搅拌16小时。加入水和乙酸乙酯,淬灭该反应。分离各层,并用额外的乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,减压浓缩,得到标题化合物,其不用进一步纯化,在随后的步骤中直接使用。
2.22.9. 3-(1-((3-(2-(((((e)-3-(3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苯基)烯丙基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向环境温度的实施例2.22.8(137mg)的甲醇(3ml)溶液中加入2m氢氧化锂溶液(0.66ml)。将该反应混合物在35℃下搅拌两个小时,并加入乙酸(0.18ml),淬灭该反应。将该反应浓缩至干,并将残余物用甲醇稀释。用反相hplc纯化粗品,使用gilson系统和c1825x100mm柱,用20-75%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物的三氟乙酸盐。ms(esi)m/e1220.3(m+na)+。
2.22.10. 4-[(1e)-3-({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)丙-1-烯-1-基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.22.9的三氟乙酸盐(41.9mg)的n,n-二甲基甲酰胺(1ml)溶液中加入n-琥珀酰亚胺基6-马来酰亚胺基(maleimido)己酸酯(9.84mg)和n,n-二异丙基乙胺(0.010ml),并将该反应在室温下搅拌16小时。用反相hplc纯化粗品反应物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.86(bs,2h),9.03(s,1h),8.25(bs,1h),8.03(d,1h),7.97-7.85(m,1h),7.79(d,1h),7.64-7.59(m,1h),7.56-7.39(m,3h),7.40-7.32(m,2h),7.28(s,1h),7.14-7.06(m,1h),7.04(d,1h),6.98(s,2h),6.95(d,1h),6.60-6.52(m,1h),6.22-6.12(m,1h),4.95(bs,2h),4.90-4.75(m,1h),4.63(d,2h),4.24-4.05(m,1h),4.08-3.62(m,8h),3.50-3.24(m,10h),3.04-2.97(m,2h),2.92-2.82(m,3h),2.11-2.06(m,3h),2.03(t,j=7.4hz,2h),1.53-1.39(m,4h),1.41-0.73(m,23h)。ms(esi)m/e1413.3(m+na)+。
2.23. 合成4-{(1e)-3-[({2-[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙氧基]乙基}氨基甲酰基)氧基]丙-1-烯-1-基}-2-({n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子dm)
2.23.1. 3-(1-((3-(2-(2-(((((e)-3-(3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苯基)烯丙基)氧基)羰基)氨基)乙氧基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例2.22.7(94mg)和实施例1.4.10(90mg)的溶液中加入n,n-二异丙胺(0.054ml)。将该反应慢慢地加热至室温,并搅拌过夜。加入水和乙酸乙酯,淬灭该反应。分离各层,并用额外的乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,并减压浓缩。将粗品溶于四氢呋喃/甲醇/水(2:1:1,8ml)中,向其中加入氢氧化锂一水合物(40mg)。将该反应混合物搅拌过夜。真空浓缩该混合物,用三氟乙酸酸化,并溶于二甲亚砜/甲醇中。用反相hplc纯化该溶液,使用gilson系统和c18柱,用10-85%的乙腈/水中的0.1%三氟乙酸洗脱,得到标题化合物。ms(esi)m/e1228.1(m+h)+。
2.23.2. 4-{(1e)-3-[({2-[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙氧基]乙基}氨基甲酰基)氧基]丙-1-烯-1-基}-2-({n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.23.1(20mg)和2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯(5.5mg)的n,n-二甲基甲酰胺(2ml)溶液中加入n,n-二异丙基乙胺(0.054ml)。将该反应搅拌过夜。将该反应混合物用甲醇(2ml)稀释,并用三氟乙酸酸化。用反相hplc纯化该溶液,使用gilson系统和c18柱,用10-85%的乙腈/水中的0.1%三氟乙酸洗脱,得到标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h),9.03(s,1h),8.24(s,1h),7.95-8.11(m,2h),7.79(d,1h),7.61(d,1h),7.32-7.52(m,5h),7.28(s,1h),7.02-7.23(m,3h),6.91-6.96(m,3h),6.57(d,1h),6.05-6.24(m,1h),4.95(s,2h),4.87(d,1h),4.59(d,2h),3.78-3.95(m,4h),3.13(q,2h),3.01(t,2h),2.51-2.57(m,2h),2.27-2.39(m,3h),2.11(s,3h),0.92-1.43(m,16h),0.83(s,6h)。ms(esi)m/e1379.2(m+h)+。
2.24. 合成4-{(1e)-3-[({2-[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙氧基]乙基}氨基甲酰基)氧基]丙-1-烯-1-基}-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子dl)
向实施例2.23.1(20mg)和2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(6.5mg)的n,n-二甲基甲酰胺(2ml)溶液中加入n,n-二异丙基乙胺(0.054ml)。将该反应混合物搅拌过夜。将该反应混合物用甲醇(2ml)稀释,并用三氟乙酸酸化。用反相hplc纯化该混合物,使用gilson系统和c18柱,用10-85%的乙腈/水中的0.1%三氟乙酸洗脱,得到标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h),9.03(s,1h),8.24(s,1h),8.03(d,1h),7.87(t,1h),7.78(s,1h),7.61(d,1h),7.32-7.55(m,5h),6.90-7.19(m,5h),6.56(d,1h),6.08-6.24(m,1h),4.91-4.93(m,1h),4.86(s,1h),4.59(d,2h),3.27-3.46(m,14h),3.13(q,3h),2.96-3.02(m,2h),2.50-2.59(m,3h),2.09(s,3h),2.00-2.05(m,3h),0.94-1.54(m,20h),0.83(s,6h)。ms(esi)m/e1421.2(m+h)+。
2.25. 合成4-[(1e)-14-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-6-甲基-5-氧代-4,9,12-三氧杂-6-氮杂十四碳-1-烯-1-基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子dr)
2.25.1. 3-(1-((3-(((e)-14-(3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苯基)-9-甲基-10-氧代-3,6,11-三氧杂-9-氮杂十四碳-13-烯-1-基)氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例2.22.7(90mg)和实施例1.2.11(92mg)溶液中加入n,n-二异丙胺(0.050ml)。除去冰浴,并将该反应搅拌过夜。加入水和乙酸乙酯,淬灭该反应。分离各层,并用额外的乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,减压浓缩,提供标题化合物,其不用进一步纯化,在随后的步骤中直接使用。ms(esi)m/e1648.2(m+h)+。
2.25.2. 3-(1-((3-(((e)-14-(3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苯基)-9-甲基-10-氧代-3,6,11-三氧杂-9-氮杂十四碳-13-烯-1-基)氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例2.25.1(158mg)的甲醇(2.0ml)溶液中加入2m氢氧化锂水溶液(0.783ml)。将该反应搅拌4小时,并加入乙酸(0.1ml),淬灭该反应。将该反应浓缩至干,并将残余物色谱分离,使用biotageisoleraone系统和反相c1840g柱,用10-85%的乙腈/水中的0.1%三氟乙酸洗脱。将含有产物的级分冷冻干燥,得到标题化合物的固体。ms(esi)m/e1286.2(m+h)+。
2.25.3. 4-[(1e)-14-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-6-甲基-5-氧代-4,9,12-三氧杂-6-氮杂十四碳-1-烯-1-基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向环境温度的实施例2.25.2(9.03mg)的n,n-二甲基甲酰胺(1.0ml)溶液中加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(4mg)和n,n-二异丙胺(0.020ml),并将该反应搅拌过夜。用二甲亚砜和甲醇稀释该反应,用rp-hplc(在biotageisolera色谱上,40gc18柱)纯化,用梯度10至75%的乙腈/水(包含0.1%v/v三氟乙酸)洗脱。通过冷冻干燥,将含有产物的级分浓缩,得到标题化合物的固体。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h),8.04(d,1h),7.99(t,1h),7.79(d,1h),7.60(d,1h),7.53-7.41(m,3h),7.40-7.32(m,2h),7.28(s,1h),6.99(s,2h),6.98-6.92(m,1h),4.95(bs,2h),3.92-3.85(m,1h),3.81(s,2h),3.63-3.55(m,4h),3.55-3.31(m,28h),3.18-3.10(m,2h),3.05-2.98(m,2h),2.97(s,2h),2.80(s,2h),2.59-2.50(m,1h),2.32(t,2h),2.10(s,3h),1.39-1.34(m,2h),1.31-1.18(m,4h),1.20-0.92(m,6h),0.84(s,6h)。ms(esi)m/e1479.3(m+h)+。
2.26. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子dz)
2.26.1. (2s,3r,4s,5s,6s)-2-(4-甲酰基-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向2,4-二羟基苯甲醛(15g)和(2s,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(10g)的乙腈溶液中加入碳酸银(10g),并将该反应加热至40℃。搅拌4小时之后,冷却该反应,过滤,并浓缩。将粗品悬浮在二氯甲烷中,通过硅藻土过滤,并浓缩。用硅胶色谱纯化残余物,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.26.2. (2s,3r,4s,5s,6s)-2-(3-羟基-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.26.1(16.12g)的四氢呋喃(200ml)和甲醇(200ml)溶液冷却至0℃,并分批加入硼氢化钠(1.476g)。将该反应搅拌20分钟,然后用水∶饱和碳酸氢钠溶液的1:1混合物(400ml)淬灭。滤出所得到的固体,并用乙酸乙酯冲洗。分离各相,并用乙酸乙酯提取水层四次。将合并的有机层用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e473.9(m+nh4)+。
2.26.3. (2s,3r,4s,5s,6s)-2-(4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在-5℃,向实施例2.26.2(7.66g)和叔丁基二甲基甲硅烷基氯(2.78g)/二氯甲烷(168ml)中加入咪唑(2.63g),并将该反应混合物搅拌过夜,使得反应的内部温度升至12℃。将该反应混合物倒入饱和氯化铵水溶液中,并用二氯甲烷提取四次。将合并的有机物用盐水洗涤,用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e593.0(m+na)+。
2.26.4. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.26.3(5.03g)和在甲苯(88ml)中的三苯基膦(4.62g)中加入偶氮二甲酸二叔丁基酯(4.06g),并将该反应混合物搅拌30分钟。加入(9h-芴-9-基)甲基(2-(2-羟乙氧基)乙基)氨基甲酸酯,并将该反应再搅拌1.5小时。将反应物直接加载到硅胶上,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.26.5. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.26.4(4.29g)在3∶1∶1的乙酸∶水∶四氢呋喃溶液(100ml)中搅拌过夜。将该反应混合物倒入饱和碳酸氢钠水溶液中,并用乙酸乙酯提取。将有机层用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.26.6. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.26.5(0.595g)和双(4-硝基苯基)碳酸酯(0.492g)的n,n-二甲基甲酰胺(4ml)溶液中加入n,n-二异丙胺(0.212ml)。1.5小时之后,在高真空下浓缩该反应。用硅胶色谱纯化残余物,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e922.9(m+na)+。
2.26.7. 3-(1-((3-(2-((((2-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向实施例1.1.17(0.106g)和实施例2.26.6(0.130g)的n,n-二甲基甲酰胺(1.5ml)溶液中加入n,n-二异丙胺(0.049ml)。6小时之后,再加入n,n-二异丙胺(0.025ml),并将该反应搅拌过夜。用乙酸乙酯(50ml)稀释该反应,并用水(10ml)洗涤,而后用盐水(15ml)洗涤四次。用硫酸镁干燥有机层,过滤,并浓缩,得到标题化合物,其不用进一步纯化,在下一步中直接使用。
2.26.8. 3-(1-((3-(2-((((2-(2-(2-氨基乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例2.26.7(0.215g)的甲醇(2ml)悬浮液用2.0m氢氧化锂水溶液(1ml)处理。搅拌1小时之后,加入乙酸(0.119ml),淬灭该反应。用二甲亚砜(1ml)稀释所得到的悬浮液,并用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。
2.26.9. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.26.8(0.050g)的n,n-二甲基甲酰胺(1ml)溶液中加入n,n-二异丙胺(0.037ml),而后加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(0.017g),并在室温下搅拌该反应。搅拌1小时之后,用水稀释该反应,并用反相hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.86(s,1h),8.03(d,1h),7.82-7.77(m,2h),7.62(d,1h),7.53-7.41(m,3h),7.40-7.33(m,2h),7.28(s,1h),7.19(d,1h),6.98(s,2h),6.95(d,1h),6.66(s,1h),6.60(d,1h),5.06(t,1h),5.00-4.93(m,4h),4.18-4.04(m,2h),3.95-3.85(m,2h),3.85-3.77(m,2h),3.71(t,2h),3.41-3.30(m,4h),3.30-3.23(m,4h),3.19(q,2h),3.01(t,2h),2.85(d,3h),2.09(s,3h),2.02(t,2h),1.53-1.40(m,4h),1.40-0.78(m,24h)。ms(esi)m/e1380.5(m-h)-。
2.27. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子ea)
向实施例2.26.8(0.031g)的n,n-二甲基甲酰胺(1ml)溶液中加入n,n-二异丙胺(0.023ml),而后加入2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯(9mg),并在室温下搅拌该反应。搅拌1小时之后,用水稀释该反应,并用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.84(s,1h),8.03(d,1h),8.00(t,1h),7.79(d,1h),7.61(d,1h),7.54-7.41(m,3h),7.40-7.32(m,2h),7.28(s,1h),7.19(d,1h),6.97(s,2h),6.95(d,1h),6.66(s,1h),6.60(d,1h),5.11-5.02(m,1h),4.96(s,4h),4.18-4.02(m,2h),3.96-3.84(m,2h),3.80(s,2h),3.71(t,2h),3.43-3.22(m,12h),3.17(q,2h),3.01(t,2h),2.85(d,3h),2.33(t,2h),2.09(s,3h),1.44-0.76(m,18h)。ms(esi)m/e1338.5(m-h)-。
2.28. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[({[3-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)-4-(β-d-吡喃半乳糖基氧基)苄基]氧基}羰基)(甲基)氨基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子eo)
2.28.1. (2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-溴四氢-2h-吡喃-3,4,5-三基三乙酸酯
将干燥的100ml圆底烧瓶用氮气吹扫,加入(2s,3r,4s,5s,6r)-6-(乙酰氧基甲基)四氢-2h-吡喃-2,3,4,5-四基四乙酸酯(5g),并在氮气氛中,用橡胶隔片覆盖。加入溴化氢的冰醋酸溶液(33%wt,11.06ml),并将该反应在室温下搅拌两个小时。将该反应混合物用二氯甲烷(75ml)稀释,并倒入250ml冰冷的水中。分离各层,并将有机层进一步用冰冷的水(3x100ml)和饱和碳酸氢钠水溶液(100ml)洗涤。将有机层用mgso4干燥,过滤,并减压浓缩。通过与甲苯(3x50ml)共沸,除去残余的乙酸。减压浓缩溶剂,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e429.8(m+nh4)+。
2.28.2. (2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-甲酰基-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.28.1(5.13g)溶于乙腈(100ml)中。加入氧化银(i)(2.89g),并将该反应搅拌20分钟。加入4-羟基-3-硝基苯甲醛(2.085g),并将该反应混合物在室温下搅拌四小时,而后通过millipore0.22μm过滤器真空过滤,除去银盐。减压浓缩溶剂,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e514.9(m+nh4)+。
2.28.3. (2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(4-(羟甲基)-2-硝基苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向氮气吹扫的干燥1l圆底烧瓶中加入实施例2.28.2(5.0g,)细磨的粉末,并保持在氮气氛中。加入四氢呋喃(70ml),并将该溶液超声处理两分钟,得到悬浮液。加入甲醇(140ml),并将该悬浮液再超声处理3分钟。将该悬浮液放在冰浴中,并在氮气氛围中搅拌20分钟,达到平衡(0℃)。用20分钟分批加入硼氢化钠(0.380g),并将该冷的(0℃)反应搅拌30分钟。将乙酸乙酯(200ml)加入到该反应混合物中,并放在冰浴中,加入300ml饱和氯化铵溶液,而后加入200ml水,淬灭该反应。用乙酸乙酯(3x300ml)提取该反应混合物,用盐水(300ml)洗涤,用mgso4干燥,过滤,并减压浓缩溶剂,得到标题化合物。ms(esi)m/e516.9(m+nh4)+。
2.28.4. (2r,3s,4s,5r,6s)-2-(乙酰氧基甲基)-6-(2-氨基-4-(羟甲基)苯氧基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在实施例2.22.3中,用实施例2.28.3替代实施例2.22.2,并取消研磨步骤,制备标题化合物。产物不用进一步纯化,在下一步中直接使用。ms(esi)m/e469.9(m+h)+。
2.28.5. (2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(羟甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在实施例2.22.5中,用实施例2.28.4替代实施例2.22.3,制备标题化合物。通过在二氯甲烷和水之间分配,淬灭该反应。分离各层,并用乙酸乙酯提取水层两次。将合并的有机层用1n盐酸水溶液和盐水洗涤,用na2so4干燥,过滤,并减压浓缩。用硅胶色谱纯化产物,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e762.9(m+h)+。
2.28.6. (2s,3r,4s,5s,6r)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(乙酰氧基甲基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向环境温度下的实施例2.28.5(3.2g)和双(4-硝基苯基)碳酸酯(1.914g)的n,n-二甲基甲酰胺(20ml)溶液中逐滴加入n,n-二异丙基乙胺(1.10ml)。将该反应在室温下搅拌1.5小时。减压浓缩溶剂。用硅胶色谱纯化粗品,用梯度10-100%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e927.8(m+h),950.1(m+na)+。
2.28.7. 3-(1-((3-(2-((((3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(((2s,3r,4s,5s,6r)-3,4,5-三乙酰氧基-6-(乙酰氧基甲基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.22.8中,用实施例2.28.6替代实施例2.22.7,制备标题化合物。ms(esi)m/e1548.3(m+h)+。
2.28.8. 3-(1-((3-(2-((((3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5r,6r)-3,4,5-三羟基-6-(羟甲基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.22.8中,用实施例2.28.7替代实施例2.22.7,制备标题化合物。ms(esi)m/e1158.3(m+h)+。
2.28.9. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[({[3-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)-4-(β-d-吡喃半乳糖基氧基)苄基]氧基}羰基)(甲基)氨基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸
在实施例2.22.9中,用实施例2.28.8替代实施例2.22.8,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(bs,1h),9.13(bs,1h),8.19(bs,1h),8.03(d,1h),7.88(d,1h),7.79(d,1h),7.62(d,1h),7.55-7.39(m,3h),7.41-7.30(m,2h),7.28(s,1h),7.14(d,1h),7.05-6.88(m,4h),4.96(bs,4h),3.57-3.48(m,1h),3.49-3.09(m,11h),3.08-2.57(m,7h),2.33(d,1h),2.14-1.97(m,6h),1.55-0.90(m,20h),0.86-0.79(m,6h)。ms(esi)m/e1351.3(m+h)+。
2.29. 合成2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-5-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子fb)
2.29.1. 4-(2-(2-溴乙氧基)乙氧基)-2-羟基苯甲醛
将2,4-二羟基苯甲醛(1.0g)、1-溴-2-(2-溴乙氧基)乙烷(3.4g)和碳酸钾(1.0g)的乙腈(30ml)溶液加热至75℃,保持2天。冷却该反应,用乙酸乙酯(100ml)稀释,用水(50ml)和盐水(50ml)洗涤,用硫酸镁干燥,过滤,并浓缩。用硅胶色谱纯化残余物,用梯度5-30%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(elsd)m/e290.4(m+h)+。
2.29.2. 4-(2-(2-叠氮基乙氧基)乙氧基)-2-羟基苯甲醛
向实施例2.29.1(1.26g)的n,n-二甲基甲酰胺(10ml)溶液中加入叠氮化钠(0.43g),并将该反应在室温下搅拌过夜。用乙醚(100ml)稀释该反应,用水(50ml)和盐水(50ml)洗涤,用硫酸镁干燥,过滤,并浓缩。用硅胶色谱纯化残余物,用梯度5-30%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(elsd)m/e251.4(m+h)+。
2.29.3. (2s,3r,4s,5s,6s)-2-(5-(2-(2-叠氮基乙氧基)乙氧基)-2-甲酰基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.29.2(0.84g)、(3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(1.99g)和氧化银(i)(1.16g)的乙腈(15ml)溶液一起搅拌。搅拌过夜之后,用二氯甲烷(20ml)稀释该反应。加入硅藻土,过滤该反应,并浓缩。用硅胶色谱纯化残余物,用梯度5-75%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.29.4. (2s,3r,4s,5s,6s)-2-(5-(2-(2-叠氮基乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.9.3(0.695g)的甲醇(5ml)和四氢呋喃(2ml)溶液冷却至0℃。加入硼氢化钠(0.023g),并将该反应升温至室温。总共搅拌1小时之后,将反应物倒入乙酸乙酯(75ml)和水(25ml)的混合物中,并加入饱和碳酸氢钠水溶液(10ml)。分离有机层,用盐水(50ml)洗涤,用硫酸镁干燥,过滤,并浓缩。用硅胶色谱纯化残余物,用梯度5-85%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(elsd)m/e551.8(m-h2o)-。
2.29.5. (2s,3r,4s,5s,6s)-2-(5-(2-(2-氨基乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在50ml耐压瓶中,向四氢呋喃(20ml)中的实施例2.29.4(0.465g)中加入5%pd/c(0.1g),并将该混合物在30psi氢气氛围中摇动16小时。过滤该反应,并浓缩,得到标题化合物,其不用进一步纯化,直接使用。ms(elsd)m/e544.1(m+h)+。
2.29.6. (2s,3r,4s,5s,6s)-2-(5-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-2-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.29.5(0.443g)的二氯甲烷(8ml)溶液冷却至0℃,然后加入n,n-二异丙胺(0.214ml)和(9h-芴-9-基)甲基氯甲酸酯(0.190g)。1小时之后,浓缩该反应。用硅胶色谱纯化残余物,用梯度5-95%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(elsd)m/e748.15(m-oh)-。
2.29.7. (2s,3r,4s,5s,6s)-2-(5-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-2-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.29.6(0.444g)的n,n-二甲基甲酰胺(5ml)溶液中加入n,n-二异丙胺(0.152ml)和双(4-硝基苯基)碳酸酯(0.353g),并在室温下搅拌该反应。5小时之后,浓缩该反应。用硅胶色谱纯化残余物,用梯度5-90%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.29.8. 3-(1-((3-(2-((((4-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-2-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向实施例1.1.17(0.117g)和实施例2.29.7(0.143g)的n,n-二甲基甲酰胺(1.5m)溶液中加入n,n-二异丙胺(0.134ml),并将该反应搅拌过夜。用乙酸乙酯(75ml)稀释该反应,然后用水(20ml)洗涤,而后用盐水(4x20ml)洗涤。用硫酸镁干燥有机层,过滤,并浓缩,得到标题化合物,其不用进一步纯化,直接使用。
2.29.9. 3-(1-((3-(2-((((4-(2-(2-氨基乙氧基)乙氧基)-2-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例2.29.8(0.205g)的甲醇(2ml)悬浮液用氢氧化锂水合物(0.083g)的水溶液(1ml)处理。搅拌1小时之后,通过加入乙酸(0.113ml)淬灭该反应,用二甲亚砜稀释,并用制备hplc纯化,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。
2.29.10. 2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-5-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.29.9(0.080g)的n,n-二甲基甲酰胺(1ml)溶液中加入n,n-二异丙胺(0.054ml),而后加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(0.025g),并在室温下搅拌该反应。搅拌1小时之后,用水(0.5ml)稀释该反应,并用制备hplc纯化(gilson系统),用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.86(s,1h),8.03(d,1h),7.86-7.81(m,1h),7.79(d,1h),7.62(d,1h),7.52-7.41(m,3h),7.39-7.32(m,2h),7.28(s,1h),7.19(d,1h),6.99(s,2h),6.95(d,1h),6.68(d,1h),6.59(d,1h),5.09-4.99(m,3h),4.96(s,2h),4.05(s,2h),3.94(d,1h),3.88(t,2h),3.81(d,2h),3.47-3.24(m,15h),3.19(q,2h),3.01(t,2h),2.86(d,3h),2.09(s,3h),2.03(t,2h),1.51-1.41(m,4h),1.41-0.78(m,18h),ms(esi)m/e1382.2(m+h)+。
2.30. 合成2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]-5-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子kx)
2.30.1. 3-(1-((3-(2-((((4-(2-(2-氨基乙氧基)乙氧基)-2-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向实施例1.3.7(0.071g)和实施例2.29.7(0.077g)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙胺(0.072ml),并将该反应搅拌3小时。浓缩该反应,将得到的油溶于四氢呋喃(0.5ml)和甲醇(0.5ml)中,并用氢氧化锂一水合物(0.052g)在水(0.5ml)中的溶液处理。搅拌1小时之后,用n,n-二甲基甲酰胺(1ml)稀释该反应,并用制备hplc纯化(使用gilson系统),用10-75%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。ms(esi)m/e1175.2(m+h)+。
2.30.2. 2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]-5-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.30.1(0.055g)和2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯(0.012g)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙胺(0.022ml),并将在室温下搅拌该反应。搅拌1小时之后,用n,n-二甲基甲酰胺和水的1:1溶液(2ml)稀释该反应,并用制备hplc纯化(使用gilson系统),用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.85(s,1h),8.07-8.00(m,2h),7.79(d,1h),7.62(d,1h),7.55-7.41(m,3h),7.40-7.32(m,2h),7.28(s,1h),7.20(d,1h),7.11(t,1h),6.98(s,2h),6.95(d,1h),6.66(s,1h),6.60(dd,1h),5.04(d,1h),5.00(s,2h),4.96(s,2h),4.10-4.03(m,2h),3.95(d,2h),3.88(t,2h),3.70(t,2h),3.59(t,2h),3.46-3.38(m,4h),3.36-3.25(m,4h),3.17(q,2h),3.08-2.98(m,4h),2.33(t,2h),2.10(s,3h),1.37(s,2h),1.25(q,4h),1.18-0.93(m,6h),0.84(s,6h),ms(esi)m/e1325.9(m+h)+。
2.31. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-(3-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}丙氧基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子ff)
2.31.1. (2s,3r,4s,5s,6s)-2-(3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙氧基)-4-甲酰基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在0℃,向(9h-芴-9-基)甲基(3-羟丙基)氨基甲酸酯(0.245g)和三苯基膦(0.216g)的四氢呋喃(2ml)溶液中逐滴加入偶氮二甲酸二异丙基酯(0.160ml)。搅拌15分钟之后,加入实施例2.26.1(0.250g),除去冰浴,并将该反应升温至室温。2小时之后,浓缩该反应。用硅胶色谱纯化残余物,用梯度5-70%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(apci)m/e512.0(m-fmoc)-。
2.31.2. (2s,3r,4s,5s,6s)-2-(3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙氧基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.31.1(0.233g)的甲醇(3ml)和四氢呋喃(1ml)悬浮液中加入硼氢化钠(6mg)。30分钟之后,将反应物倒入乙酸乙酯(50ml)和水(25ml)中,而后加入饱和碳酸氢钠水溶液(5ml)。分离有机层,用盐水(25ml)洗涤,用硫酸镁干燥,过滤,并浓缩。用硅胶色谱纯化残余物,用梯度5-80%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(apci)m/e718.1(m-oh)-。
2.31.3. (2s,3r,4s,5s,6s)-2-(3-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.31.2(0.140g)和双(4-硝基苯基)碳酸酯(0.116g)的n,n-二甲基甲酰胺(1ml)溶液中加入n,n-二异丙胺(0.050ml)。1.5小时之后,在高真空下浓缩该反应。用硅胶色谱纯化残余物,用梯度10-70%的乙酸乙酯/庚烷洗脱,得到标题化合物。
2.31.4. 3-(1-((3-(2-((((2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙氧基)-4-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向实施例1.1.17(0.065g)和实施例2.31.3(0.067g)的n,n-二甲基甲酰胺(0.75ml)溶液中加入n,n-二异丙胺(0.065ml)。6小时之后,再加入n,n-二异丙胺(0.025ml),并将该反应混合物搅拌过夜。用乙酸乙酯(50ml)稀释该反应,并用水(20ml)洗涤,而后用盐水(20ml)洗涤。用硫酸镁干燥乙酸乙酯层,过滤,并浓缩,得到标题化合物,其不用进一步纯化,在下一步中直接使用。
2.31.5. 3-(1-((3-(2-((((2-(3-氨基丙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例2.31.4(0.064g)溶于甲醇(0.75ml)中,并用氢氧化锂一水合物(0.031g)在水(0.75ml)中的溶液处理。搅拌2小时之后,用n,n-二甲基甲酰胺(1ml)稀释该反应,并用三氟乙酸(0.057ml)淬灭。用制备hplc纯化该溶液,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。
2.31.6. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-(3-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}丙氧基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.31.5(0.020g)和2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(5.8mg)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙胺(0.014ml)。搅拌2小时之后,用n,n-二甲基甲酰胺(1.5ml)和水(0.5ml)稀释该反应。用制备hplc纯化该溶液,使用gilson系统,用10-75%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.83(s,1h),8.03(d,1h),7.83(t,1h),7.79(d,1h),7.62(d,1h),7.54-7.42(m,3h),7.37(d,1h),7.34(d,1h),7.28(s,1h),7.19(d,1h),6.98(s,2h),6.95(d,1h),6.64(d,1h),6.59(d,1h),5.05(t,1h),4.96(d,4h),4.02-3.94(m,2h),3.88(t,2h),3.46-3.22(m,14h),3.18(q,2h),3.01(t,2h),2.85(d,3h),2.09(s,3h),2.02(t,2h),1.81(p,2h),1.54-1.41(m,4h),1.41-0.78(m,18h)。ms(esi)m/e1350.5(m-h)-。
2.32. 合成1-o-({4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基}氨基甲酰基)-β-d-吡喃葡萄糖醛酸(glucopyranuronicacid)(合成子fu)
2.32.1. 2-氨基-5-(羟甲基)苯酚
在-78℃,用5分钟将二异丁基氢化铝(1m,在二氯甲烷中,120ml)加入到4-氨基-3-羟基苯甲酸甲酯(10g)/50ml二氯甲烷中,并将该溶液升温至0℃。将该反应混合物搅拌2小时。再加入60ml二异丁基氢化铝(1m,在二氯甲烷中),并将该反应在0℃下再搅拌一小时。小心地加入甲醇(40ml)。加入饱和酒石酸钾钠溶液(100ml),并将该混合物搅拌过夜。用乙酸乙酯提取该混合物两次,将合并的提取物浓缩至大约100ml的体积,并过滤该混合物。收集固体,将溶液浓缩至很少的体积,并过滤。将合并的固体干燥,得到标题化合物。
2.32.2. 2-(2-叠氮基乙氧基)乙基4-甲基苯磺酸酯
向环境温度的2-(2-叠氮基乙氧基)乙醇(4.85g)、三乙胺(5.16ml)和n,n-二甲基吡啶-4-胺(0.226g)的二氯甲烷(123ml)溶液中加入4-甲苯-1-磺酰氯(7.05g)。将该反应搅拌过夜,通过加入二氯甲烷和饱和氯化铵水溶液,淬灭该反应。分离各层,并用盐水洗涤有机层两次。将有机层用无水硫酸钠干燥,过滤,减压浓缩,提供标题化合物,其不用进一步纯化,在随后的反应中直接使用。ms(esi)m/e302.9(m+nh4)+。
2.32.3. (4-氨基-3-(2-(2-叠氮基乙氧基)乙氧基)苯基)甲醇
向环境温度的实施例2.32.1(0.488g)的n,n-二甲基甲酰胺(11.68ml)溶液中加入氢化钠(0.140g)。将该混合物搅拌0.5小时,并加入实施例2.32.2(1.0g)的n,n-二甲基甲酰胺(2.0ml)溶液。将该反应加热至50℃过夜。加入水和乙酸乙酯,淬灭该反应混合物。分离各层,并用乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,并减压浓缩。用硅胶色谱纯化残余物,用梯度25-100%的乙酸乙酯洗脱,得到标题化合物。ms(esi)m/e253.1(m+h)+。
2.32.4. 2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯胺
向环境温度的实施例2.32.3(440mg)和咪唑(178mg)的四氢呋喃(10.6ml)溶液中加入叔丁基二甲基氯硅烷(289mg)。将该反应混合物搅拌16个小时,并加入乙酸乙酯(30ml)和饱和碳酸氢钠水溶液(20ml),淬灭该反应。分离各层,并用乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,并减压浓缩。用硅胶色谱纯化残余物,用梯度0至50%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e366.9(m+h)。
2.32.5. (2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在50ml干燥圆底烧瓶中,在高真空下,将实施例2.32.4(410mg)干燥过夜。向冷(0℃浴温)的实施例2.32.4(410mg)和三乙胺(0.234ml)的甲苯(18ml)溶液中加入光气(0.798ml,1m,在二氯甲烷中)。将该反应慢慢地升温至室温,并搅拌一小时。将该反应冷却(0℃浴温),并加入(3r,4s,5s,6s)-2-羟基-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(411mg)和三乙胺(0.35ml)的甲苯(5ml)溶液。将该反应升温至室温,并加热到50℃,保持2小时。加入饱和碳酸氢盐水溶液和乙酸乙酯,淬灭该反应。分离各层,并用乙酸乙酯提取水层两次。将合并的有机层用无水硫酸钠干燥,过滤,并减压浓缩。用硅胶色谱纯化残余物,用梯度0-40%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e743.9(m+nh4)+。
2.32.6. (2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-(羟甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.32.5(700mg)的甲醇(5ml)溶液中加入对甲苯磺酸一水合物(18.32mg)的甲醇(2ml)溶液。将该反应在室温下搅拌1小时。加入饱和碳酸氢钠水溶液和二氯甲烷,淬灭该反应。分离各层,并用额外的二氯甲烷提取水层。将合并的有机物用mgso4干燥,过滤,减压蒸发溶剂,得到标题化合物,其不用进一步纯化,在随后的步骤中直接使用。ms(esi)m/e629.8(m+nh4)+。
2.32.7. (2s,3r,4s,5s,6s)-2-(((2-(2-(2-叠氮基乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯基)氨基甲酰基)氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将n,n-二异丙基乙胺(0.227ml)逐滴加入到环境温度的实施例2.32.6(530mg)和双(4-硝基苯基)碳酸酯(395mg)的n,n-二甲基甲酰胺(4.3ml)溶液中。将该反应混合物在环境温度下搅拌1.5小时。减压浓缩溶剂。用硅胶色谱纯化残余物,用梯度0-50%的乙酸乙酯/庚烷洗脱,得到标题化合物。ms(esi)m/e794.9(m+nh4)+。
2.32.8. 3-(1-((3-(2-((((3-(2-(2-叠氮基乙氧基)乙氧基)-4-(((((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)羰基)氨基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例1.1.17的三氟乙酸盐(111mg)和实施例2.32.7(98.5mg)的n,n-二甲基甲酰胺(3.5ml)溶液中加入n,n-二异丙基乙胺(0.066ml)。将该反应慢慢地升温至室温,并搅拌16小时。加入水和乙酸乙酯,淬灭该反应。分离各层,并用乙酸乙酯提取水层两次。将合并的有机物用无水硫酸钠干燥,过滤,减压浓缩,得到标题化合物,其不用进一步纯化,在随后的步骤中直接使用。ms(esi)m/e1398.2(m+h)+。
2.32.9. 3-(1-((3-(2-((((3-(2-(2-叠氮基乙氧基)乙氧基)-4-(((((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)羰基)氨基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向冷(0℃)的实施例2.32.8(150mg)的甲醇(3.0ml)溶液中加入2m氢氧化锂溶液(0.804ml)。将该反应搅拌1小时,并在0℃,加入乙酸(0.123ml),淬灭该反应。用反相hplc纯化该粗品反应溶液,使用带有c18柱的gilson系统,用梯度10-100%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将含有产物的级分冷冻干燥,得到标题化合物。ms(esi)m/e1258.2(m+h)+。
2.32.10. 3-(1-((3-(2-((((3-(2-(2-氨基乙氧基)乙氧基)-4-(((((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)羰基)氨基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向溶于2∶1的四氢呋喃∶水(0.3ml)中的实施例2.32.9(45mg)溶液中加入三(2-羧乙基))膦盐酸盐溶液(51.3mg,在0.2ml水中)。将该反应在室温下搅拌16小时。部分地减压浓缩溶剂,除去大部分四氢呋喃。用反相hplc纯化粗品反应物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物的三氟乙酸盐。ms(esi)m/e1232.3(m+h)+。
2.32.11. 1-o-({4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]氨基}乙氧基)乙氧基]苯基}氨基甲酰基)-β-d-吡喃葡萄糖醛酸(glucopyranuronicacid)
向实施例2.32.10的三氟乙酸盐(15mg)的1mln,n-二甲基甲酰胺溶液中加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(4.12mg)和n,n-二异丙基乙胺(0.010ml),并将该反应在室温下搅拌16小时。用反相hplc纯化该粗品反应混合物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.84(s,1h),8.58(d,1h),8.03(d,1h),7.79(t,2h),7.68(s,1h),7.61(d,1h),7.40-7.54(m,3h),7.36(q,2h),7.27(s,1h),7.05(s,1h),6.97(s,2h),6.93(t,2h),5.41(d,hz,1h),5.38(d,1h),5.27(d,1h),4.85-5.07(m,4h),4.11(t,2h),3.87(t,2h),3.80(s,2h),3.71-3.77(m,3h),3.46(s,3h),3.22(d,2h),3.00(t,2h),2.86(d,3h),2.08(s,3h),2.01(t,2h),1.44(dd,4h),1.34(d,2h),0.89-1.29(m,16h),0.82(d,7h),3.51-3.66(m,3h)。ms(esi)m/e1447.2(m+na)+。
2.33. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[3-(2-{[({3-[(n-{[2-({n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-3-磺基-d-丙氨酰}氨基)乙氧基]乙酰基}-β-丙氨酰)氨基]-4-(β-d-吡喃半乳糖基氧基)苄基}氧基)羰基](甲基)氨基}乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸(合成子gh)
2.33.1. (r)-28-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-7,10,26-三氧代-8-(磺基甲基)-3,13,16,19,22-五氧杂-6,9,25-三氮杂二十八烷-1-酸
使用本文所描述的固相肽合成方法,合成标题化合物。将2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙酸(1543mg)溶于10ml二噁烷中,并减压浓缩溶剂(将该过程重复两次)。将该物质冷冻干燥过夜。将二噁烷-干燥的氨基酸溶于20ml分子筛干燥的二氯甲烷中,并向其中加入n,n-二异丙基乙胺(4.07ml)。将该溶液加入到预先用分子筛干燥的二氯甲烷洗涤(两次)的2-氯三苯甲基固体载体树脂(8000mg)中。将树脂和氨基酸的混合物在环境温度下摇动4小时,排出,用17∶2∶1的二氯甲烷∶甲醇∶n,n-二异丙基乙胺洗涤,并用n,n-二甲基甲酰胺洗涤三次。然后,将该混合物反复用分子筛干燥的二氯甲烷和甲醇交替再洗涤三次。将加载的树脂在真空烘箱中、在40℃下干燥。利用定量fmoc加载试验,将通过用20%哌啶/n,n-二甲基甲酰胺处理而去保护已知数量的树脂所获得的溶液,在301nm下测定吸光度,测定树脂加载量。通过用20%哌啶/n,n-二甲基甲酰胺处理树脂20分钟,进行所有的fmoc脱保护步骤,而后进行n,n-二甲基甲酰胺洗涤步骤。在n,n-二甲基甲酰胺中用4当量((1h-苯并[d][1,2,3]三唑-1-基)氧基)三(吡咯烷-1-基)鏻六氟磷酸盐(v)和8当量n,n-二异丙基乙胺(n,n-diidopropylethylamine)将4当量氨基酸活化一分钟,而后用树脂培养一小时,使氨基酸(r)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-磺基丙酸与随后的1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸缀合。通过用5%三氟乙酸/二氯甲烷处理30分钟,使标题化合物从树脂上断裂下来。滤出树脂,并将滤液减压浓缩,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e669.0(m+h)+。
2.33.2. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[3-(2-{[({3-[(n-{[2-({n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-3-磺基-d-丙氨酰}氨基)乙氧基]乙酰基}-β-丙氨酰)氨基]-4-(β-d-吡喃半乳糖基氧基)苄基}氧基)羰基](甲基)氨基}乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸
将实施例2.33.1(5.09mg)与2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(2.63mg)和n,n-二异丙基乙胺(0.004ml)在1mln,n-二甲基甲酰胺中混合,并搅拌两分钟。加入实施例2.28.8(8.8mg),并将该反应混合物在室温下搅拌1.5小时。用反相hplc纯化该粗品反应混合物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(esi)m/e1806.5(m-h)-。
2.34. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[3-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰}氨基)丙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子fx)
2.34.1. 3-(1-((3-(2-((((2-(3-((r)-2-氨基-3-磺基丙酰胺基)丙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
向(r)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)-3-磺基丙酸(0.019g)和2-(3h-[1,2,3]三唑并[4,5-b]吡啶-3-基)-1,1,3,3-四甲基异脲六氟磷酸盐(v)(0.019g)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙胺(7.82µl)。搅拌2分钟之后,在室温下,将反应物加入到实施例2.31.5(0.057g)和n,n-二异丙胺(0.031ml)的n,n-二甲基甲酰胺(0.5ml)溶液中,并搅拌3小时。将二乙胺(0.023ml)加入到该反应中,并继续搅拌2小时。用水(1ml)稀释该反应,用三氟乙酸(0.034ml)淬灭,并用制备hplc纯化该溶液,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。ms(esi)m/e1310.1(m+h)+。
2.34.2. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[3-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰}氨基)丙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
向实施例2.34.1(0.0277g)和2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(7.82mg)的n,n-二甲基甲酰胺(0.5ml)溶液中加入n,n-二异丙胺(0.018ml),并在室温下搅拌该反应。用制备hplc纯化该反应,使用gilson系统,用10-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.81(s,1h),8.02(d,1h),7.89-7.81(m,2h),7.78(d,1h),7.60(d,1h),7.53-7.40(m,3h),7.39-7.31(m,2h),7.29(s,1h),7.16(d,1h),6.98-6.92(m,3h),6.63(s,1h),6.56(d,1h),5.08-4.99(m,1h),4.95(s,4h),4.28(q,2h),3.90-3.85(m,4h),3.48-3.06(m,12h),3.00(t,2h),2.88-2.64(m,8h),2.08(s,3h),2.04(t,2h),1.80(p,2h),1.51-1.39(m,4h),1.39-0.75(m,18h)。ms(esi)m/e1501.4(m-h)-。
2.35. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子h)
2.35.1. (2s,3r,4s,5s,6s)-2-(4-甲酰基-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向(2r,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(4g)的乙腈(100ml)溶液中加入氧化银(i)(10.04g)和4-羟基-3-硝基苯甲醛(1.683g)。将该反应混合物在室温下搅拌4小时,并过滤。浓缩滤液,用硅胶色谱纯化残余物,用5-50%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(esi)m/e(m+18)+。
2.35.2. (2s,3r,4s,5s,6s)-2-(4-(羟甲基)-2-硝基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向在氯仿(75ml)和异丙醇(18.75ml)的混合物中的实施例2.35.1(6g)溶液中加入0.87g硅胶。将得到的混合物冷却到0℃,加入nabh4(0.470g),并将得到的悬浮液在0℃下搅拌45分钟。将该反应混合物用二氯甲烷(100ml)稀释,并通过硅藻土过滤。用水和盐水洗涤滤液,浓缩,得到粗品,其不用进一步纯化,直接使用。ms(esi)m/e(m+nh4)+。
2.35.3. (2s,3r,4s,5s,6s)-2-(2-氨基-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
使用10%pd/c(1.535g)作为催化剂,将实施例2.35.2(7g)的乙酸乙酯(81ml)搅拌溶液在20℃、在1大气压h2中氢化12小时。通过硅藻土过滤该反应混合物,并将溶剂减压蒸发。用硅胶色谱纯化残余物,用95/5的二氯甲烷/甲醇洗脱,得到标题化合物。
2.35.4. 3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酸
在500ml烧瓶中,将3-氨基丙酸(4.99g)溶于10%na2co3水溶液(120ml)中,并用冰浴冷却。向所得到的溶液中逐渐地加入(9h-芴-9-基)甲基氯甲酸酯(14.5g)/1,4-二噁烷(100ml)。将该反应混合物在室温下搅拌4小时,然后加入水(800ml)。将水相层与反应混合物分离,并用乙醚(3x750ml)洗涤。将水层用2nhcl水溶液酸化到ph2,并用乙酸乙酯(3x750ml)提取。将有机层合并,并浓缩,获得粗品。将粗品在乙酸乙酯∶己烷(1:2,300ml)的混合溶剂中重结晶,得到标题化合物。
2.35.5. (9h-芴-9-基)甲基(3-氯-3-氧代丙基)氨基甲酸酯
向实施例2.35.4的二氯甲烷(160ml)溶液中加入亚硫酰氯(50ml)。将该混合物在60℃下搅拌1小时。将该混合物冷却,并浓缩,得到标题化合物。
2.35.6. (2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.35.3(6g)的二氯甲烷(480ml)溶液中加入n,n-二异丙基乙胺(4.60ml)。加入实施例2.35.5(5.34g),并将该混合物在室温下搅拌30分钟。将该混合物倒入饱和碳酸氢钠水溶液中,并用乙酸乙酯提取。将合并的提取物用水和盐水洗涤,并用硫酸钠干燥。过滤,浓缩,得到残余物,通过径向色谱纯化残余物,使用0-100%的乙酸乙酯/石油醚作为流动相,得到标题化合物。
2.35.7. (2s,3r,4s,5s,6s)-2-(2-(3-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酰胺基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.35.6(5.1g)和n,n-二甲基甲酰胺(200ml)的混合物中加入双(4-硝基苯基)碳酸酯(4.14g)和n,n-二异丙基乙胺(1.784ml)。将该混合物在室温下搅拌16小时,并减压浓缩。将粗品溶于二氯甲烷中,并直接吸到1mm径向色谱板上,用50-100%的乙酸乙酯/己烷洗脱,得到标题化合物。ms(esi)m/e(m+h)+。
2.35.8. 3-(1-((3-(2-((((3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在0℃,向实施例1.1.17(325mg)和实施例2.35.7(382mg)的n,n-二甲基甲酰胺(9ml)溶液中加入n,n-二异丙胺(49.1mg)。将该反应混合物在0℃下搅拌5小时,并加入乙酸(22.8mg)。将所得到的混合物用乙酸乙酯稀释,并用水和盐水洗涤。用na2so4干燥有机层,过滤,并浓缩。将残余物溶于四氢呋喃(10ml)和甲醇(5ml)的混合物中。在0℃,向此溶液中加入1m氢氧化锂水溶液(3.8ml)。将得到的混合物在0℃下搅拌1小时,用乙酸酸化,并浓缩。将浓缩物冷冻干燥,提供粉末。将该粉末溶于n,n-二甲基甲酰胺(10ml)中,在冰浴中冷却,并在0℃下加入哌啶(1ml)。将该混合物在0℃下搅拌15分钟,并加入1.5ml乙酸。通过反相hplc纯化该溶液,使用gilson系统,用30-80%的乙腈/水(包含0.1%v/v三氟乙酸)洗脱,提供标题化合物。ms(esi)m/e1172.2(m+h)+。
2.35.9. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
在0℃,向n,n-二甲基甲酰胺(5ml)中的实施例2.35.8(200mg)中加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(105mg)和n,n-二异丙基乙胺(0.12ml)。将该混合物在0℃下搅拌15分钟,升温至室温,并在gilson系统上用反相hplc纯化,使用100gc18柱,用30-80%的乙腈/水(包含0.1%v/v三氟乙酸)洗脱,提供标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.85(s,2h)9.07(s,1h)8.18(s,1h)8.03(d,1h)7.87(t,1h)7.79(d,1h)7.61(d,1h)7.41-7.53(m,3h)7.36(q,2h)7.28(s,1h)7.03-7.09(m,1h)6.96-7.03(m,3h)6.94(d,1h)4.95(s,4h)4.82(t,1h)3.88(t,3h)3.80(d,2h)3.01(t,2h)2.86(d,3h)2.54(t,2h)2.08(s,3h)2.03(t,2h)1.40-1.53(m,4h)1.34(d,2h)0.90-1.28(m,12h)0.82(d,6h)。ms(esi)m/e1365.3(m+h)+。
2.36. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[19-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-17-氧代-4,7,10,13-四氧杂-16-氮杂十九烷-1-酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子i)
使用实施例2.35.9的方法,用2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm8.95(s,1h)8.16(s,1h)7.99(d,1h)7.57-7.81(m,4h)7.38-7.50(m,3h)7.34(q,2h)7.27(s,1h)7.10(d,1h)7.00(d,1h)6.88-6.95(m,2h)4.97(d,4h)4.76(d,2h)3.89(t,2h)3.84(d,2h)3.80(s,2h)3.57-3.63(m,4h)3.44-3.50(m,4h)3.32-3.43(m,6h)3.29(t,2h)3.16(q,2h)3.02(t,2h)2.87(s,3h)2.52-2.60(m,2h)2.29-2.39(m,3h)2.09(s,3h)1.37(s,2h)1.20-1.29(m,4h)1.06-1.18(m,4h)0.92-1.05(m,2h)0.83(s,6h)。ms(esi)m/e1568.6(m-h)-。
2.37. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丁酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子kq)
使用实施例2.35.9的方法,用2,5-二氧代吡咯烷-1-基4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丁酸酯替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.86(s,3h)9.08(s,2h)8.17(s,1h)8.03(d,1h)7.89(t,1h)7.79(d,1h)7.61(d,1h)7.46-7.53(m,1h)7.41-7.46(m,1h)7.31-7.40(m,1h)7.28(s,1h)7.03-7.10(m,1h)6.91-7.03(m,2h)4.69-5.08(m,4h)3.83-3.95(m,2h)3.74-3.83(m,2h)3.21-3.47(m,12h)2.95-3.08(m,1h)2.86(d,2h)1.98-2.12(m,3h)1.62-1.79(m,2h)0.90-1.43(m,8h)0.82(d,3h)。ms(esi)m/e1337.2(m+h)+。
2.38. 合成4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]-2-{[n-({2-[2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基]乙氧基}乙酰基)-β-丙氨酰]氨基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子kp)
2.38.1. 3-(1-((-((1-(3-(3-氨基丙酰胺基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苯基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-12-基)氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.35.8中,用实施例1.2.11替代实施例1.1.17,制备标题化合物。
2.38.2. 4-[12-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)-4-甲基-3-氧代-2,7,10-三氧杂-4-氮杂十二烷-1-基]-2-{[n-({2-[2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基]乙氧基}乙酰基)-β-丙氨酰]氨基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
在实施例2.35.9中,用实施例2.38.1替代实施例2.35.8,用2,5-二氧代吡咯烷-1-基2-(2-(2-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)乙氧基)乙氧基)乙酸酯替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm8.92(s,1h),8.12-8.15(m,1h),7.97(d,1h),7.76(d,1h),7.61(d,1h),7.28-7.49(m,6h),7.25(s,1h),7.09(d,1h),6.97-7.02(m,1h),6.88-6.94(m,2h),4.97(d,4h),4.75(d,1h),3.76-3.93(m,9h),3.47-3.60(m,16h),3.32-3.47(m,15h),2.88(s,3h),2.59(t,2h),2.08(s,3h),1.38(s,2h),0.93-1.32(m,11h),0.84(s,6h)。ms(esi)m/e1485.2(m+h)+。
2.39. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-[(n-{6-[(乙烯基磺酰基)氨基]己酰基}-β-丙氨酰)氨基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子ha)
2.39.1. 6-(乙烯基磺酰胺基)己酸甲酯
在0℃向6-甲氧基-6-氧代己-1-铵氯化物(0.3g)和三乙胺(1.15ml)的二氯甲烷溶液中逐滴加入乙磺酰氯(0.209g)。将该反应混合物升温至室温,并搅拌1小时。将该混合物用二氯甲烷稀释,并用盐水洗涤。用硫酸钠干燥有机层,过滤,并浓缩,提供标题化合物。ms(esi)m/e471.0(2m+h)+。
2.39.2. 6-(乙烯基磺酰胺基)己酸
将在四氢呋喃(1ml)和水(1ml)的混合物中的实施例2.39.1(80mg)和氢氧化锂一水合物(81mg)溶液搅拌2小时,然后用水(20ml)稀释,并用乙醚(10ml)洗涤。用1nhcl水溶液将水层酸化至ph4,并用二氯甲烷(3x10ml)提取。用盐水(5ml)洗涤有机层,用硫酸钠干燥,过滤,浓缩,提供标题化合物。
2.39.3. 2,5-二氧代吡咯烷-1-基6-(乙烯基磺酰胺基)己酸酯
将实施例2.39.2(25mg)、1-乙基-3-[3-(二甲基氨基)丙基]-碳二亚胺盐酸盐(43.3mg)和1-羟基吡咯烷-2,5-二酮(15.6mg)在二氯甲烷(8ml)中的混合物搅拌过夜,用饱和氯化铵水溶液和盐水洗涤,并浓缩,提供标题化合物。
2.39.4. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-[(n-{6-[(乙烯基磺酰基)氨基]己酰基}-β-丙氨酰)氨基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
使用实施例2.35.9的方法,用实施例2.39.3替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.85(s,2h)9.07(s,1h)8.18(s,1h)8.03(d,1h)7.87(t,1h)7.79(d,1h)7.61(d,1h)7.41-7.53(m,3h)7.33-7.39(m,2h)7.28(s,1h)7.17(t,1h)7.04-7.08(m,1h)6.98-7.03(m,1h)6.95(d,1h)6.65(dd,1h)5.91-6.04(m,2h)4.96(s,4h)4.82(s,1h)3.22-3.48(m,11h)3.01(t,2h)2.86(d,3h)2.73-2.80(m,2h)2.51-2.57(m,2h)1.99-2.12(m,5h)1.29-1.52(m,6h)0.90-1.29(m,12h)0.82(d,6h)。ms(esi)m/e1375.3(m+h)+。
2.40. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[6-(乙烯基磺酰基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子hb)
2.40.1. 6-((2-羟乙基)硫代)己酸乙酯
将6-溴己酸乙酯(3g)、2-巯基乙醇(0.947ml)和k2co3(12g)在乙醇(100ml)中的混合物搅拌过夜,并过滤。浓缩滤液。将残余物溶于二氯甲烷(100ml)中,并用水和盐水洗涤。用na2so4干燥有机层,过滤,浓缩,提供标题化合物。
2.40.2. 6-((2-羟乙基)硫代)己酸
使用实施例2.39.2的方法,用实施例2.40.1替代实施例2.39.2,制备标题化合物。ms(esi)m/e175.1(m-h2o)-。
2.40.3. 6-((2-羟乙基)磺酰基)己酸
向在水(40ml)和1,4-二噁烷(160ml)的混合物中的实施例2.40.2(4g)的搅拌溶液中加入oxone®(38.4g)。将该混合物搅拌过夜。过滤该混合物,并将滤液浓缩。用二氯甲烷提取剩余的水层。将提取物合并,用na2so4干燥,过滤,浓缩,提供标题化合物。
2.40.4. 6-(乙烯基磺酰基)己酸
在氩气氛围中,在0℃,向实施例2.40.3(1g)的二氯甲烷(10ml)搅拌溶液中加入三乙胺(2.8ml),而后加入甲磺酰氯(1.1ml)。将该混合物搅拌过夜,并用水和盐水洗涤。用硫酸钠干燥有机层,过滤,并浓缩,提供标题化合物。
2.40.5. 2,5-二氧代吡咯烷-1-基6-(乙烯基磺酰基)己酸酯
向实施例2.40.4(0.88g)的二氯甲烷(10ml)搅拌溶液中加入1-羟基吡咯烷-2,5-二酮(0.54g)和n,n'-二环己基碳二亚胺(methanediylidenedicyclohexanamine)(0.92g)。将该混合物搅拌过夜,并过滤。浓缩滤液,并用快速色谱纯化,用10-25%的乙酸乙酯/石油醚洗脱,提供标题化合物。ms(esi)m/e304.1(m+h)+。
2.40.6. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-2-({n-[6-(乙烯基磺酰基)己酰基]-β-丙氨酰}氨基)苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
使用实施例2.35.9的方法,用实施例2.40.5替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.84(s,2h)9.07(s,1h)8.18(s,1h)8.03(d,1h)7.89(t,1h)7.79(d,1h)7.61(d,1h)7.41-7.53(m,3h)7.32-7.40(m,2h)7.28(s,1h)7.04-7.11(m,1h)6.98-7.03(m,1h)6.88-6.97(m,2h)6.17-6.26(m,2h)4.95(s,4h)4.82(s,1h)3.74-3.99(m,8h)3.41-3.46(m,8h)3.24-3.41(m,8h)2.97-3.08(m,4h)2.86(d,3h)2.54(t,2h)2.00-2.13(m,5h)1.43-1.64(m,4h)0.89-1.40(m,15h)0.82(d,6h)。ms(esi)m/e1360.2(m+h)+。
2.41. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]-3-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子lb)
2.41.1. 3-(1-((3-(2-((((2-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-3,4,5-三乙酰氧基-6-(甲氧羰基)四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.26.7中,用实施例1.6.13替代实施例1.1.17,制备标题化合物。
2.41.2. 3-(1-((3-(2-((((2-(2-(2-氨基乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.26.8中,用实施例2.41.1替代实施例2.26.7,制备标题化合物。ms(esi)m/e1193(m+h)+,1191(m-h)-。
2.41.3. 4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-5-氟-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]-3-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
在实施例2.27中,用实施例2.41.2替代实施例2.26.8,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.88(bs,1h),8.03(d,1h),8.02(t,1h),7.78(d,1h),7.73(1h),7.53(d,1h),7.47(td,1h),7.35(td,1h),7.29(s,1h),7.26(t,1h),7.26(t,1h),7.19(d,1h),7.02(d,1h),6.98(s,1h),6.65(d,1h),6.59(dd,1h),5.07(d,1h),5.01(s,1h),4.92(1h),4.08(m,2h),3.94(t,2h),3.90(d,2h),3.87(s,2h),3.70(m,6h),3.60(m,6h),3.44(t,2h),3.39(t,2h),3.32(t,1h),3.28(dd,1h),3.17(q,2h),3.03(q,2h),2.92(t,2h),2.33(t,2h),2.10(s,3h),1.37(s,2h),1.25(q,4h),1.11(q,4h),1.00(dd,2h),0.83(s,6h)。ms(esi)m/e1366(m+na)+,1342(m-h)-。
2.42. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-{2-[2-({n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-3-磺基-l-丙氨酰}氨基)乙氧基]乙氧基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子nf)
2.42.1. (2s,3r,4s,5s,6s)-2-(4-甲酰基-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将2,4-二羟基苯甲醛(15g)和(2s,3r,4s,5s,6s)-2-溴-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯(10g)溶于乙腈中,而后加入碳酸银(10g),并将该反应加热至49℃。搅拌4小时之后,冷却该反应,过滤,并浓缩。将粗品标题化合物悬浮在二氯甲烷中,通过硅藻土过滤,并浓缩。用硅胶色谱纯化残余物,用1-100%的乙酸乙酯/庚烷洗脱,提供标题化合物。
2.42.2. (2s,3r,4s,5s,6s)-2-(3-羟基-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.42.1(16.12g)的四氢呋喃(200ml)和甲醇(200ml)溶液冷却至0℃,并分批加入硼氢化钠(1.476g)。将该反应搅拌20分钟,并用水∶饱和碳酸氢钠溶液的1:1混合物(400ml)淬灭。滤出所得到的固体,并用乙酸乙酯冲洗。分离各相,并用乙酸乙酯提取水层四次。将合并的有机层用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品标题化合物,用1-100%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(esi)m/e473.9(m+nh4)+。
2.42.3. (2s,3r,4s,5s,6s)-2-(4-(((叔丁基二甲基甲硅烷基)氧基)甲基)-3-羟基苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
在-5℃,向实施例2.42.2(7.66g)和在二氯甲烷(168ml)中的叔丁基二甲基甲硅烷基氯(2.78g)中加入咪唑(2.63g),并将该反应混合物搅拌过夜,使得反应的内部温度升至12℃。将该反应混合物倒入饱和氯化铵水溶液中,并用二氯甲烷提取四次。将合并的有机物用盐水洗涤,用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品标题化合物,用1-50%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(esi)m/e593.0(m+na)+。
2.42.4. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(((叔丁基二甲基甲硅烷基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.42.3(5.03g)和在甲苯(88ml)中的三苯基膦(4.62g)中加入偶氮二甲酸二叔丁基酯(4.06g),并将该反应搅拌30分钟。加入(9h-芴-9-基)甲基(2-(2-羟乙氧基)乙基)氨基甲酸酯,并将该反应再搅拌1.5小时。将反应物直接加载到硅胶上,用1-50%的乙酸乙酯/庚烷洗脱,提供标题化合物。
2.42.5. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-(羟甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
将实施例2.42.4(4.29g)在3∶1∶1的乙酸∶水∶四氢呋喃溶液(100ml)中搅拌过夜。将反应物倒入饱和碳酸氢钠水溶液中,并用乙酸乙酯提取。将有机层用硫酸镁干燥,过滤,并浓缩。通过硅胶色谱纯化粗品标题化合物,用1-50%的乙酸乙酯/庚烷洗脱,提供标题化合物。
2.42.6. (2s,3r,4s,5s,6s)-2-(3-(2-(2-((((9h-芴-9-基)甲氧基)羰基)氨基)乙氧基)乙氧基)-4-((((4-硝基苯氧基)羰基)氧基)甲基)苯氧基)-6-(甲氧羰基)四氢-2h-吡喃-3,4,5-三基三乙酸酯
向实施例2.42.5(0.595g)和双(4-硝基苯基)碳酸酯(0.492g)的n,n-二甲基甲酰胺(4ml)溶液中加入n-乙基-n-异丙基丙-2-胺(0.212ml)。1.5小时之后,在高真空下浓缩该反应。将反应物直接加载到硅胶上,使用1-50%的乙酸乙酯/庚烷洗脱,提供标题化合物。ms(esi)m/e922.9(m+na)+。
2.42.7. 3-(1-((3-(2-((((2-(2-(2-氨基乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例1.1.17(92mg)溶于二甲基甲酰胺(0.6ml)中。加入实施例2.42.6(129mg)和n-乙基-n-异丙基丙-2-胺(0.18ml)。将该反应在室温下搅拌一小时。然后,浓缩该反应,并将残余物溶于四氢呋喃(0.6ml)和甲醇(0.6ml)中。加入lioh水溶液(1.94n,0.55ml),并将该混合物在室温下搅拌一小时。用反相色谱纯化(c18柱),用10-90%的乙腈/水中的0.1%tfa洗脱,提供标题化合物的三氟乙酸盐。ms(esi)m/e1187.4(m-h)-。
2.42.8. 3-(1-((3-(2-((((2-(2-(2-((r)-2-氨基-3-磺基丙酰胺基)乙氧基)乙氧基)-4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.34.1中,用实施例2.26.8替代实施例2.31.5,制备标题化合物。ms(esi)m/e1338.4(m-h)-。
2.42.9. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(1-((3-(2-((((4-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)-2-(2-(2-((r)-2-(3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰胺基)-3-磺基丙酰胺基)乙氧基)乙氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸
在实施例2.34.2中,用实施例2.42.2替代实施例2.34.1,用2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm8.06(d,1h),8.02(d,1h),7.80(m,2h),7.61(d,1h),7.52(d,1h),7.45(m,2h),7.36(m,2h),7.30(s,1h),7.18(d,1h),6.97(s,2h),6.96(m,2h),6.66(d,1h),6.58(dd,1h),5.06(brm,1h),4.96(s,4h),4.31(m,1h),4.09(m,2h),3.88(m,3h),3.80(m,2h),3.71(m,2h),3.59(t,2h),3.44(m,6h),3.28(m,4h),3.19(m,2h),3.01(m,2h),2.82(brm,3h),2.72(m,1h),2.33(m,2h),2.09(s,3h),1.33(brm,2h),1.28-0.90(m,10h),0.84,0.81(boths,total6h)。ms(esi-)m/e1489.5(m-1)。
2.43. 合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-{2-[2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰}氨基)乙氧基]乙氧基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)(合成子ng)
在实施例2.34.2中,用实施例2.42.1替代实施例2.34.1,制备标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm8.02(d,1h),7.87(d,1h),7.80(m,2h),7.61(d,1h),7.52(d,1h),7.45(m,2h),7.36(m,2h),7.30(s,1h),7.18(d,1h),6.97(s,2h),6.96(m,2h),6.66(d,1h),6.58(dd,1h),5.06(brm,1h),4.96(s,4h),4.31(m,1h),4.09(m,2h),3.88(m,3h),3.80(m,2h),3.71(m,2h),3.59(t,2h),3.44(m,6h),3.28(m,4h),3.19(m,2h),3.01(m,2h),2.82(brm,3h),2.72(m,1h),2.09(s,3h),2.05(t,2h),1.46(brm,4h),1.33(brm,2h),1.28-0.90(m,12h),0.84,0.81(两个都是s,共6h)。ms(esi-)m/e1531.5(m-1)。
2.44. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{[22-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-甲基-4,20-二氧代-7,10,13,16-四氧杂-3,19-二氮杂二十二烷-1-基]氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子as)
向实施例1.1.17(56.9mg)和n,n-二异丙基乙胺(0.065ml)的n,n-二甲基甲酰胺(1.0ml)溶液中加入2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯(50mg)。将该反应搅拌过夜,并用反相hplc纯化该溶液,使用gilson系统,用20-80%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz二甲亚砜-d6)δppm12.85(s,1h),8.08-7.95(m,1h),7.79(d,1h),7.62(d,1h),7.55-7.40(m,3h),7.40-7.32(m,2h),7.28(s,1h),7.01-6.89(m,3h),4.95(s,2h),3.89(s,2h),3.81(s,2h),3.55-3.25(m,23h),3.14(d,2h),2.97(t,4h),2.76(d,2h),2.57(s,1h),2.31(d,1h),2.09(s,3h),1.35(s,2h),1.30-0.93(m,12h),0.85(d,6h)。ms(esi)m/e1180.3(m+na)+。
2.45. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{[28-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-9-甲基-10,26-二氧代-3,6,13,16,19,22-六氧杂-9,25-二氮杂二十八烷-1-基]氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子at)
向实施例1.2.11(50mg)和n,n-二异丙基乙胺(0.051ml)的n,n-二甲基甲酰胺(1.0ml)溶液中加入2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯(39mg)。将该反应搅拌过夜,并用反相hplc纯化,使用gilson系统,用20-80%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz二甲亚砜-d6)δppm12.85(s,1h),8.04(d,1h),7.99(t,1h),7.79(d,1h),7.60(d,1h),7.53-7.41(m,3h),7.40-7.32(m,2h),7.28(s,1h),6.99(s,2h),6.98-6.92(m,1h),4.95(bs,2h),3.92-3.85(m,1h),3.81(s,2h),3.63-3.55(m,4h),3.55-3.31(m,28h),3.18-3.10(m,2h),3.05-2.98(m,2h),2.97(s,2h),2.80(s,2h),2.59-2.50(m,1h),2.32(t,2h),2.10(s,3h),1.39-1.34(m,2h),1.31-1.18(m,4h),1.20-0.92(m,6h),0.84(s,6h)。ms(esi)m/e1268.4(m+na)+。
2.46. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{2-[2-(2-{[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基](甲基)氨基}乙氧基)乙氧基]乙氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子au)
向实施例1.2.11(50mg)和n,n-二异丙基乙胺(0.051ml)的n,n-二甲基甲酰胺(1.0ml)溶液中加入2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯(18mg)。将该反应搅拌过夜,并用反相hplc纯化,使用gilson系统,用20-80%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将目标级分合并,冷冻干燥,提供标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm12.92-12.82(m,1h),8.03(d,1h),7.79(d,1h),7.62(d,1h),7.53-7.41(m,3h),7.40-7.32(m,2h),7.28(s,1h),7.01-6.97(m,2h),6.98-6.92(m,1h),4.95(bs,2h),4.04-3.84(m,3h),3.86-3.75(m,3h),3.49-3.32(m,10h),3.01(s,2h),2.95(s,2h),2.79(s,2h),2.31-2.19(m,2h),2.10(s,3h),1.52-1.40(m,4h),1.36(s,2h),1.31-0.94(m,14h),0.84(s,6h)。ms(esi)m/e1041.3(m+h)+。
2.47. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[3-(2-{[4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2-磺基丁酰基](甲基)氨基}乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸(合成子bk)
2.47.1. 4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-1-((2,5-二氧代吡咯烷-1-基)氧基)-1-氧代丁烷-2-磺酸酯
在用氮气吹扫的100ml烧瓶中,将1-羧基-3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙烷-1-磺酸酯溶于二甲基乙酰胺(20ml)中。向此溶液中加入n-羟基琥珀酰亚胺(440mg,)和1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(1000mg),并将该反应在氮气氛围中、在室温下搅拌16小时。减压浓缩溶剂,并将残余物用硅胶色谱纯化,用1-2%的甲醇/二氯甲烷(在溶剂中含有0.1%v/v乙酸)的梯度进行洗脱,得到标题化合物的~80%活化酯和20%酸的混合物,其不用进一步纯化,在下一步中直接使用。ms(esi)m/e360.1(m+h)+。
2.47.2. 6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-(1-{[3-(2-{[4-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-2-磺基丁酰基](甲基)氨基}乙氧基)-5,7-二甲基三环[3.3.1.13,7]癸-1-基]甲基}-5-甲基-1h-吡唑-4-基)吡啶-2-甲酸
向实施例1.1.17(5mg)和实施例2.47.1(20.55mg)的n,n-二甲基甲酰胺(0.25ml)溶液中加入n,n-二异丙基乙胺(0.002ml),并将该反应在室温下搅拌16小时。用反相hplc纯化该粗品反应混合物,使用gilson系统和c1825x100mm柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm8.01-7.95(m,1h),7.76(d,1h),7.60(dd,1h),7.49-7.37(m,3h),7.37-7.29(m,2h),7.28-7.22(m,1h),6.92(d,1h),6.85(s,1h),4.96(bs,2h),3.89(t,2h),3.80(s,2h),3.35(bs,5h),3.08-2.96(m,3h),2.97-2.74(m,2h),2.21(bs,1h),2.08(s,4h),1.42-1.38(m,2h),1.31-1.23(m,4h),1.23-1.01(m,6h),0.97(d,1h),0.89-0.79(m,6h)。ms(esi)m/e1005.2(m+h)+。
2.48. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{[34-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-甲基-4,32-二氧代-7,10,13,16,19,22,25,28-八氧杂-3,31-二氮杂三十四烷-1-基]氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子bq)
如实施例2.44所述,用2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16,19,22,25,28-八氧杂-4-氮杂三十一烷-31-酸酯(mal-dpeg8-nhs-酯)替代2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯,制备标题化合物。ms(esi)m/e1334.3(m+h)+。
2.49. 合成6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-3-{1-[(3-{[28-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-甲基-4,26-二氧代-7,10,13,16,19,22-六氧杂-3,25-二氮杂二十八烷-1-基]氧基}-5,7-二甲基三环[3.3.1.13,7]癸-1-基)甲基]-5-甲基-1h-吡唑-4-基}吡啶-2-甲酸(合成子br)
如实施例2.44所述,用2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16,19,22-六氧杂-4-氮杂二十五烷-25-酸酯(mal-dpeg6-nhs-酯)替代2,5-二氧代吡咯烷-1-基1-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)-3-氧代-7,10,13,16-四氧杂-4-氮杂十九烷-19-酸酯,制备标题化合物。ms(esi)m/e1246.3(m+h)+。
2.50 合成2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-5-{2-[2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰}氨基)乙氧基]乙氧基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
2.50.1. 3-(1-((3-(2-((((4-(2-(2-氨基乙氧基)乙氧基)-2-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.30.1中,用实施例1.1.17替代实施例1.3.7,制备标题化合物。ms(esi)m/e1189.5(m+h)+。
2.50.2. 3-(1-((3-(2-((((4-(2-(2-((r)-2-氨基-3-磺基丙酰胺基)乙氧基)乙氧基)-2-(((2s,3r,4s,5s,6s)-6-羧基-3,4,5-三羟基四氢-2h-吡喃-2-基)氧基)苄基)氧基)羰基)(甲基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在实施例2.34.1中,用实施例2.50.1替代实施例2.31.5,制备标题化合物。ms(esi)m/e1339.5(m+h)+。
2.50.3. 2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-5-{2-[2-({n-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-3-磺基-l-丙氨酰}氨基)乙氧基]乙氧基}苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
在实施例2.34.2中,用实施例2.50.2替代实施例2.34.1,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.83(s,2h);8.01(dd,1h),7.86(d,1h),7.80-7.71(m,2h),7.60(dd,1h),7.52-7.26(m,7h),7.16(d,1h),6.94(d,3h),6.69(d,1h),6.61-6.53(m,1h),5.09-4.91(m,5h),3.46-3.08(m,14h),2.99(t,2h),2.88-2.63(m,5h),2.13-1.94(m,5h),1.52-0.73(m,27h)。ms(esi)m/e1531.4(m-h)-。
2.51 合成n2-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-n6-(37-氧代-2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂三十七烷-37-基)-l-赖氨酰基-l-丙氨酰-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
2.51.1. (s)-6-((((9h-芴-9-基)甲氧基)羰基)氨基)-2-((叔丁氧羰基)氨基)己酸
向冷(冰浴)的(s)-6-氨基-2-((叔丁氧羰基)氨基)己酸(8.5g)溶液(在5%nahco3水溶液(300ml)和1,4-二噁烷(40ml)的混合物中)中逐滴加入(9h-芴-9-基)甲基吡咯烷-1-基碳酸酯(11.7g)的1,4-二噁烷(40ml)溶液。将该反应混合物升温至室温,并搅拌24小时。额外准备三个上述管瓶。反应完成之后,将四个反应混合物合并,并真空除去有机溶剂。用盐酸水溶液(1n)将水层酸化至ph3,并用乙酸乙酯(3×500ml)提取。将合并的有机层用盐水洗涤,用硫酸镁干燥,过滤,并真空浓缩,得到粗品化合物,用甲基叔丁基醚重结晶,得到标题化合物。1hnmr(400mhz,氯仿-d)δ11.05(br..s.,1h),7.76(d,2h),7.59(d,2h),7.45-7.27(m,4h),6.52-6.17(m,1h),5.16-4.87(m,1h),4.54-4.17(m,4h),3.26-2.98(m,2h),1.76-1.64(m,1h),1.62-1.31(m,14h)。
2.51.2. 17-羟基-3,6,9,12,15-五氧杂十七烷-1-酸叔丁基酯
向3,6,9,12-四氧杂十四烷-1,14-二醇(40g)的甲苯(800ml)溶液中分批加入叔丁醇钾(20.7g)。将该混合物在室温下搅拌30分钟。将2-溴乙酸叔丁基酯(36g)逐滴加入到该混合物中。将该反应在室温下搅拌16小时。额外准备两个上述管瓶。反应完成之后,将三个反应混合物合并。将水(500ml)加入到合并的混合物中,并将体积浓缩至1升。用二氯甲烷提取该混合物,并用1n叔丁醇钾水溶液(1l)洗涤。将有机层用无水硫酸钠干燥,过滤,并减压浓缩。用硅胶柱色谱纯化残余物,用50∶1的二氯甲烷∶甲醇洗脱,获得标题化合物。1hnmr(400mhz,氯仿-d)δ4.01(s,2h),3.75-3.58(m,21h),1.46(s,9h)。
2.51.3. 17-(甲苯磺酰基氧基)-3,6,9,12,15-五氧杂十七烷-1-酸叔丁基酯
在0℃,在氮气氛围中,向实施例2.51.2(30g)的二氯甲烷(500ml)溶液中逐滴加入4-甲苯-1-磺酰氯(19.5g)和三乙胺(10.3g)的二氯甲烷(500ml)溶液。将该混合物在室温下搅拌18小时,并倒入水(100ml)中。用二氯甲烷(3×150ml)提取该溶液,并将有机层用盐酸(6n,15ml)洗涤,然后用nahco3(5%水溶液,15ml)洗涤,而后用水(20ml)洗涤。用无水硫酸钠干燥有机层,过滤,浓缩,获得残余物,用硅胶柱色谱纯化残余物,用石油醚∶乙酸乙酯(10∶1)至二氯甲烷∶甲醇(5∶1)洗脱,获得标题化合物。1hnmr(400mhz,氯仿-d)δ7.79(d,2h),7.34(d,2h),4.18-4.13(m,2h),4.01(s,2h),3.72-3.56(m,18h),2.44(s,3h),1.47(s,9h)。
2.51.4. 2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂三十七烷-37-酸
在0℃,向实施例2.51.3(16g)的四氢呋喃(300ml)溶液中加入氢化钠(1.6g)。将该混合物在室温下搅拌4小时。在室温下,将2,5,8,11,14,17-六氧杂十九烷-19-醇(32.8g)的四氢呋喃(300ml)溶液逐滴加入到该反应混合物中。将得到的反应混合物在室温下搅拌16小时,并加入水(20ml)。将该混合物在室温下再搅拌3小时,完成叔丁基酯的水解。将最终反应混合物减压浓缩,除去有机溶剂。将水性残余物用二氯甲烷(2×150ml)提取。将水层酸化至ph3,而后用乙酸乙酯(2×150ml)提取。最后,浓缩水层,获得粗品,用硅胶柱色谱纯化,用石油醚∶乙酸乙酯(1∶1)至二氯甲烷∶甲醇(5∶1)的梯度进行洗脱,获得标题化合物。1hnmr(400mhz,氯仿-d)δ4.19(s,2h),3.80-3.75(m,2h),3.73-3.62(m,40h),3.57(dd,2h),3.40(s,3h)。
2.51.5. (43s,46s)-43-((叔丁氧羰基)氨基)-46-甲基-37,44-二氧代-2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂-38,45-二氮杂四十七烷-47-酸
使用标准fmoc固相肽合成方法和2-氯三苯甲基树脂,合成实施例2.51.5。具体地说,在14℃下,将2-氯三苯甲基树脂(12g),(s)-2-((((9h-芴-9-基)甲氧基)羰基)氨基)丙酸(10g)和n,n-二异丙基乙胺(44.9ml)在无水、分子筛干燥的二氯甲烷(100ml)中摇动24小时。过滤该混合物,并将滤饼用二氯甲烷(3×500ml)、n,n-二甲基甲酰胺(2×250ml)和甲醇(2×250ml)洗涤(每个步骤5分钟)。向上述树脂中加入20%哌啶/n,n-二甲基甲酰胺(100ml),除去fmoc基团。向该混合物中鼓入氮气15分钟,并过滤。再用20%哌啶/n,n-二甲基甲酰胺(100ml)洗涤树脂五次(每个洗涤步骤5分钟),用n,n-二甲基甲酰胺(5×100ml)洗涤,得到脱保护的l-ala加载的树脂。
向实施例2.51.1(9.0g)的n,n-二甲基甲酰胺(50ml)溶液中加入羟基苯并三唑(3.5g)、2-(6-氯-1h-苯并三唑-1-基)-1,1,3,3-四甲基铵六氟磷酸盐(9.3g)和n,n-二异丙基乙胺(8.4ml)。将该混合物在20℃下搅拌30分钟。将上述混合物加入到l-ala加载的树脂中,并在室温下鼓入氮气90分钟,进行混合。过滤该混合物,并用n,n-二甲基甲酰胺(每个步骤5分钟)洗涤树脂。向上述树脂中加入大约20%哌啶/n,n-二甲基甲酰胺(100ml),除去fmoc基团。向该混合物中鼓入氮气15分钟,并过滤。用20%哌啶/n,n-二甲基甲酰胺(100mlx5)和n,n-二甲基甲酰胺(100mlx5)洗涤树脂(每个洗涤步骤5分钟)。
向实施例2.51.4(11.0g)的n,n-二甲基甲酰胺(50ml)溶液中加入羟基苯并三唑(3.5g)、2-(6-氯-1h-苯并三唑-1-基)-1,1,3,3-四甲基铵六氟磷酸盐(9.3g)和n,n-二异丙基乙胺(8.4ml),并将该混合物加入到树脂中,在室温下鼓入氮气3小时,进行混合。过滤该混合物,用n,n-二甲基甲酰胺(5×100ml)、二氯甲烷(8×100ml)洗涤残余物(每个步骤5分钟)。
向最终的树脂中加入1%三氟乙酸/二氯甲烷(100ml),并鼓入氮气5分钟,进行混合。过滤该混合物,并收集滤液。将这种裂解操作重复四次。使用nahco3,将合并的滤液调到ph7,并用水洗涤。将有机层用无水硫酸钠干燥,过滤,并浓缩,获得标题化合物。1hnmr(400mhz,甲醇-d4)δ4.44-4.33(m,1h),4.08-4.00(m,1h),3.98(s,2h),3.77-3.57(m,42h),3.57-3.51(m,2h),3.36(s,3h),3.25(t,2h),1.77(br..s.,1h),1.70-1.51(m,4h),1.44(s,9h),1.42-1.39(m,3h)。
2.51.6. 3-(1-((3-(2-((((4-((s)-2-((s)-2-氨基-3-甲基丁酰胺基)丙酰胺基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
在室温下,将实施例1.3.7的三氟乙酸盐(0.102g)、实施例2.21.4(0.089g)和n,n-二异丙基乙胺(0.104ml)在n,n-二甲基甲酰胺(1ml)中的溶液搅拌16小时。加入二乙胺(0.062ml),并将该反应在室温下搅拌2小时。用水(1ml)稀释该反应,用三氟乙酸(0.050ml)淬灭,并用反相hplc纯化,使用gilson系统和c18柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(lc-ms)m/e1066.5(m+h)+。
2.51.7. 6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)-3-(1-((3-(2-((((4-((43s,46s,49s,52s)-43-((叔丁氧羰基)氨基)-49-异丙基-46,52-二甲基-37,44,47,50-四氧代-2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂-38,45,48,51-四氮杂五十三烷酰胺基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)吡啶甲酸
将实施例2.51.5(16.68mg)与1-[双(二甲基氨基)亚甲基]-1h-1,2,3-三唑并[4,5-b]吡啶鎓3-氧化物六氟磷酸盐(7.25mg)和n,n-二异丙基乙胺(0.015ml)在n-甲基吡咯烷酮(1ml)中混合10分钟,并加入到实施例2.51.6(25mg)和n,n-二异丙基乙胺(0.015ml)的n-甲基吡咯烷酮(1.5ml)溶液中。将该反应混合物在室温下搅拌两个小时。用反相hplc纯化该反应混合物,使用gilson系统和c18柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。ms(esi)m/e961.33(2m+h)2+。
2.51.8. 3-(1-((3-(2-((((4-((43s,46s,49s,52s)-43-氨基-49-异丙基-46,52-二甲基-37,44,47,50-四氧代-2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂-38,45,48,51-四氮杂五十三烷酰胺基)苄基)氧基)羰基)氨基)乙氧基)-5,7-二甲基金刚烷-1-基)甲基)-5-甲基-1h-吡唑-4-基)-6-(8-(苯并[d]噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基)吡啶甲酸
将实施例2.51.7(25mg)用1ml三氟乙酸处理5分钟。利用温和的氮气流,除去溶剂。将残余物用1:1的乙腈∶水冷冻干燥,得到标题化合物,其不用进一步纯化,在下一步中直接使用。ms(lc-ms)m/e1822.0(m+h)+。
2.51.9. n2-[6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酰基]-n6-(37-氧代-2,5,8,11,14,17,20,23,26,29,32,35-十二氧杂三十七烷-37-基)-l-赖氨酰基-l-丙氨酰-l-缬氨酰-n-{4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基]氨基甲酰基}氧基)甲基]苯基}-l-丙氨酰胺
向实施例2.51.8,(23mg)、n-琥珀酰亚胺基6-马来酰亚胺基(maleimido)己酸酯(4.40mg)和羟基苯并三唑(0.321mg)的n-甲基吡咯烷酮(1.5ml)溶液中加入n,n-二异丙基乙胺(8.28μl)。将该反应混合物在室温下搅拌16小时。用反相hplc纯化该反应混合物,使用gilson系统和c18柱,用5-85%的乙腈/水(含有0.1%v/v三氟乙酸)洗脱。将产物级分冷冻干燥,得到标题化合物。1hnmr(400mhz,二甲亚砜-d6)δppm7.76(dq,3h),7.64-7.51(m,5h),7.45(dd,4h),7.35(td,hz,3h),4.97(d,5h),3.95-3.79(m,8h),3.57(d,46h),3.50-3.30(m,14h),1.58-0.82(m,59h)。ms(lc-ms)m/e1007.8(2m+h)2+。
2.52合成2-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-5-[2-(2-{[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]氨基}乙氧基)乙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
在实施例2.30.2中,用实施例2.50.1替代实施例2.30.1,制备标题化合物。1hnmr(500mhz,二甲亚砜-d6)δppm12.87(s,2h);8.06-7.98(m,1h),7.78(d,1h),7.61(dd,1h),7.52-7.41(m,2h),7.39-7.26(m,2h),7.18(d,1h),7.01-6.91(m,2h),6.68(d,1h),6.59(d,1h),5.08-4.98(m,2h),4.95(s,1h),3.59(t,1h),3.46-3.36(m,3h),3.34-3.22(m,2h),3.16(q,1h),3.01(t,1h),2.85(d,2h),2.32(t,1h),2.09(s,2h),1.44-0.71(m,10h)。ms(esi)m/e1338.4(m-h)-。
2.53合成4-[({[2-({3-[(4-{6-[8-(1,3-苯并噻唑-2-基氨基甲酰基)-3,4-二氢异喹啉-2(1h)-基]-2-羧基吡啶-3-基}-5-甲基-1h-吡唑-1-基)甲基]-5,7-二甲基三环[3.3.1.13,7]癸-1-基}氧基)乙基](甲基)氨基甲酰基}氧基)甲基]-3-[3-({n-[3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酰基]-3-磺基-l-丙氨酰}氨基)丙氧基]苯基β-d-吡喃葡糖苷酸(glucopyranosiduronicacid)
如实施例2.34.2所述,用2,5-二氧代吡咯烷-1-基3-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)丙酸酯替代2,5-二氧代吡咯烷-1-基6-(2,5-二氧代-2,5-二氢-1h-吡咯-1-基)己酸酯,n-甲基-2-吡咯烷酮替代n,n-二甲基甲酰胺,制备标题化合物。ms(esi)m/e1458.0(m-h)-。
实施例3.合成示范性的bcl-xl抑制性的adc
使用如下所述的四个示范性方法中的一个方法,合成示范性的adc。表1涉及合成每个示范性的adc所使用的方法。
方法a.将tcep溶液(10mm,0.017ml)加入到预热至37℃的抗体溶液(10mg/ml,1ml)中。将该反应混合物在37℃下保持1小时。将还原的抗体溶液加入到连接子-弹头有效载荷(linker-warheadpayload)溶液(3.3mm,0.160ml,在dmso中)中,并平缓地混合30分钟。将该反应溶液加载到脱盐柱(pd10,在使用之前,用dpbs洗涤三次)上,而后使用dpbs(1.6ml),并用额外的dpbs(3ml)洗脱。将纯化的adc溶液通过0.2微米的结合了低蛋白的13mm针筒式滤器过滤,并在4℃下保存。
方法b.将tcep溶液(10mm,0.017ml)加入到预热至37℃的抗体溶液(10mg/ml,1ml)中。将该反应混合物在37℃下保持1小时。加入硼酸缓冲剂(0.05ml,0.5m,ph8),将还原的抗体的溶液调节至ph=8,加入到连接子-弹头有效载荷溶液(3.3mm,0.160ml,在dmso中)中,并平缓地混合4小时。将该反应溶液加载到脱盐柱(pd10,在使用之前,用dpbs洗涤三次)上,而后使用dpbs(1.6ml),并用额外的dpbs(3ml)洗脱。将纯化的adc溶液通过0.2微米的结合了低蛋白的13mm针筒式滤器过滤,并在4℃下保存。
方法c.使用perkinelmerjanus(ajl8m01部分)机器人液体处理系统进行缀合,所述处理系统配备有i235/96尖端模块化分配技术(mdt)、包含夹臂(7400358部分)的一次性头部(70243540部分)和8个尖端varispan移液臂(7002357部分)(在延伸板上)。使用winprep版本的4.8.3.315软件,控制perkinelmerjanus系统。
使用mdt,用100μl1xdpbs预先湿润pall滤板5052。给滤板抽真空10秒钟,而后放空5秒钟,以便从滤板上除去dpbs。将50%的a蛋白树脂(gemabselectsure)的dpbs浆液倒入配备有磁性球的8孔储液器中,并通过传递储液器板下面的移动磁铁,使树脂混合。使用配备有1ml传导端的8尖端varispan臂,吸出树脂(250μl),并转入96孔滤板中。进行2个循环的抽真空,除去大部分缓冲剂。使用mdt,吸出150μl1xpbs,并分配到容纳树脂的96孔滤板中。进行抽真空,从树脂中除去缓冲剂。将冲洗/抽真空循环重复3次。将2ml的96孔采集板安装在janus面板上,mdt将450μl的5xdpbs转移至采集板上,用于随后的使用。如上面条件a所述,制备还原的抗体(2mg)的dpbs溶液(200μl),并预加载到96孔板中。将还原的抗体的溶液转入到含有树脂的滤板孔中,并通过100μl体积在孔内的重复抽吸/分配(每个循环45秒钟),用mdt将该混合物混合。在5分钟时间内,将抽吸/分配循环总共重复5次。对滤板进行2个循环的抽真空,由此除去过量的抗体。用水冲洗mdt端部,冲洗5个循环(200μl,总体积1ml)。使mdt吸出150μldpbs,并分配到含有树脂结合的抗体的滤板孔中,并进行两个循环的抽真空。将洗涤和抽真空程序再重复两次。最后一次抽真空循环之后,将100μl的1xdpbs分配到含有树脂结合的抗体的孔中。然后,mdt收集各30μl3.3mm的合成子的二甲亚砜溶液(涂覆在96孔板上),并分配到含有树脂结合的抗体(在dpbs中)的滤板上。通过在孔内的100μl体积的重复抽吸/分配,将含有缀合混合物的孔用mdt混合,每个循环45秒钟。在5分钟时间内,将抽吸/分配程序总共重复5次。进行2个循环的抽真空,除去过量的合成子至废料。用水冲洗mdt端部,冲洗5个循环(200μl,总体积1ml)。mdt吸出dpbs(150μl),并分配到缀合混合物中,进行两个循环的抽真空。将洗涤和抽真空程序再重复两次。然后,mdt夹钳移动滤板,并夹持到支持工作站(holdingstation)。mdt放置2ml采集板,板中含有450μl的10xdpbs(在真空歧管内部)。通过放置滤板,并夹持,mdt重新装配真空歧管。用水冲洗mdt端部,冲洗5个循环(200μl,总体积1ml)。mdt吸出100μl的igg洗脱缓冲剂3.75(pierce),并分配到缀合混合物中。一分钟之后,进行2个循环的抽真空,并将洗脱液收集在含有450μl5xdpbs的接收板中。将抽吸/分配程序再重复3次,输送adc样品,浓度在1.5-2.5mg/ml范围内(ph7.4,在dpbs中)。
方法d.使用perkinelmerjanus(ajl8m01部分)机器人液体处理系统进行缀合,所述处理系统配备有i235/96尖端模块化分配技术(mdt)、包含夹臂(7400358部分)的一次性头部(70243540部分)和8个尖端varispan移液臂(7002357部分)(在延伸板上)。使用winprep版本的4.8.3.315软件,控制perkinelmerjanus系统。
使用mdt,用100μl1xdpbs预先湿润pall滤板5052。给滤板抽真空10秒钟,而后放空5秒钟,以便从滤板上除去dpbs。将50%的a蛋白树脂(gemabselectsure)的dpbs浆液倒入配备有磁性球的8孔储液器中,并通过传递储液器板下面的移动磁铁,使树脂混合。使用配备有1ml传导端的8尖端varispan臂,吸出树脂(250μl),并转入96孔滤板中。对滤板进行2个循环的抽真空,除去大部分缓冲剂。mdt吸出150μl的dpbs,并分配到含有树脂的滤板的孔中。将洗涤和抽真空程序再重复两次。将2ml的96孔采集板安装在janus面板上,mdt将450μl的5xdpbs转移至采集板上,随后使用。如上面条件a所述,制备还原的抗体(2mg)的dpbs溶液(200μl),并分配到96孔板中。然后,mdt收集各30μl3.3mm的合成子的二甲亚砜溶液(涂覆在96孔板上),并分配到加载了还原的抗体(在dpbs中)的滤板上。通过在孔内的100μl体积的重复抽吸/分配,将该混合物用mdt混合。五分钟之后,将缀合反应混合物(230μl)转移到含有树脂的96孔滤板中。通过在孔内的100μl体积的重复抽吸/分配,将含有缀合混合物和树脂的孔用mdt混合,每个循环45秒钟。在5分钟时间内,将抽吸/分配程序总共重复5次。进行2个循环的抽真空,除去过量的合成子和蛋白至废料。用水冲洗mdt端部,冲洗5个循环(200μl,总体积1ml)。mdt吸出dpbs(150μl),并分配到缀合混合物中,进行两个循环的抽真空。将洗涤和抽真空程序再重复两次。然后,mdt夹钳移动滤板,并夹持到支持工作站(holdingstation)。mdt放置2ml采集板,板中含有450μl的10xdpbs(在真空歧管内部)。通过放置滤板,并夹持,mdt重新装配真空歧管。用水冲洗mdt端部,冲洗5个循环(200μl,总体积1ml)。mdt吸出100μl的igg洗脱缓冲剂3.75(p),并分配到缀合混合物中。一分钟之后,进行2个循环的抽真空,并将洗脱液收集在含有450μl5xdpbs的接收板中。将抽吸/分配程序再重复3次,输送adc样品,浓度在1.5-2.5mg/ml范围内(ph7.4,在dpbs中)。
方法e.在室温下,将tcep溶液(10mm,0.017ml)加入到抗体溶液(10mg/ml,1ml)中。将该反应混合物加热到37℃,保持75分钟。将还原的抗体的溶液冷却到室温,并加入到合成子溶液(10mm,0.040ml,在dmso中)中,而后加入硼酸缓冲剂(0.1ml,1m,ph8)。在室温下,将该反应溶液静置3天,加载到脱盐柱(pd10,在使用之前,用3x5mldpbs洗涤)上,而后使用dpbs(1.6ml),并用额外的dpbs(3ml)洗脱。将纯化的adc溶液通过0.2微米的结合了低蛋白的13mm针筒式过滤器过滤,并在4℃下保存。
下面的表1表明说明了通过哪种示范性的方法来合成哪种示范性的adc。下列文献描述了被称为epcam(ing-1)的epcam的单克隆抗体:studnicka等人,1994,proteinengineering,7:805-814,以及ammons等人,2003,neoplasia5:146-154。下列文献描述了被称为n901的ncam-1抗体:roguska等人,1994,procnatlacadsciusa91:969-973。wo2009/134776(参见120页)描述了被称为ab033的egfr抗体。
实施例4.示范性的bcl-xl抑制剂结合bcl-xl
使用时间分辨的荧光共振能量转移(tr-fret)试验,证明实施例1.1、1.2、1.3、1.4、1.5、1.6、1.7和1.8(分别为化合物w1.01-w1.08)的示范性的bcl-xl抑制剂结合bcl-xl的能力。tb-抗gst抗体购买于invitrogen(目录编号:pv4216)。
4.1探针合成
4.1.1试剂
所有的试剂使用供应商提供的形式,除非另作说明。包括二异丙基乙胺(diea)、二氯甲烷(dcm)、n-甲基吡咯烷酮(nmp)、2-(1h-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸盐(hbtu)、n-羟基苯并三唑(hobt)和哌啶的肽合成试剂,是从appliedbiosystems,inc.(abi),fostercity,ca或americanbioanalytical,natick,ma获得的。
从abi或anaspec,sanjose,ca获得预加载的9-芴甲氧羰基(fmoc)氨基酸柱(fmoc-ala-oh、fmoc-cys(trt)-oh、fmoc-asp(tbu)-oh、fmoc-glu(tbu)-oh、fmoc-phe-oh、fmoc-gly-oh、fmoc-his(trt)-oh、fmoc-ile-oh、fmoc-leu-oh、fmoc-lys(boc)-oh、fmoc-met-oh、fmoc-asn(trt)-oh、fmoc-pro-oh、fmor-gln(trt)-oh、fmoc-arg(pbf)-oh、fmoc-ser(tbu)-oh、fmoc-thr(tbu)-oh、fmoc-val-oh、fmoc-trp(boc)-oh、fmoc-tyr(tbu)-oh)。
从novabiochem,sandiego,ca获得肽合成树脂(fmoc-rink酰胺mbha树脂)和fmoc-lys(mtt)-oh。
从anaspec获得单一异构体6-羧基荧光素琥珀酰亚胺基酯(6-fam-nhs)。
从oakwoodproducts,westcolumbia,sc获得三氟乙酸(tfa)。
从aldrichchemicalco.,milwaukee,wi获得苯甲硫醚、苯酚、三异丙基硅烷(tis)、3,6-二氧杂-1,8-辛烷二硫醇(dodt)和异丙醇。
在appliedbiosystemsvoyagerde-proms)上记录基质辅助的激光解吸电离质谱(maldi-ms)。
在finniganssq7000(finnigancorp.,sanjose,ca)上,以阳离子和阴离子两种模式记录电喷雾质谱(esi-ms)。
4.1.2.固相肽合成(spps)的一般方法
在abi433a肽合成仪上,使用250µmol规模的fastmoc™缀合循环,每个容器使用至多250µmol的预加载的wang树脂,合成肽。使用含有1mmol标准fmoc-氨基酸的预加载的柱,荧光团的连接位置除外,其中,将1mmolfmoc-lys(mtt)-oh放置在所述柱中,并用传导性反馈监测。在标准偶合条件下,在柱中使用1mmol乙酸,实现n端乙酰化。
4.1.3.从赖氨酸上除去4-甲基三苯甲基(mtt)
使用二氯甲烷,将源自于合成器的树脂洗涤三次,并保持湿润。用30分钟,使150ml95:4:1的二氯甲烷∶三异丙基硅烷∶三氟乙酸流过树脂床。该混合物变成深黄色,然后褪色至浅黄色。用15分钟使100mldmf流过该床。然后,用dmf洗涤树脂三次,并过滤。茚三酮试验显示了对于伯胺的强信号。
4.1.4.用6-羧基荧光素-nhs(6-fam-nhs)标记树脂
在1%diea/dmf中,用2当量的6-fam-nhs处理树脂,并在环境温度下搅拌或摇动过夜。当完成时,沥干树脂,用dmf洗涤三次,用(1×二氯甲烷和1×甲醇)洗涤三次,干燥,提供橙色树脂,茚三酮试验显示该树脂呈阴性。
4.1.5.树脂结合的肽的断裂和脱保护的一般方法
在环境温度下,在由80%tfa、5%水、5%苯甲硫醚、5%苯酚、2.5%tis和2.5%edt组成的断裂混合物(1ml/0.1g树脂)中,摇动3小时,使肽从树脂上断裂下来。过滤取出树脂,并用tfa冲洗两次。从滤液中蒸发出tfa,用醚(10ml/0.1g树脂)沉淀出产物,离心回收产物,用醚(10ml/0.1g树脂)洗涤两次,并干燥,得到粗品肽。
4.1.6.纯化肽的一般方法
在运行unipoint®分析软件(gilson,inc.,middleton,wi)的gilson制备hplc系统的径向压缩柱上纯化粗品肽,所述柱包括两个25x100mm部分,柱中填充了delta-paktmc1815μm颗粒,孔径100å,并用下文列出的梯度法之一进行洗脱。每次注射纯化一至两毫升粗品肽溶液(10mg/ml,在90%dmso/水中)。收集每次操作的含有产物的峰,并冷冻干燥。在20ml/min的条件下进行所有的制备操作,洗脱液为缓冲剂a∶0.1%tfa-水和缓冲剂b∶乙腈。
4.1.7.分析hplc的一般方法
利用具有二极管阵列检测器和hewlett-packard1046a荧光检测器的hewlett-packard1200串联系统进行分析hplc,运行hplc3dchemstation软件版本a.03.04(hewlett-packard.paloalto,ca),使用4.6x250mmymc柱,柱中填充有ods-aq5μm颗粒,孔径120å,在起始条件下预先平衡7分钟之后,用下文列出的梯度法之一洗脱。洗脱液是缓冲剂a∶0.1%tfa-水和缓冲剂b∶乙腈。所有梯度的流速是1ml/min。
4.1.8.合成探针f-bak
如下所述,合成结合bcl-xl的肽探针f-bak。在n端,将探针f-bak乙酰化,在c端酰胺化,并且具有氨基酸序列gqvgrqlaiigdkinr。使用6-fam,将它在赖氨酸残基(k)处荧光素化。探针f-bak可以缩写如下∶乙酰基-gqvgrqlaiigdk(6-fam)inr-nh2。
为了制备探针f-bak,使用常规肽合成方法,使fmoc-rink酰胺mbha树脂延长,提供保护的树脂结合的肽(1.020g)。除去mtt基团,用6-fam-nhs标记,并按照上文描述的方法,进行断裂和脱保护,提供粗品的橙色固体(0.37g)。利用rp-hplc,将产物纯化。利用分析rp-hplc,检验整个主峰的级分,分离出纯的级分,并冷冻干燥,主峰提供标题化合物(0.0802g)的黄色固体;maldi-msm/z=2137.1[(m+h)+]。
4.1.9.肽探针f-bak的另一种合成方法
在另一种方法中,在运行fastmoctm缀合循环的appliedbiosystems433a自动肽合成仪上,在0.25mmolfmoc-rink酰胺mbha树脂(novabiochem)上组装保护的肽,使用预加载的1mmol氨基酸柱,荧光素(6-fam)标记的赖氨酸除外,其中将1mmolfmoc-lys(4-甲基三苯甲基)称到柱中。在柱中放入1mmol乙酸,并如上文所述进行缀合,引入n端乙酰基。使95:4:1的dcm:tis:tfa(v/v/v)溶液流过树脂15分钟,而后用二甲基甲酰胺流淬灭,选择性除去4-甲基三苯甲基。在1%diea/dmf中,使单一异构体6-羧基荧光素-nhs与赖氨酸侧链反应,并通过茚三酮试验证明反应完成。通过用80:5:5:5:2.5:2.5的tfa/水/苯酚/苯甲硫醚/三异丙基硅烷∶3,6-二氧杂-1,8-辛烷二硫醇(v/v/v/v/v/v)处理,使肽从树脂上断裂并进行侧链脱保护,并通过用乙醚沉淀,回收粗品肽。通过反相高效液相色谱纯化粗品肽,并利用分析反相高效液相色谱和基质辅助的激光解吸质谱,证明它的纯度和特征(m/z=2137.1((m+h)+)。
4.2.时间分辨的荧光共振能量转移(tr-fret)试验
使用时间分辨的荧光共振能量转移(tr-fret)结合试验,说明示范性的bcl-xl抑制剂w1.01-w1.08与探针f-bak竞争结合bcl-xl的能力。
4.2.1.方法
对于该试验,将试验化合物在dmso中连续稀释,起始浓度为50μm(2x起始浓度;10%dmso),并将10μl转移到384孔板中。然后,将10μl的蛋白/探针/抗体混合物加入到每个孔中,最终浓度如下∶
然后,将样品在振荡器上混合1分钟,并在室温下再培养2小时。对于每个试验板,分别包括探针/抗体和蛋白/抗体/探针混合物作为阴性和阳性对照。使用340/35nm激发滤片以及520/525(f-bak)和495/510nm(tb标记的抗his抗体)发射滤片,在envision(perkinelmer)上测定荧光。使用wang's方程式,测定解离常数(ki)(wang,1995,febslett.360:111-114)。在不同浓度的人血清(hs)或胎牛血清(fbs)的存在下,可以进行tr-fret试验。在没有hs和存在1%hs的两种条件下,检验化合物。
4.2.2.结果
结合试验的结果(ki,纳摩尔)提供于下面表2中∶
实施例5.示范性的bcl-xl抑制剂在molt-4细胞活力试验中抑制bcl-xl
在基于细胞的杀伤试验中,使用各种细胞系和小鼠肿瘤模型,可以测定示范性的bcl-xl抑制剂的能力。例如,可以利用培养的致瘤和非致瘤细胞系,以及原代小鼠或人类细胞群体,评估它们对于细胞活力的活性。在使用了molt-4细胞的细胞活力试验中,证明了示范性的bcl-xl抑制剂的bcl-xl抑制活性。
5.1.方法
在一组示范性的条件下,将molt-4(atcc,manassas,va)人类急性淋巴母细胞性白血病细胞涂覆在384孔组织培养板(corning,corning,ny)中,每个孔12,500个细胞,组织培养基的总体积为25µl,补充有10%人类血清(sigma-aldrich,st.louis,mo),并用感兴趣的化合物的3倍连续稀释物处理(从10µm至0.0005µm)。每个浓度(一式两份)至少单独的检验3次。使用celltiter-glo®发光法细胞活力检测,按照生产商的建议(promegacorp.,madison,wi),测定化合物处理48小时之后的活细胞数目。在10%hs的存在下,检验化合物。
5.2.结果
对于实施例1.1-1.6的示范性的bcl-xl抑制剂(分别为化合物w1.01-w1.08),在10%hs的存在下进行的molt-4细胞活力试验的结果(ec50,纳摩尔)提供于下面表3中。
nt=没有检验。
实施例6.示范性的adc的dar和聚集
分别利用lc-ms和分子排阻色谱(sec),测定按照上面实施例3合成的示范性的adc的dar和聚集百分比。
6.1.lc-ms常规方法
使用与agilentlc/msdtof6220esi质谱仪连接的agilent1100hplc系统,进行lc-ms分析。使用5mm(最后浓度)bond-breaker®tcep溶液(thermoscientific,rockford,il),将adc还原,加载到proteinmicrotrap(michrombioresorces,auburn,ca)脱盐柱上,并在环境温度下,用10%b至75%b的梯度洗脱0.2分钟。流动相a是水(含有0.1%甲酸(fa)),流动相b是乙腈(含有0.1%fa),流速是0.2ml/min。使用agilentmasshunter(tm)acquisition软件,获得共同洗脱轻和重链的电喷雾电离飞行时间质谱。使用themaximumentropyfeatureofmasshunter软件将提取强度vs.m/z光谱进行解卷积(deconvoluted),测定每个还原抗体片段的质量。通过轻链和重链的裸峰和修饰峰的强度加和,通过强度乘以所连接的药物数目来进行归一化,由解卷积(deconvoluted)的光谱计算dar。将加和的、归一化强度除以强度的和,两个轻链和两个重链的加和结果产生全部adc的最终平均dar值。
6.2.尺寸排阻色谱常规方法
在含有0.25mm氯化钾和15%ipa的0.2m磷酸钾(ph6.2)中,流速0.75ml/min,使用shodexkw802.5柱,进行尺寸排阻色谱。通过将曲线下的面积积分,测定在280nm下每个高分子量和单体洗脱液的峰面积吸光度。通过用高分子量洗脱液的峰面积吸光度(280nm)除以280nm下高分子量和单体洗脱液的峰面积吸光度的和,再乘以100%,测定缀合样品的%聚集部分。
6.3.结果
表4报道了上述lc-ms方法测定的平均dar值,以及示范性的adc的%聚集部分。在试验中,评价靶向epcam的包含单克隆抗体ing-1的adc(参见,studnicka等人,1994,proteinengineering,7:805-814,和ammons等人,2003,neoplasia5:146-154)。wo2009/134776描述了靶向egfr的抗体ab033。roguska等人(1994,procnatlacadsciusa91:969-973)描述了单克隆抗体n901(靶向ncam-1)。
实施例7.靶向egfr的adc体外抑制癌细胞的生长
评价某些包含抗体ab033的示范性的adc。抗体ab033靶向人类egfr。wo2009/134776(参见120页)描述了抗体ab033的可变的重和轻链序列。使用mcl-1-/-小鼠胚胎纤维母(mef)细胞,表明抗体ab033抑制癌细胞生长的能力。mcl-1-/-mef的存活依赖于bcl-xl(lessene等人,2013,naturechemicalbiology9:390-397)。为了评价示范性的靶向ab033的bcl-xl-adc的效果,使人类egfr在mcl-1-/-mef中过度表达。
7.1.方法
使用fugene6转染试剂(rochemolecularbiochemicals,mannheim,germany),用含有huegfr序列的逆转录病毒结构plvc-ires-hygro(clontech)或空载体转染gp2-293包装细胞系(clontech),制备逆转录病毒上清液。培养48小时之后,采集含有病毒的上清液,并在75cm2培养瓶中,在聚凝胺(8µg/ml;sigma)的存在下,施加于mcl-1-/-mef(每个烧瓶0.5x106个)中,并进一步培养48小时。3天之后,使用250μg/ml潮霉素b(invitrogen)(在完全补充的培养基中),洗涤mcl-1-/-mef,并选择。利用流式细胞术,确定huegfr的表达,并与母体细胞系或空载体转染的细胞系相比较。
在含有10%fbs的dmem中,用靶向ab033的bcl-xl-adc、单独的ab033或靶向msl109的bcl-xl-adc处理表达huegfr的mcl-1-/-mef或plvx空载体(vct对照)96小时。对于该试验,将细胞涂覆在384孔组织培养板(corning,corning,ny)的总体积25µl的试验培养基(dmem和10%hifbs)中,每个孔250个细胞。利用echo550声波液体处理器(labcyte),将涂覆的细胞用感兴趣的抗体药物缀合物的4倍连续稀释物(从1µm至1pm)处理。在mcl-1-/-mefhuegfr细胞系的12个平行测定和mcl-1-/-mef载体细胞系的6个平行测定中,检验每个浓度。使用celltiter-glo®发光法细胞活力检测,按照生产商的建议(promegacorp.,madison,wi),在37℃和5%co2中,在抗体药物缀合物处理96小时之后,测定活细胞部分。使用荧光方案,积分时间0.5sec,将板在perkinelmerenvision中读数。将每个稀释点的平行测定值平均,使用graphpadprism5(graphpadsoftware,inc.),使用线性回归y=((底部-顶部)/(1+((x/k)n)))+顶部,将数据与s形曲线模型拟合,得到抗体药物缀合物的ec50值,其中,y是测定的响应值,x是化合物浓度,n是hill斜率,k是ec50值,底部和顶部分别是较低和较高的渐近线。肉眼观察曲线,用于证实曲线拟合结果。由walterandelizahallinstituteofmedical的davidc.s.huang,获得mcl-1-/-mef。
7.2.结果
代表性的实施例的细胞活力试验结果(ec50值,纳摩尔)提供于下面表5中。
nt=没有检验。
代表性的实施例3.28、3.29和3.35针对mcl-1-/-mef载体细胞系的细胞活力试验结果(ec50值,纳摩尔)分别是67nm、69nm和249nm。
实施例8.靶向epcam-和ncam1-的抗体药物缀合物体外抑制癌细胞的生长
使用表达内源性epcam蛋白的ncc38细胞,人类乳腺癌细胞系,表明含有靶向人类细胞粘附分子(epcam)的抗体的某些示范性的adc抑制bcl-xl和诱导细胞程序死亡的能力。在表达内源性ncam-1的nci-h146细胞(人类小细胞肺癌系)中,表明靶向人类神经细胞粘着分子ncam1的某些示范性的adc的细胞毒性。
8.1.方法
对于该试验,在含有10%fbs的rpmi1640培养基(invitrogen,#11995)中,培养hcc38和nci-h146两个细胞系。在试验之前,将细胞再悬浮在培养基中,4x104个细胞/ml,而后加入到96孔组织培养板,75µl细胞/孔,最后浓度为3,000个细胞/孔。然后,将试验板在37℃和5%co2条件下培养过夜。在第二天,将epcam、ncam-1或阴性对照(msl109)adc在培养基中连续稀释,并加入到试验板中,25µl/孔。然后,将试验板在37℃和5%co2条件下培养72小时。利用celltiterglo®发光法细胞活力检测试剂盒(promega,#g7573),测定细胞活力。
8.2.结果
使用graphpadprism软件,分析数据。ic50值(达到细胞的最大生长抑制的50%时的adc的浓度)分别报道于表6和7中。
如表6所示,靶向epcam的adc有效地杀伤hcc38乳腺癌细胞(ic50≤0.4nm),而阴性对照adcmsl109-db的活性弱。如表7所示,ncam1-db和ncam1-hadc对于nci-146小细胞肺癌细胞显示了特异性活性(ic50值~20nm)。
实施例9.靶向egfr的bcl-xl抑制性抗体药物缀合物(adc),单独以及与多西他赛联合,体内抑制非小细胞肺癌(nsclc)异种移植物的生长
在两个nsclc异种移植模型nci-h1650和ebc-1中,说明靶向肿瘤相关抗原(taa)(例如,egfr)的示范性的bcl-xl抑制性adc(本文还称为bcl-xliadc)的效果和选择性。正如免疫组织化学所揭示的那样,在下面表8中,举例说明了抗原在异种移植肿瘤上的膜表达。
实施例10.靶向egfr的bcl-xl抗体药物缀合物体内抑制nsclc肿瘤细胞的生长
在由nsclc细胞系、nci-h1650和ebc-1获得的异种移植模型中,说明某些示范性的靶向egfr的adc在小鼠中选择性抑制表达egfr的肿瘤细胞的能力。
10.1.方法
nsclc细胞系nci-h1650购买于americantypeculturecollection(atcc,manassas,va)。在补充有胎牛血清(fbs,hyclone,logan,ut)的rpmi1640培养基(invitrogen,carlsbad,ca)中,将细胞培养成为单层细胞。从japanesecollectionofresearchbiosources(jcrb,osaka,japan)获得鳞状肺癌细胞系ebc-1。在补充有10%胎牛血清(fbs,hyclone,logan,ut)的mem培养基(invitrogen,carlsbad,ca)中,将细胞培养成为单层细胞。为了产生异种移植物,将5*106个活细胞皮下接种到免疫缺乏的雌性scid/bg小鼠(charlesriverlaboratories,wilmington,ma)的右肋部。注射剂体积是0.2ml,并且由s-mem和基质胶(bd,franklinlakes,nj)的1:1混合物组成。使肿瘤大小匹配为大约200mm3。在磷酸盐缓冲盐水(pbs)中,配制抗体和缀合物,并腹膜内注射。注射体积不超过400µl。在肿瘤大小匹配之后24小时之内,开始治疗。在治疗开始时,小鼠重量大约25g。每周评估两至三次肿瘤体积。通过电子卡钳,测定肿瘤的长度(l)和宽度(w),并按照下列方程式计算体积:v=lxw2/2。当肿瘤体积达到3,000mm3或出现皮肤溃疡时,使小鼠安乐死。每个笼中放置八个至十只小鼠。食品和水不限量。在开始实验之前,使小鼠适应动物设备至少一周时间。在12小时照明∶12小时黑暗计划(在06:00开始照明)的照明阶段,检验动物。按照abbvie'sinstitutionalanimalcareandusecommittee以及nationalinstitutesofhealthguideforcareanduseoflaboratoryanimals的原则,在associationfortheassessmentandaccreditationoflaboratoryanimalcare认可的设备中,进行所有实验。
10.2.结果说明和分析
为了指出治疗剂的效果,使用治疗剂响应的幅度(tgimax)、持久性(tgd)和响应频率(cr、pr、or)的参数。
tgimax是在实验期间的最大肿瘤生长抑制。利用100*(1-tv/cv)计算肿瘤生长抑制,其中tv和cv分别是治疗和对照组的平均肿瘤体积。
tgd或肿瘤生长延迟是所治疗的肿瘤达到1cm3体积需要延长的时间(相对于对照组)。通过100*(tt/ct-1)来计算tgd,其中,tt和ct分别是治疗和对照组的达到1cm3的中值时间周期。
响应幅度在特定组中的分布是通过完全响应者(cr)、部分响应者(pr)和全部响应者的频率得到的。cr是在肿瘤负荷为25mm3(至少三次测定)的组内的小鼠的百分比。pr是在肿瘤负荷大于25mm3但小于治疗开始时的一半体积(至少三次测定)的组内的小鼠的百分比。or是cr和pr的和。
双尾student's检验和kaplan-meier对数秩检验分别用于测定tgimax和tgd的差异的显著性。
10.3.肺腺癌模型nci-h1650(下文称为h1650)的生长抑制
10.3.1.靶向egfr的adc
对于靶向egfr的adc,分别如实施例3.2、3.35和3.36所述,将实施例2.2(合成子d)、实施例2.35(合成子h)和实施例2.36(合成子i)的合成子与靶向egfr的抗体ab033缀合,分别得到adcab033-d、ab033-h和ab033-i。合成子h、db或i与靶向巨细胞病毒(cmv)的抗体msl109的缀合物(msl109-h、msl109-db或msl-109-i)用作被动式靶向对照物。这些缀合物下文还称为‘非靶向’adc,这是由于载体抗体不能识别肿瘤相关抗原。msl109描述于下列文献中:drobyski等人,1991,transplantation51:1190-1196,以及美国专利us5,750,106。靶向破伤风类毒素的抗体(抗体ab095)用作给予igg的效果的对照物。参见larrick等人,1992,immunologicalreviews69-85。
10.3.2.靶向egfr的adc的结果
下面图1和表9说明了靶向egfr的adc抑制h1650异种移植物生长的效果和选择性。在图1中,图例中列举了每次给药的数量。在接种肿瘤细胞后的第16天,开始治疗。治疗的肿瘤的平均尺寸是210mm3。ab095、ab033-h、ab033-i、msl109-h和msl109-i的方案是q4dx6。使用7.5mg/kg剂量(qdx1)的标准疗法(standard-of-care)化学治疗剂多西他赛(dtx),作为单一药剂使用,或与adc联用。腹膜内注射抗体和缀合物。静脉内给予dtx。曲线的每个点代表5个肿瘤的平均值。误差线描述了平均值的标准误差。
通过57%的tgimax和78%的tgd(表9)可以说明,给予小分子bcl-xl抑制剂a-1331852(leverson等人,2015,sci.transl.med.7:279ra40)显著地抑制异种移植物生长。与非靶向的adcmsl109h和裸的抗egfr抗体相比,以单一药剂形式给予靶向egfr的bcl-xliadc(30mg/kg,q4dx6),抑制肿瘤生长(tgimax)大约1.5倍。通过非靶向的adcmsl109-h和msl109-i,增强dtx治疗的生长抑制和响应的持久性。靶向egfr的adcab033-h和ab033-i,当加入到dtx中时,与加入非靶向adc相比,导致更持久的肿瘤衰退(表9和图1)。
前面的实验表明,靶向egfr的bcl-xl抑制性adc显著地抑制h1650的生长。因此,这个模型适于进一步探索这些缀合物的最小有效剂量,以及鉴定其它有效的bcl-xl抑制剂。表10和图2说明了靶向egfr的bcl-xliadc(ab033-h和ab033-db)的剂量响应关系。在接种肿瘤细胞后的第12天,开始治疗。治疗的肿瘤的平均尺寸是215mm3。抗体和缀合物的方案是q4dx6。图例中列举了每次给药的数量。腹膜内注射抗体和缀合物。图2的曲线的每个点代表10个肿瘤的平均值。误差线描述了平均值的标准误差。
虽然载体抗体ab033抑制肿瘤生长,但adc的bcl-xl抑制剂-连接子部分主要负责缀合物的效果。ab033的抑制的持久性是剂量响应性的,并且在q4dx6方案中,在3mg/kg和30mg/kg剂量下,tgd位于44和80%之间。与ab033-h和ab033-db的效果相比较,这个效果显著地更低。剂量为3mg/kg时,这些缀合物(ab033-db)的最低的tgd是160%。此外,用ab033治疗不会产生可测的响应率,而bcl-xladc,即使在3mg/kg条件下,也会引起最小90%的总响应率。靶向egfr的缀合物的效果也不太可能由消极靶点的效果所引起,这是由于,使用msl109作为载体抗体,在没有任何响应率的情况下,只会产生微不足道的生长抑制。
表11总结了单一剂量的、与ab033连接的各种bcl-xl抑制剂对于h1650异种移植物生长的抑制。在接种肿瘤细胞后的第11天,开始治疗。治疗的肿瘤的平均尺寸是216mm3。抗体和缀合物的方案是qdx1。给予10mg/kg的单剂量。腹膜内给予抗体和缀合物。每个治疗组由8只小鼠组成。
靶向egfr的bcl-xliadc一致地比对照物缀合物更有效。尽管给予单一剂量,如所示,但靶向adc引起持久的响应,tgd的范围是133%至>507%。
在单独的实验中,评价了与ab033连接的两个其它bcl-xl抑制剂对于h1650异种移植物生长的抑制(表12)。在接种肿瘤细胞后的第12天,开始治疗。治疗的肿瘤的平均尺寸是213mm3。抗体和缀合物的方案是qdx1,给予的剂量为10mg/kg。腹膜内给予抗体和缀合物。每个治疗组由8只小鼠组成。
10.4.鳞状nsclc模型ebc-1的生长抑制
10.4.1.靶向egfr的adc的方法
形成异种移植物、监测肿瘤生长和数据分析的一般方法提供于实施例2中。对于靶向egfr的adc,分别如实施例3.2、3.35和3.36所述,将实施例2.2(合成子d)、实施例2.35(合成子h)和实施例2.36(合成子i)的合成子与靶向egfr的抗体ab033缀合,分别得到adcab033-d、ab033-h和ab033-i。合成子h与靶向cmv的抗体msl109的缀合物(msl109-h)用作消极靶点对照物。靶向破伤风类毒素的抗体(抗体ab095)用作给予igg的效果的对照物。
10.4.2.使用靶向egfr的adc的结果
当治疗鳞状细胞癌ebc-1的异种移植模型时,也观察到靶向egfr的adc的治疗效能。使用靶向egfr的adc的这个实验的结果示于下面图3和表13中。在图3中,图例列举了每次给药的剂量。曲线的每个点代表5个肿瘤的平均值。误差线描述了平均值的标准误差。在接种肿瘤细胞后的第9天,开始治疗。治疗的肿瘤的平均尺寸是215mm3。ab095、ab033、ab033-d、ab033-h、ab033-i和msl109-h的方案是q4dx6。在qdx1条件下,给予dtx。腹膜内注射抗体和缀合物。静脉内给予dtx。
靶向cmv的adc(msl109-h,剂量:30mg/kg)抑制肿瘤生长(43%),由此显示出与消极靶点相关的抗肿瘤活性。在整个实验中,使用7.5mg/kg剂量的标准疗法化学治疗剂多西他赛(dtx),作为单一药剂使用,或与adc联用。作为单一药剂,dtx产生76%的tgi。在与dtx的联用药中,msl109-h(30mg/kg)使tgi从76%升高至83%,和tgd从44%升高至89%(表13,图3a)。被动式靶向达到的效果不如使用egfr靶向抗体ab033的adc所看到的效果。靶向egfr的adc(ab033-h,30mg/kg)的tgimax是64%(图3c)。向dtx中加入ab033-h(30mg/kg),使tgimax从76%升高至98%,tgd从44%升高至178%。给予ab033-i,引起66%的最大肿瘤生长抑制(图3d)。当给予联用药时,这种缀合物分别使dtx的tgimax和tgd升至98%和178%。当以单一药剂形式给予ab033-d时,ab033-d的效果有限(tgimax=49%)。尽管如此,向dtx中加入这种缀合物,能够使dtx的tgimax和tgd分别升高至94%和156%(表13,图3b)。值得注意的是,给予10mg/kg的靶向egfr的缀合物,也会提高dtx的效果(表13)。向dtx中加入靶向的bcl-xliadc的治疗益处是,持久性提高,而不是增加响应的幅度。
实施例11.单独和与bcl-2-选择性抑制剂联用的靶向ncam-1的bcl-xl抑制性adc,体内抑制小细胞肺癌(sclc)肿瘤的生长
在两个小细胞肺癌(sclc)异种移植模型nci-h146和nci-h1963.fp5中,单独和与化合物abt-199(bcl-2的小分子选择性抑制剂)联用的靶向ncam1的示范性的bcl-xliadc抑制肿瘤细胞生长的效果。
11.1.方法
sclc细胞系nci-h146(下文称为h146)购买于americantypeculturecollection(atcc,manassas,va)。在补充有10%胎牛血清(fbs,hyclone,logan,ut)的rpmi-1640培养基(invitrogen,carlsbad,ca)中,将细胞培养成为单层细胞。sclc癌细胞系nci-h1963.fp5(下文称为h1963.fp5)源自于在免疫受损小鼠的肋部经过5次连续传代后的nci-h1963(atcc)。在补充有胎牛血清(fbs,hyclone,logan,ut)的rpmi-1640培养基(invitrogen,carlsbad,ca)中,将细胞培养成为单层细胞。为了产生异种移植物,将5*106个活细胞皮下接种到免疫缺乏的雌性scid/bg小鼠(charlesriverlaboratories,wilmington,ma)的右肋部。注射体积是0.2ml,并且由s-mem和基质胶(bd,franklinlakes,nj)的1:1混合物组成。使肿瘤大小匹配为大约200mm3。在磷酸盐缓冲盐水(pbs)中,配制抗体和缀合物,并腹膜内注射。注射体积不超过400µl。在肿瘤大小匹配之后24小时之内,开始治疗。在治疗开始时,小鼠称重大约25g。如实施例10所述,进行肿瘤体积确定和动物管理。
对于该试验,使用靶向ncam1的adcα-ncam1-h(也称为n901-h)。所评价的adc包含单克隆抗体ncam-1(n901)(参见roguska等人,1994,procnatlacadsciusa91:969-973)。靶向的肿瘤相关抗原ncam1在h146和h1963.fp5细胞的表面上表达。对于先前的实验,抗体ab095用作阴性对照。给予单一药剂和与abt-199联用的bcl-xliadc。
11.2.结果
11.2.1.靶向ncam-1的adc对于h146sclc异种移植物的生长的效果
在接种肿瘤细胞后的第10天,开始治疗。治疗的肿瘤的平均尺寸是216mm3。ab095和α-ncam-1-h的方案是q4dx6,abt-199的方案是qdx21。实验的结果示于图4和表14中。给予单一药剂和与abt-199联用的bcl-xladc。腹膜内注射抗体和缀合物。口服给予abt-199。在图4中,图例列举了每次给药的数量。曲线的每个点代表10个肿瘤的平均值,只不过包含7只小鼠的msl109-h治疗组除外。误差线描述了平均值的标准误差。
当以单一药剂形式给予时,靶向ncam1的bcl-xliadc抑制肿瘤生长。靶向的adcα-ncam1-h抑制肿瘤生长(88%),并且使肿瘤达到1cm3体积的时间提高了144%(图4,表14)。ncam1-hadc的效果具有选择性,这是因为它的效果显著地高于裸抗体α-ncam1或非靶向对照物msl109-h的效果(表14)。另外,当用α-ncam1-h治疗与给予选择性bcl-2抑制剂abt-199联用时,出现显著的加合效应。tgi、tgd和完全响应率分别从88、144和0%提高至98、204和80%。
11.2.2.靶向ncam-1的adc对于h1963.fp5sclc异种移植物的生长的效果
使用h1963异种移植物,进行其它异种移植物实验。这些异种移植物表达ncam1。在接种肿瘤细胞后的第15天,开始治疗。治疗的肿瘤的平均尺寸是233mm3。ab095和α-ncam1-h的方案是q4dx6,abt-199的方案是qdx21。实验的结果示于图5和表15中。给予单一药剂和与abt-199联用的bcl-xliadc。腹膜内注射抗体和缀合物。口服给予abt-199。在图5中,图例列举了每次给药的剂量。曲线的每个点代表9个肿瘤的平均值,只不过包含5只小鼠的αmsl109-h治疗组除外。误差线描述了平均值的标准误差。
11.2.3.结果
h1963.fp5异种移植物实验的结果提供于下面表15中。当以单一药剂形式给药时,靶向ncam1的缀合物抑制肿瘤生长。α-ncam1-h(n901-h)抑制肿瘤生长(74%,图5,表15)。α-ncam1-h(也称为n901-h)的效果具有选择性,这是因为它的效果显著地高于裸抗体或非靶向对照物msl109-h的效果(表15)。与在h146异种移植物中所观察到的效果相似,当用α-ncam1-h治疗与给予选择性bcl-2抑制剂abt-199联用时,出现显著的加合效应。
实施例12.bcl-xl抗体-药物缀合物减轻全身性毒性
12.1.防止血小板减少
给予bcl-xliadc作为抗体药物缀合物,可以通过选择性地靶向肿瘤,规避小分子的全身性毒性。用这样的方式,通过两种合适的机制,adc可以避开全身性毒性,并且达到肿瘤特异性效果。第一,对于具有细胞膜渗透性bcl-xl抑制剂的adc,与载体抗体的结合,可以限制与小分子的全身性接触。第二,adc可以促进非渗透性bcl-xl抑制剂的内化,并由此选择性地影响携带靶向抗原的肿瘤细胞。第一个机制的证据提供于图6中。
12.1.1.方法和结果
在小鼠中,单一腹膜内注射之后,检验两种bcl-xl抑制性adc(bcl-xliadc)对于循环血小板的数目的影响(抑制性adc含有抗egfr抗体ab033,并且被称为ab033-h和ab033-i)。抗破伤风类毒素抗体ab095用作阴性对照。navitoclax(abt-263,双重bcl-2和bcl-xl抑制剂)、a-1331852(选择性的bcl-xl抑制剂,leverson等人,2015,sci.transl.med.7:279ra40.)和非缀合的bcl-xl抑制剂导致血小板减少,在注射化合物之后6小时,血小板减少达到最大值。0.61mg/kg的剂量(等同于在30mg/kg的bcl-xladc中存在的bcl-xl抑制剂的数量),使血小板数量降低100倍,从大约6*105个/mm3的正常数降低至6*103个/mm3。
相比之下,给药之后6小时(表16),或在14天的观察期的任何时间点,bcl-xl抑制性adc都没有使血小板实质性地减少。随后的观察结果提出:由adc缓慢释放抑制剂所引起的血小板减少的诱导作用是不太可能的。
尽管已经说明和描述了各个具体实施方案,但应理解,在不背离本公开的精神和范围的条件下,可以进行各种改变。
进一步地,本发明涉及如下各项的技术方案:
项项1.抗体药物缀合物(adc),其包含通过连接子与抗体连接的药物,其中所述药物是按照结构式(iia)的bcl-xl抑制剂∶
或其可药用盐,其中∶
ar选自
z1选自n、ch和c-cn;
z2选自nh、ch2、o、s、s(o)和s(o2);
r1选自甲基、氯和氰基;
r2选自氢、甲基、氯和氰基;
r4是氢、c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基或c1-4羟烃基,其中r4的c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基和c1-4羟烃基任选被一个或多个独立地选自och3、och2ch2och3和och2ch2nhch3的取代基取代;
r10a、r10b和r10c各自彼此独立地选自氢、卤素、c1-6烷基、c2-6烯基、c2-6炔基和c1-6卤代烃基;
r11a和r11b各自彼此独立地选自氢、甲基、乙基、卤代甲基、羟基、甲氧基、卤素、cn和sch3;
n是0、1、2或3;和
#代表与连接子l的连接点。
2.项1的adc,或其可药用盐,它的药物对抗体比例为1-10。
3.项1的adc,或其可药用盐,其中连接子是通过溶酶体酶可断裂的。
4.项3的adc,或其可药用盐,其中溶酶体酶是组织蛋白酶b。
5.项3的adc,或其可药用盐,其中连接子包含按照结构式(iva)、(ivb)或(ivc)的片段∶
或其盐,其中∶
肽代表通过溶酶体酶可断裂的肽(举例说明的n→c,其中肽包括氨基和羧基“端”);
t代表含有一个或多个乙二醇单元或亚烃基链或其组合的聚合物;
ra选自氢、烃基、磺酸酯和磺酸甲酯;
p是0至5的整数;
q是0或1;
x是0或1;
y是0或1;
*代表与连接子的其余部分的连接点。
6.项5的adc,或其可药用盐,其中,肽选自∶val-cit;cit-val;ala-ala;ala-cit;cit-ala;asn-cit;cit-asn;cit-cit;val-glu;glu-val;ser-cit;cit-ser;lys-cit;cit-lys;asp-cit;cit-asp;ala-val;val-ala;phe-lys;lys-phe;val-lys;lys-val;ala-lys;lys-ala;phe-cit;cit-phe;leu-cit;cit-leu;ile-cit;cit-ile;phe-arg;arg-phe;cit-trp;和trp-cit,或其盐。
7.项3的adc,或其可药用盐,其中溶酶体酶是β-葡糖苷酸酶或β-半乳糖苷酶。
8.项7的adc,或其可药用盐,其中连接子包含按照结构式(va)、(vb)、(vc)或(vd)的片段∶
或其盐,其中∶
q是0或1;
r是0或1;
x1是o或nh;
*代表与连接子的其余部分的连接点。
9.项1的adc,或其可药用盐,其中连接子包含具有1至6个乙二醇单元的聚乙二醇片段。
10.项1的adc,或其可药用盐,其中抗体结合肿瘤细胞上表达的细胞表面受体或肿瘤相关抗原。
11.项10的adc,或其可药用盐,其中抗体结合选自egfr、epcam和ncam1的细胞表面受体或肿瘤相关抗原中的一种。
12.项11的adc,或其可药用盐,其中抗体结合egfr、epcam或ncam1。
13.项11的adc,或其可药用盐,其中抗体结合epcam。
14.项1的adc,或其可药用盐,其是按照结构式(i)的化合物∶
或其盐,其中∶
d是药物;
l是连接子;
ab是抗体;
lk代表连接连接子l与抗体ab的共价连接基;和
m是1至20的整数。
15.项14的adc,或其可药用盐,其中m是1-8的整数。
16.项14的adc,或其可药用盐,其中m是2、3或4。
17.项14的adc,或其可药用盐,其中ar选自∶
18.项14的adc,或其可药用盐,其中ar是
19.项14的adc,或其可药用盐,其中z1是n。
20.项14的adc,或其可药用盐,其中z1是ch。
21.项14的adc,或其可药用盐,其中z2是o。
22.项14的adc,或其可药用盐,其中r1选自甲基和氯。
23.项14的adc,或其可药用盐,其中r2选自氢和甲基。
24.项14的adc,或其可药用盐,其中r2是氢。
25.项14的adc,或其可药用盐,其中r10a是卤素,且r10b和r10c各自是氢。
26.项14的adc,或其可药用盐,其中r10a是氟,且r10b和r10c各自是氢。
27.项14的adc,或其可药用盐,其中r10a、r10b和r10c各自是氢。
28.项14的adc,或其可药用盐,其中r11a和r11b是相同的。
29.项14的adc,或其可药用盐,其中r11a和r11b各自是甲基。
30.项14的adc,或其可药用盐,其中n是0或1。
31.项14的adc,其中d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08,以及其可药用盐。
32.项14的adc,其中连接子l选自iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其可药用盐。
33.项14的adc,或其可药用盐,其中lk是与抗体ab上的氨基形成的连接基。
34.项33的adc,或其可药用盐,其中lk是酰胺或硫脲。
35.项14的adc,或其可药用盐,其中lk是与抗体ab上的巯基形成的连接基。
36.项35的adc,或其可药用盐,其中lk是硫醚。
37.项14的adc,或其可药用盐,其中抗体ab结合egfr或ncam1。
38.项14的adc,或其可药用盐,其中抗体ab结合egfr。
39.项14的adc,其中∶
d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08,以及其可药用盐;
l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其盐;
lk选自酰胺、硫脲和硫醚;和
m是1至8的整数。
40.项39的adc,或其可药用盐,其中抗体ab结合egfr或ncam1。
41.组合物,其包含按照项1-40的任一项的adc和载体、赋形剂和/或稀释剂。
42.项41的组合物,其被配制为在人类中用于药物用途。
43.项42的组合物,其是单位剂型。
44.按照结构式d-l-rx的合成子,或其可药用盐,其中∶
d是药物;
l是连接子;和
rx是包含能够共价连接合成子与抗体的官能团的部分,
和进一步的,其中药物d是按照下列结构式的bcl-xl抑制剂∶
或其可药用盐,其中∶
ar选自
z1选自n、ch和c-cn;
z2选自nh、ch2、o、s、s(o)和s(o2);
r1选自甲基、氯和氰基;
r2选自氢、甲基、氯和氰基;
r4是氢、c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基或c1-4羟烃基,其中r4的c1-4烷基、c2-4烯基、c2-4炔基、c1-4卤代烃基和c1-4羟烃基任选被一个或多个独立地选自och3、och2ch2och3和och2ch2nhch3的取代基取代;
r10a、r10b和r10c各自彼此独立地选自氢、卤素、c1-6烷基、c2-6烯基、c2-6炔基和c1-6卤代烃基;
r11a和r11b各自彼此独立地选自氢、甲基、乙基、卤代甲基、羟基、甲氧基、卤素、cn和sch3;
n是0、1、2或3;和
#代表与连接子l的连接点。
45.项44的合成子,或其可药用盐,其中连接子是通过溶酶体酶可断裂的。
46.项45的合成子,或其可药用盐,其中溶酶体酶是组织蛋白酶b。
47.项44的合成子,或其可药用盐,其中连接子包含按照结构式(iva)、(ivb)或(ivc)的片段∶
其中∶
肽代表通过溶酶体酶可断裂的肽(举例说明的n→c,其中,肽包括氨基和羧基“端”);
t代表含有一个或多个乙二醇单元或亚烃基链或其组合的聚合物;
ra选自氢、烃基、磺酸酯和磺酸甲酯;
p是0至5的整数;
q是0或1;
x是0或1;
y是0或1;
*代表与连接子的其余部分的连接点。
48.项47的合成子,或其可药用盐,其中肽选自∶val-cit;cit-val;ala-ala;ala-cit;cit-ala;asn-cit;cit-asn;cit-cit;val-glu;glu-val;ser-cit;cit-ser;lys-cit;cit-lys;asp-cit;cit-asp;ala-val;val-ala;phe-lys;lys-phe;val-lys;lys-val;ala-lys;lys-ala;phe-cit;cit-phe;leu-cit;cit-leu;ile-cit;cit-ile;phe-arg;arg-phe;cit-trp;和trp-cit。
49.项44的合成子,或其可药用盐,其中溶酶体酶是β-葡糖苷酸酶或β-半乳糖苷酶。
50.项49的合成子,其中,连接子包含按照结构式(va)、(vb)或(vc)的片段∶
或其可药用盐,其中∶
q是0或1;
r是0或1;
x1是o或nh;
*代表与连接子的其余部分的连接点。
51.项44的合成子,或其可药用盐,其中连接子包含具有1至6个乙二醇单元的聚乙二醇片段。
52.项44的合成子,或其可药用盐,其中ar选自∶
53.项44的合成子,或其可药用盐,其中ar是
54.项44的合成子,或其可药用盐,其中z1是n。
55.项44的合成子,或其可药用盐,其中z1是ch。
56.项44的合成子,或其可药用盐,其中z2是o。
57.项44的合成子,或其可药用盐,其中r1选自甲基和氯。
58.项44的合成子,或其可药用盐,其中r2选自氢和甲基。
59.项44的合成子,或其可药用盐,其中r2是氢。
60.项44的合成子,或其可药用盐,其中r10a是卤素,且r10b和r10c各自是氢。
61.项44的合成子,或其可药用盐,其中r10a是氟,且r10b和r10c各自是氢。
62.项44的合成子,或其可药用盐,其中r10a、r10b和r10c各自是氢。
63.项44的合成子,或其可药用盐,其中r11a和r11b相同。
64.项44的合成子,或其可药用盐,其中r11a和r11b各自是甲基。
65.项44的合成子,或其可药用盐,其中n是0或1。
66.项44的合成子,其中d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08,以及其可药用盐。
67.项44的合成子,以及其可药用盐,其中连接子l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其可药用盐。
68.项44的合成子,以及其可药用盐,其中rx包含能够连接合成子与抗体上的氨基的官能团。
69.项68的合成子,以及其可药用盐,其中rx包含nhs-酯或异硫氰酸酯。
70.项66的合成子,以及其可药用盐,其中rx包含能够连接合成子与抗体上的巯基的官能团。
71.项70的合成子,以及其可药用盐,其中rx包含卤代乙酰基或马来酰亚胺。
72.项66的合成子,以及其可药用盐,其中∶
d选自w1.01、w1.02、w1.03、w1.04、w1.05、w1.06、w1.07和w1.08,以及其可药用盐;
l选自连接子iva.1-iva.7、ivb.1-ivb.15、ivc.1-ivc.2、va.1-va.12、vb.1-vb.4、vc.1-vc.9、vd.1-vd.2、via.1、v1c.1-v1c.2、v1d.1-v1d.3和其盐;和
rx包含选自nhs-酯、异硫氰酸酯、卤代乙酰基和马来酰亚胺的官能团。
73.如下形成的adc:在合成子共价连接抗体的条件下,使结合肿瘤细胞上表达的细胞表面受体或肿瘤相关抗原的抗体与按照项44-72的任一项的合成子接触。
74.项73的adc,其中在adc具有2、3或4的dar的条件下进行接触步骤。
75.组合物,其包含按照项73或74的adc和赋形剂、载体和/或稀释剂。
76.项75的组合物,其被配制为在人类中用于药物用途。
77.项76的组合物,其是单位剂型。
78.制备adc的方法,所述方法包括:在合成子共价连接抗体的条件下,接触按照项63-69的任一项的合成子。
79.在表达bcl-xl的细胞中抑制bcl-xl活性的方法,所述方法包括:在adc结合细胞的条件下,使细胞与能够结合细胞的按照项1-40和73-74的任一项的adc接触。
80.在表达bcl-xl的细胞中诱导细胞程序死亡的方法,所述方法包括:在adc结合细胞的条件下,使细胞与能够结合细胞的按照项1-40和73-74的任一项的adc接触。
81.治疗涉及内在的细胞程序死亡失调的疾病的方法,所述方法包括:给予患有涉及细胞程序死亡失调的疾病的患者有效提供治疗益处数量的按照项1-40和73-74的任一项的adc,其中adc的抗体结合内在的细胞程序死亡失调的细胞上的细胞表面受体。
82.治疗癌症的方法,所述方法包括:给予患有癌症的患者有效提供治疗益处数量的按照1-40和73-74的任一项的adc,所述adc能够结合在癌细胞的表面上表达的细胞表面受体或肿瘤相关抗原。
83.项74的方法,其中以单一疗法给予adc。
84.项74的方法,其中所治疗的癌症是致瘤性癌症。
85.项74的方法,其中辅助于另一种化学治疗剂放射治疗给予adc。
86.项85的方法,其中与开始化学疗法和/或放射治疗同时给予adc。
87.项85的方法,其中在开始化学疗法和/或放射治疗之前给予adc。
88.项85-87的任一项的方法,其中给予有效使肿瘤细胞对于标准化学疗法和/或放射治疗敏感数量的adc。
89.使肿瘤对于标准细胞毒素药剂和/或放射疗法敏感的方法,所述方法包括:使肿瘤与有效使肿瘤细胞对于标准细胞毒素药剂和/或放射疗法敏感数量的、能够结合肿瘤的按照项1-40和73-74的任一项的adc接触。
90.项89的方法,其中所述肿瘤对于标准细胞毒素药剂和/或放射疗法已变得耐受。
91.项89的方法,其中所述肿瘤预先没有接触过标准细胞毒素药剂和/或放射治疗。
92.选自合成子实施例2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.10、2.12、2.17、2.18、2.21、2.22、2.23、2.24、2.25、2.26、2.27、2.28、2.29、2.30、2.31、2.32、2.33、2.34、2.35、2.36、2.37、2.38、2.39、2.40、2.41、2.42、2.43、2.44、2.45、2.46、2.47、2.48、2.49、2.50、2.51、2.52、2.53和其可药用盐的合成子。
93.抗体药物缀合物(adc),或其可药用盐,其包含与抗体缀合的、按照项92的合成子。
- 还没有人留言评论。精彩留言会获得点赞!