一种湿性敷料及其制备方法与流程
1.本发明涉及一种湿性敷料及其制备方法,属于化学技术领域。
背景技术:
2.1962年,伦敦大学的winter博士首先用动物实验(猪)证实,湿性环境的伤口愈合速度比干性伤口愈合快一倍。1963年,hinman用人体研究证实湿性愈合的科学性。2000年8月,美国食品与药品管理局(a)在新颁布的创面医疗用品(外用药和敷料)的行业指南中特别强调,保持创面的温润环境是标准的处理方法。
3.湿性环境伤口愈合的优点包括:为伤口提供适度湿润的环境,加速伤口愈合,能显著缩短伤口的治疗时间;湿性环境不会形成伤口干痂,避免再次换药时机械性损伤带来的痛苦,减少瘢痕形成,使患者感觉舒适美观;减少换药频次,节省敷料,缩短患者住院时间,降低了医疗费用,同时减少护理工作量;使用安全方便,疗效显著,大大降低了感染几率。
4.目前,常见的湿性敷料包括水凝胶敷料、水胶体敷料、硅凝胶敷料、藻酸盐类敷料、泡沫类敷料、离子敷料等。但是,这些常见湿性敷料仍存在一定的缺陷,例如,这些常见湿性敷料均为固定形状,因伤口形状不一致,导致敷料在使用过程中,需使用者的手工裁剪,以使敷料和伤口有效充分的贴合,这极易导致敷料污染的风险,并且,使用不便;这些常见湿性敷料均为固定厚度,因伤口的深度的不一致,导致敷料不能和伤口充分贴合,进而导致受力不能均衡,降低敷料使用效果;另外,在伤口实际的治疗中,常常会有如因外力造成的贯穿伤,及瘘道、瘘管等深度伤口,无匹配的敷料直接使用,只能通过对伤口敷料多次裁剪,然后对伤口进行填塞,造成敷料更不易贴合伤口,使用极为不方便。
5.因此,亟需找到一种使用方便,可与伤口贴合一致,可实现伤口的自由填充,并且,可加快伤口的愈合速度的,特别适用于急救用贯穿伤,有瘘道、瘘管的各类伤口的湿性敷料。
技术实现要素:
6.为解决上述问题,本发明提供了一种制备湿性敷料的方法,所述方法为将星状亲水性聚氨酯与发泡组分混合后进行发泡,得到湿性敷料;所述发泡组分包含发泡剂、溶剂、界面活性剂、多胺以及催化剂;所述发泡剂包括碳酸氢钠、碳酸钙或碳酸镁中的一种或一种以上;所述界面活性剂包括聚二甲硅氧烷、聚氧化烯或聚二甲硅氧烷
‑
聚氧化烯共聚物中的一种或一种以上;所述多胺包括1,2
‑
乙二胺、1,4
‑
丁二胺、1,6
‑
己二胺、三伸乙四胺或聚醚二胺中的一种或一种以上;所述催化剂包括异辛酸锌、三伸乙二胺、二甲基环己胺、二甲基乙醇胺、三乙胺、1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯或五甲基二伸乙三胺中的一种或一种以上。
[0007]
在本发明的一种实施方式中,当发泡剂包括碳酸氢钠时,所述溶剂为水;当发泡剂包括碳酸钙或碳酸镁中的一种或一种以上时,所述溶剂为醇类。
[0008]
在本发明的一种实施方式中,所述发泡剂为碳酸氢钠;所述界面活性剂为聚二甲
硅氧烷
‑
聚氧化烯共聚物;所述多胺为乙二胺;所述催化剂为异辛酸锌。
[0009]
在本发明的一种实施方式中,所述发泡组分中,发泡剂、溶剂、界面活性剂、多胺以及催化剂的摩尔比为1~5:1~10:1~10:1~5:1~2。
[0010]
在本发明的一种实施方式中,所述发泡组分中,发泡剂、溶剂、界面活性剂、多胺以及催化剂的摩尔比为1:4:5:1:1。
[0011]
在本发明的一种实施方式中,所述星状亲水性聚氨酯与发泡组分的混合质量比为1:1~10。
[0012]
在本发明的一种实施方式中,所述星状亲水性聚氨酯与发泡组分的混合质量比为1:2~3。
[0013]
在本发明的一种实施方式中,所述发泡的方法为:于800~1000rpm的条件下搅拌30~60s。
[0014]
在本发明的一种实施方式中,所述星状亲水性聚氨酯的制备方法为:先将第一聚醚多元醇与多异氰酸酯混合后进行第一次反应,得到反应产物,然后将反应产物、第二聚醚多元醇与亲水剂混合后进行第二次反应,得到星状亲水性聚氨酯。
[0015]
在本发明的一种实施方式中,所述第一聚醚多元醇包括聚丙二醇或聚三醇中的一种或一种以上;所述多异氰酸酯为1,6
‑
己二异氰酸酯、伸甲基二环己基二异氰酸酯或异佛酮二异氰酸酯中的一种或一种以上;所述第二聚醚多元醇包括聚乙二醇、2
‑
乙基
‑2‑
噁唑啉、聚乙二醇或聚丁二醇中的一种或一种以上;所述亲水剂包括透明质酸或透明质酸纳中的一种或一种以上。
[0016]
在本发明的一种实施方式中,所述所述第一聚醚多元醇为聚丙二醇;所述多异氰酸酯为1,6
‑
己二异氰酸酯;所述第二聚醚多元醇包括聚乙二醇;所述亲水剂包括透明质酸。
[0017]
在本发明的一种实施方式中,所述聚丙二醇的重量平均分子量为2000~8000;所述聚乙二醇的重量平均分子量为1000~6000;所述透明质酸的重量平均分子量为500000~2500000。
[0018]
在本发明的一种实施方式中,所述第一聚醚多元醇、多异氰酸酯、第二聚醚多元醇与亲水剂的质量比为0.3~1.3:0.1~0.5:0.15~1:15~50。
[0019]
在本发明的一种实施方式中,所述第一聚醚多元醇、多异氰酸酯、第二聚醚多元醇与亲水剂的质量比为1:0.15:0.35:33。
[0020]
在本发明的一种实施方式中,所述第一次反应的温度为40~120℃、时间为1~4h。
[0021]
在本发明的一种实施方式中,所述第一次反应的温度为80℃、时间为1h。
[0022]
在本发明的一种实施方式中,所述第二次反应的温度为40~100℃、时间为1~12h。
[0023]
在本发明的一种实施方式中,所述第二次反应的温度为80℃、时间为6h。
[0024]
本发明还提供了一种湿性敷料,所述湿性敷料是使用上述方法制备得到的。
[0025]
本发明技术方案,具有如下优点:
[0026]
1、本发明提供了一种制备湿性敷料的方法,所述方法为将星状亲水性聚氨酯与发泡组分混合后进行发泡,得到湿性敷料,其中,发泡组分包含发泡剂、溶剂、界面活性剂、多胺以及催化剂,发泡剂包括碳酸氢钠、碳酸钙或碳酸镁中的一种或一种以上,界面活性剂包括聚二甲硅氧烷、聚氧化烯或聚二甲硅氧烷
‑
聚氧化烯共聚物中的一种或一种以上,多胺包
括1,2
‑
乙二胺、1,4
‑
丁二胺、1,6
‑
己二胺、三伸乙四胺或聚醚二胺中的一种或一种以上,催化剂包括异辛酸锌、三伸乙二胺、二甲基环己胺、二甲基乙醇胺、三乙胺、1,8
‑
二氮杂二环[5.4.0]十一碳
‑7‑
烯或五甲基二伸乙三胺中的一种或一种以上;使用此方法制备得到的湿性敷料使用方便,可与伤口贴合一致,可实现伤口的自由填充,并且,可加快伤口的愈合速度,特别适用于急救用贯穿伤,有瘘道、瘘管的各类伤口。
[0027]
进一步地,所述方法通过使用透明质酸,大大增加了湿性敷料的亲水性、保湿能力和吸收性能,使得湿性敷料与伤口之间更加贴合。
[0028]
进一步地,所述方法采用水和碳酸氢钠做发泡组份,与现有的采用丙酮溶剂作为发泡组分的制备工艺相比,采用水和碳酸氢钠做发泡组份大大减少了湿性敷料的制备总时间。
[0029]
进一步地,此方法选用由碳酸氢钠、聚二甲硅氧烷
‑
聚氧化烯共聚物、乙二胺和异辛酸锌按照1:4:5:1:1的摩尔比复配而成的发泡组分对星状亲水性聚氨酯进行发泡以制得湿性敷料;采用此发泡组分,可显著降低制得的湿性敷料的毒性,保持湿性敷料的机械强度,并保持湿性敷料的自由型变能力及液体吸收能力。
[0030]
进一步地,此方法选用重量平均分子量范围为1000~6000的聚乙二醇作为第二聚醚多元醇以制得星状亲水性聚氨酯;若聚乙二醇的重量平均分子量小于1000,会经由代谢产生生物毒性,若聚乙二醇的重量平均分子量大于6000,会因黏度过高而使交联反应的操作产生困难,但若是添加溶剂以降低黏度进行操作,则可能导致溶剂残留于填充材上而有细胞毒性的风险。
[0031]
进一步地,此方法使用的发泡组分含有多胺;多胺可显著增加制得的湿性敷料的机械强度。
[0032]
2、本发明提供了一种湿性敷料,此湿性敷料是通过将星状亲水性聚氨酯与发泡组分混合后进行发泡制得的,此湿性敷料使用方便,可与伤口贴合一致,可实现伤口的自由填充,并且,可加快伤口的愈合速度,特别适用于急救用贯穿伤,有瘘道、瘘管的各类伤口。
[0033]
进一步地,此湿性敷料的制备方法使用透明质酸,这大大增加了湿性敷料的亲水性、保湿能力和吸收性能,使得湿性敷料与伤口之间更加贴合。
[0034]
进一步地,此湿性敷料的制备方法采用水和碳酸氢钠做发泡组份,与现有的采用丙酮溶剂作为发泡组分的制备工艺相比,采用水和碳酸氢钠做发泡组份大大减少了湿性敷料的制备总时间。
[0035]
进一步地,此湿性敷料的制备方法选用由碳酸氢钠、聚二甲硅氧烷
‑
聚氧化烯共聚物、乙二胺和异辛酸锌按照1:4:5:1:1的摩尔比复配而成的发泡组分对星状亲水性聚氨酯进行发泡;采用此发泡组分,可显著降低制得的湿性敷料的毒性,保持湿性敷料的机械强度,并保持湿性敷料的自由型变能力及液体吸收能力。
[0036]
进一步地,此湿性敷料的制备方法选用重量平均分子量范围为1000~6000的聚乙二醇作为第二聚醚多元醇以制得星状亲水性聚氨酯;若聚乙二醇的重量平均分子量小于1000,会经由代谢产生生物毒性,若聚乙二醇的重量平均分子量大于6000,会因黏度过高而使交联反应的操作产生困难,但若是添加溶剂以降低黏度进行操作,则可能导致溶剂残留于填充材上而有细胞毒性的风险。
[0037]
进一步地,此湿性敷料的制备方法使用的发泡组分含有多胺;多胺可显著增加湿
性敷料的机械强度。
具体实施方式
[0038]
提供下述实施例是为了更好地进一步理解本发明,并不局限于所述最佳实施方式,不对本发明的内容和保护范围构成限制,任何人在本发明的启示下或是将本发明与其他现有技术的特征进行组合而得出的任何与本发明相同或相近似的产品,均落在本发明的保护范围之内。
[0039]
下述实施例中未注明具体实验步骤或条件者,按照本领域内的文献所描述的常规实验步骤的操作或条件即可进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规试剂产品。
[0040]
实施例1:一种湿性敷料及其制备方法
[0041]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0042]
将0.5mol ppg6000 triol(重量平均分子量为6000)与2.5mol hdi混合,在80℃下反应1h,得到反应产物后;在反应产物中加入0.85mol peg1000(重量平均分子量为1000)、0.85mol peg2000(重量平均分子量为2000)及0.1mol透明质酸(重量平均分量为1,000,000),在80℃下交联反应6h,得到星状亲水性聚氨酯;将0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合,得到发泡组分;将星状亲水性聚氨酯与发泡组分按照1:2.5的质量比混合均匀后,于800转/分钟的速度快速磁力搅拌10min进行发泡,得到湿性敷料1。
[0043]
实施例2:一种湿性敷料及其制备方法
[0044]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0045]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.4mol二甲基乙醇胺(催化剂)混合而得发泡组分,得到湿性敷料2。
[0046]
实施例3:一种湿性敷料及其制备方法
[0047]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0048]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚氧化烯(界面活性剂)、0.2mol丁二胺及0.4mol二甲基乙醇胺(催化剂)混合而得发泡组分,得到湿性敷料3。
[0049]
实施例4:一种湿性敷料及其制备方法
[0050]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0051]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.2mol丁二胺及0.4mol二甲基乙醇胺(催化剂)混合而得发泡组分,得到湿性敷料4。
[0052]
实施例5:一种湿性敷料及其制备方法
[0053]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0054]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚氧化烯(界面活性剂)、0.1mol乙二胺及0.4mol二甲基乙醇胺(催化剂)混合而得发泡组分,得到湿性敷料5。
[0055]
实施例6:一种湿性敷料及其制备方法
[0056]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0057]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸镁(发泡剂)、0.5mol聚二甲硅氧烷(界面活性剂)、0.1mol 1,6
‑
己二胺及0.3mol二甲基环己胺(催化剂)混合而得发泡组分,得到湿性敷料6。
[0058]
实施例7:一种湿性敷料及其制备方法
[0059]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0060]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸镁(发泡剂)、0.5mol聚二甲硅氧烷(界面活性剂)、0.3mol三伸乙四胺及0.4mol二甲基乙醇胺(催化剂)混合而得发泡组分,得到湿性敷料7。
[0061]
实施例8:一种湿性敷料及其制备方法
[0062]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0063]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸镁(发泡剂)、0.5mol聚二甲硅氧烷(界面活性剂)、0.05mol聚醚二胺及0.3mol三乙胺(催化剂)混合而得发泡组分,得到湿性敷料8。
[0064]
实施例9:一种湿性敷料及其制备方法
[0065]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0066]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸镁(发泡剂)、0.5mol聚二甲硅氧烷(界面活性剂)、0.05mol聚醚二胺及0.2mol 1,8
‑
二吖双环[5.4.0]十一
‑7‑
烯(催化剂)混合而得发泡组分,得到湿性敷料9。
[0067]
实施例10:一种湿性敷料及其制备方法
[0068]
本实施例提供了一种湿性敷料,所述湿性敷料的制备方法如下:
[0069]
在实施例1的基础上,将由0.1mol碳酸氢钠(发泡剂)、0.4mol水、0.5mol聚二甲硅氧烷
‑
聚氧化烯共聚物(界面活性剂)、0.1mol乙二胺及0.1mol异辛酸锌(催化剂)混合而得发泡组分替换为由0.1mol碳酸镁(发泡剂)、0.5mol聚二甲硅氧烷(界面活性剂)、0.05mol聚醚二胺及0.3mol五甲基二伸乙三胺(催化剂)混合而得发泡组分,得到湿性敷料10。
[0070]
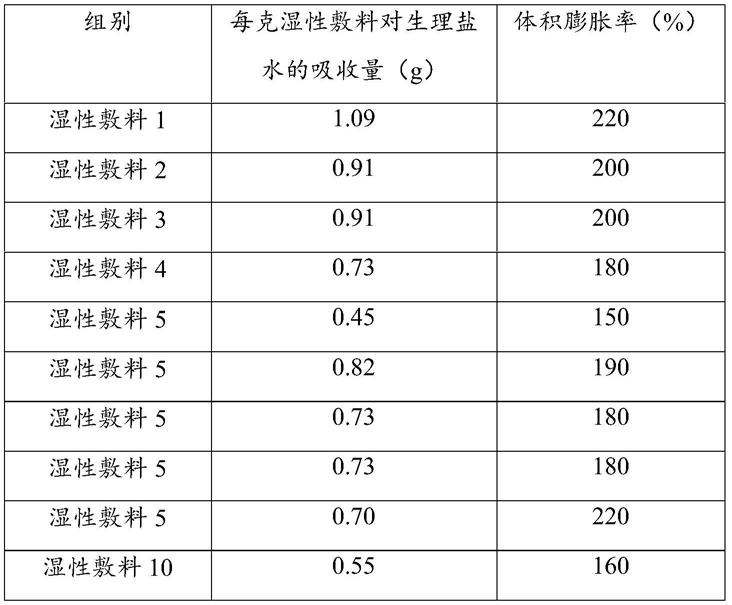
实验例1:湿性敷料性能的检测
[0071]
分别取10g实施例1~10制得的湿性敷料1~10作为待测样品,检测湿性敷料1~10
对生理盐水的吸收量和体积膨胀率(检测结果见表1);
[0072]
其中,湿性敷料对生理盐水的吸收量和体积膨胀率的检测方法如下:
[0073]
(1)准备湿性敷料:将每个湿性敷料用手术刀切分为3组样品,用电子天平称重每个样品,并记录每组样品的未吸收生理盐水的重量克数g1;
[0074]
(2)准备生理盐水:用量杯盛装20ml生理盐水,置于电热鼓风干燥箱,设定温度37℃,生理盐水温度达到37℃,且温度平衡30分钟后,可进行测试;
[0075]
(3)测试:将样品放入37℃的生理盐水量杯中,每10秒内记录体积变化,得到初始体积v1;记录好体积后,将盛有凝胶生理盐水的量杯再次至于电热鼓风干燥箱内,时间15分钟(依据前期试验数据得到的值,凝胶的吸水在10分钟左右达到平衡);15分钟后,取出样品,用吸收纸擦拭凝胶表面的少许水分后,用天平称取吸收饱和生理盐水的样品,得到重量g2;将重量称取完成后的凝胶再次放入盛装20ml 37℃生理盐水的量杯中,读取量杯的体积v2;依次循环完成共计30组样品的测试和数据记录。
[0076]
(4)计算:每克样品对生理盐水的吸收量=(g2
‑
g1)/g1(对三组样品取平均数);体积膨胀率=[(v2
‑
v1)/(v1
‑
20)]*100%(对三组样品取平均数)。
[0077]
如表1所示,湿性敷料1~10对生理盐水的吸收量和体积膨胀率均较佳,其中,湿性敷料1~3对生理盐水的吸收量和体积膨胀率最佳,远超湿性敷料4~10。
[0078]
表1湿性敷料1~10对生理盐水的吸收量和体积膨胀率
[0079][0080]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1