生物组织粘合剂的制作方法
1.本发明属于医用材料制备技术领域,特别涉及一种生物组织粘合剂,该组织粘合剂用于湿润环境组织的粘合及修复。
背景技术:
2.生物组织粘合剂是一类可以直接应用于人体的生物医用材料,可用于生物组织粘合、局部组织出血封闭和修复。在外科手术中可以替代或辅助缝合线,与常规手术缝合线相比,组织粘合剂可以更加快速的闭合伤口,尤其在面对一些不规则的创伤表面时适用性更强,并且在后期的治疗中还能够加快伤口恢复。
3.目前生物组织粘合剂的材料主要有化学类(聚氨酯和氰基丙烯酸酯等)、可降解合成大分子(聚乙烯醇和聚己内酯等)、生物大分子类(壳聚糖、海藻酸和透明质酸等)、生物大分子多肽类(聚谷氨酸、聚赖氨酸和纤维蛋白等)。但是化学类和可降解合成大分子其生物相容性、与周围组织界面的结合性及降解性都较差。生物大分子多糖和多肽具有较好的结构可调性、生物相容性和降解性等优点,然而,目前制得的生物组织粘合剂普遍存在湿态粘附性能差,与生物组织不能完全贴合以及密封性能不够,生物降解性能不可控等问题,限制了该类生物组织粘合剂的进一步应用。例如现有生物组织粘合剂在人体内降解程度不可控时,将导致粘附能力失效,产生炎症反应等。
4.现有技术为解决上述问题,开发贻贝仿生组织粘合剂,在组织粘合剂中引入儿茶酚,以提高粘附强度。一般而言,多糖链上自由羧基(
‑
cooh)在n
‑
羟基琥珀酰亚胺催化下,可以与碳二亚胺发生缩合反应接枝儿茶酚。但这种接枝反应副产物较多,产品得率低,接枝率也比较低,从而限制了粘附性能。
技术实现要素:
5.为了解决现有生物组织粘合剂湿态粘附性能差、生物降解性能不可控的问题,本发明提供一种多巴胺修饰生物组织粘合剂,接枝多巴胺,具有良好粘附性能,同时使组织粘合剂降解时间可控。
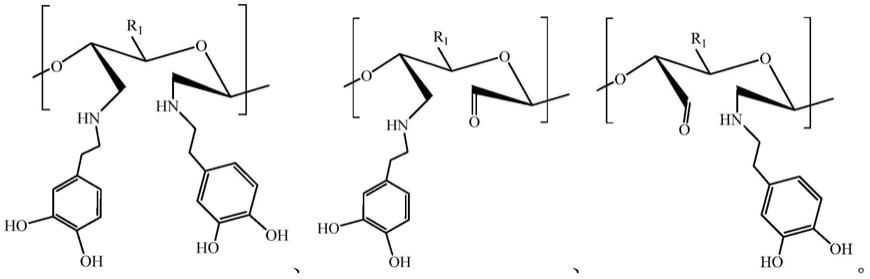
6.本发明公开了一种生物组织粘合剂,组织粘合剂包括聚合物,聚合物由多个聚合单体通过糖苷键连接而成;其中聚合单体包括第一聚合单体、第二聚合单体,第一聚合单体的结构式为:
[0007][0008]
其中r2为oh或nh2;
[0009]
第二聚合单体的结构为第一聚合单体的2位和3位碳原子之间开环,且2位和3位碳
原子中的至少一个以c
‑
n键接枝基团a,基团a的结构式为:
[0010][0011]
采用上述技术方案,聚合物以多糖为基础材料反应形成,具有良好的生物相容性;在多糖中接枝多巴胺形成贻贝仿生组织粘合剂,可以提高粘合剂在湿态环境下的粘附强度;并且第二聚合单体为开环结构接枝多巴胺,可减少分子结构的位阻,增加多巴胺的接枝率,而聚合物中多巴胺的含量与粘附强度呈正比,因此以提高粘附强度。且多巴胺与碳原子以单键相连,稳定性高,可以降低粘合剂应用于人体组织后的水解反应,延长粘合剂作用时间;因此通过调节接枝反应的接枝率,从而调节第二聚合单体在聚合物中的占比,以实现使组织粘合剂降解时间可控。
[0012]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合单体还包括第三聚合单体和第四聚合单体;第三聚合单体的结构式为:
[0013][0014]
第四聚合单体的结构为第三聚合单体的两个醛基中的至少一个替换成以亚胺键与基团b连接,基团b的结构式为:
[0015][0016]
采用上述方案,第三聚合单体为醛基化反应形成,第四聚合单体为第三聚合单体与多巴胺发生席夫碱反应形成;醛基化过程使多糖单体开环,减少环结构空间位阻对接枝多巴胺的影响,且产生足够量的醛基,利用醛基与氨基之间的席夫碱快速缩合反应,保证接枝大量多巴胺,从而可以提高聚合物的粘附强度。且在席夫碱反应后,进行还原反应,将亚胺键特异性还原成不易水解的c
‑
n键,使得整个结构体系更加稳定,后期在作为组织粘合剂使用时,可以通过调节亚胺键的还原度来控制降解时间。
[0017]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合单体中的r1为cooh、ch2oh、ch2och2cooh中的一种。
[0018]
采用上述方案,聚合物的原料可以是透明质酸、壳聚糖、海藻酸、羧甲基纤维素中的至少一种。
[0019]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合单体中的r1为cooh,r2为oh;且聚合单体还包括第五聚合单体,第五聚合单体的结构式为:
[0020][0021]
且第五聚合单体与第一聚合单体连接形成第一重复单元,与第二聚合单体连接形成第二重复单元,与第三聚合单体连接形成第三重复单元,与第四聚合单体连接形成第四重复单元。
[0022]
采用上述方案,聚合物的原料是透明质酸,具有优异生物相容性。
[0023]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合物中多巴胺的摩尔取代度为0.35~0.65,其中接枝的多巴胺中还原多巴胺摩尔比为40~65%。
[0024]
采用上述方案,聚合物用做组织粘合剂湿态粘附性能良好,且还原率达到上述比例范围时,降解时间较长。
[0025]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,第一重复单元的重复单元数占聚合物的所有重复单元的重复单元数的比例为10%
‑
25%。
[0026]
采用上述方案,氧化反应醛基化率达到75~90%。
[0027]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,组织粘合剂为聚合物的水溶液在氧化剂的参与下彼此交联生成的水凝胶。
[0028]
在凝胶粘合剂制备过程中,通过选用不同的聚合物溶度、交联剂配比以及交联时间可以得到具有差异性的组织粘合剂产品。
[0029]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,交联剂包括质量百分比浓度为2.5%
‑
10%的高碘酸钠的水溶液。
[0030]
采用上述方案,高碘酸钠作为交联剂形成的凝胶性能较好。
[0031]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合物的水溶液的质量百分比浓度为5%
‑
10%。
[0032]
根据本发明的另一具体实施方式,本发明提供的生物组织粘合剂,聚合物的水溶液与高碘酸钠的水溶液发生交联反应时的体积比为1:1,反应时间为20~40秒。
具体实施方式
[0033]
为了下面的详细描述的目的,应当理解,除了在任何操作实例中,或者以其他方式指出的情况下,表示例如说明书和权利要求中使用的成分的量的所有数字应被理解为在所有情况下被术语“约”修饰。因此,除非相反指出,否则在以下说明书和所附权利要求中阐述的数值参数是根据本申请所要获得的期望性能而变化的近似值。至少并不是试图将等同原则的适用限制在权利要求的范围内,每个数值参数至少应该根据报告的有效数字的个数并通过应用普通舍入技术来解释。
[0034]
本申请中使用的术语仅用于描述具体实施方式的目的并且不理解为限制性的。如本文中使用的,单数形式“一个(种)”和“该()”也意图包括复数形式,除非上下文清楚地另外指明。表述例如“......的至少一个(种)”当在要素列表之前或之后时修饰整个要素列表,而不修饰该列表的单独要素。
[0035]
进一步,本申请中使用的术语“包括”或“包含”当用在本说明书中时,表明存在所
陈述的特征、区域、整体、步骤、操作、元件、和/或组分,但不排除存在或增加一种或多种另外的特征、区域、整体、步骤、操作、元件、组分、和/或其集合。
[0036]
如本申请中使用的“约”或“大约”包括所描述的值并且意味着例如本领域普通技术人员考虑到所讨论的测量和与具体量的测量有关的误差(即,测量系统的限制)而确定的对于具体值的可接受的偏差范围内。除非另外指明,否则组分的所有比率均指重量百分比(重量%);除非另外指明,所公开的所有参数范围包括端点值及其间的所有值。
[0037]
在本发明的描述中,如无特殊说明,术语的含义与本领域技术人员一般理解的含义相同,但如有不同,以本发明的定义为准;如无特殊说明,试验方法均为常规方法;如无特殊说明,本发明中的所用的原料及试验材料均为可常规购买得到的。
[0038]
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的实施方式作进一步地详细描述。
[0039]
本发明提供一种生物组织粘合剂,组织粘合剂包括聚合物,聚合物由多个聚合单体通过糖苷键连接而成;其中聚合单体包括第一聚合单体和第二聚合单体,其中聚合单体包括第一聚合单体、第二聚合单体,第一聚合单体的结构式为:
[0040][0041]
其中r2为oh或nh2;
[0042]
第二聚合单体的结构式为第一聚合单体的2位和3位碳原子之间开环,且2位和3位碳原子中的至少一个以c
‑
n键接枝基团a,基团a的结构式为:
[0043][0044]
具体地,第一聚合单体为多糖单体(己糖),第二聚合单体为第一聚合单体经过反应使2位和3位碳之间开环后,2位和3位碳中的至少一个接枝多巴胺形成的结构,第二聚合单体的结构式可以为如下几种的一种:
[0045][0046]
其中r1根据多糖的原料不同而不同,具体可以为cooh、ch2oh、ch2och2cooh、ch2ocho、ch2och3、ch2nh2、ch2oso3na、ch2cooh等基团,其中羧基(cooh)也可以为钠盐的形式存在(coona)。本发明制备聚合物的原料可以为可用于组织粘合剂的多糖类如含有r2为oh
的第一聚合单体的透明质酸、羧甲基纤维素、海藻酸、β
‑
1,3
‑
葡聚糖、羧甲基淀粉等,以及含有r2为nh2的第一聚合单体的壳聚糖等,还可以为透明质酸、海藻酸、壳聚糖的改性衍生物等。需要说明的是,聚合物中除第一聚合单体和第二聚合单体还可以包括其他聚合单体,获得其他聚合单体种类以及第一聚合单体、第二聚合单体在聚合物中的占比根据原料结构以及接枝制备工艺不同而不同。接枝多巴胺的制备工艺可根据需要进行,只要能够实现形成第二聚合单体的结构就行,但一般来说达不到完全接枝,因此,聚合物中还有第一聚合单体存在。
[0047]
采用多糖为原料制备生物组织粘合剂虽然具有生物相容性好的优点,但同时存在湿态粘附性能差,与生物组织不能完全贴合以及密封性能不够的问题。因此,在多糖中接枝多巴胺形成贻贝仿生组织粘合剂,其邻苯二酚结构能够与生物组织的巯基之间生成化学键,使得该生物组织粘合剂同时具有优异密封性能和湿态粘附性能,提高粘合剂在湿态环境下的粘附强度;并且第二聚合单体为开环结构接枝多巴胺,可减少分子结构的位阻,增加多巴胺的接枝率,而聚合物中多巴胺的含量影响聚合物的粘附性,因此以提高粘附强度;且多巴胺与碳原子以单键相连,稳定性高,可以降低粘合剂应用于人体组织后的水解反应,延长粘合剂作用时间;因此通过调节接枝反应的接枝率,从而调节第二聚合单体在聚合物中的占比,以实现使组织粘合剂降解时间可控,其中第二聚合单体在聚合物中的占比越高,降解时间越长。
[0048]
根据本发明的另一具体实施方式,聚合单体还包括第三聚合单体和第四聚合单体;第三聚合单体的结构式为:
[0049][0050]
第四聚合单体的结构为第三聚合单体的两个醛基中的至少一个替换成以亚胺键与基团b连接,基团b的结构式为:
[0051][0052]
具体地,在本实施方式中,获得聚合物的制备工艺为,首先,将多糖原料通过氧化反应使部分第一聚合单体中的2位和3位碳之间开环,且使其上的羟基(
‑
oh)或氨基(
‑
nh2)转变为醛基,从而获得第三聚合单体;然后,醛基化后的多糖与多巴胺发生席夫碱反应,其中部分第三聚合单体生成第四聚合单体,第四聚合单体的结构式可以为如下几种的一种:
[0053][0054]
最后,加入还原剂,使部分第四聚合单体的亚胺键(c=n)还原为单键c
‑
n键,并且使羰基(=o)还原成羟基(
‑
oh),从而获得第二聚合单体。氧化反应所需的氧化剂可以为高碘酸盐、四乙酸铅中的至少一种;优选高碘酸钠,高碘酸钠可选择性断裂多糖单体中的邻二羟基或邻三羟基处,生成相应的多糖醛,例如透明质酸的单体d
‑
葡萄糖醛酸、海藻酸单体、壳聚糖单体都可以在高碘酸钠的氧化下,在2、3为碳原子之间开环醛基化;还原反应的还原剂可以为氢化锂铝(lialh4)、硼氢化钠(nabh4)、氰基硼氢化钠(nabh3cn)、三乙酰氧基硼氢化钠(nabh(oac)3)等中的至少一种。
[0055]
本发明中,醛基化的过程使多糖单体开环,减少环结构空间位阻对接枝多巴胺的影响,且产生足够量的醛基,利用醛基与氨基之间的席夫碱快速缩合反应,保证接枝大量多巴胺,从而可以提高聚合物的粘附强度。
[0056]
醛基化后的多糖与多巴胺发生席夫碱反应后,多巴胺与多糖单体之间依靠亚胺键连接,但亚胺键稳定性较差,易水解使多巴胺游离,从而使组织粘合剂出现粘附性能失效和生物降解不可控,同时其水解反应产物可能会使机体引发炎症反应。本发明在席夫碱反应后,进行还原反应,将亚胺键特异性还原成不易水解的c
‑
n键,使得整个结构体系更加稳定,后期在作为组织粘合剂使用时,可以通过调节亚胺键的还原度来控制降解时间;具体可以通过增加亚胺键的还原度来降低组织粘合剂应用于人体组织后的水解反应,延长粘合剂作用时间,同时使粘合剂降解时间控制更加精确。
[0057]
根据本发明的另一具体实施方式,聚合单体中的r1为cooh、ch2oh、ch2och2cooh中的一种。
[0058]
具体地,当r1为cooh、r2为oh时,第一聚合单元为己糖醛酸,可以为海藻酸、透明质酸的多糖单体;当r1为ch2oh,r2为nh2时,第一聚合单元为己糖胺,可以为壳聚糖的聚合单体;当r1为ch2och2cooh,r2为oh时,可以为羧甲基纤维素的聚合单体。
[0059]
根据本发明的另一具体实施方式,聚合单体中的r1为cooh,r2为oh;且聚合单体还包括第五聚合单体,第五聚合单体的结构式为:
[0060][0061]
且第五聚合单体与第一聚合单体连接形成第一重复单元,与第二聚合单体连接形成第二重复单元,与第三聚合单体连接形成第三重复单元,与第四聚合单体连接形成第四重复单元。
[0062]
具体地,第五聚合单体为n
‑
乙酰葡糖胺,第一聚合单体为d
‑
葡萄糖醛酸,两者之间通过β
‑
1,3
‑
糖苷键相连,形成第一重复单元为透明质酸的重复单元,重复单元之间通过β
‑
1,4
‑
糖苷键相连;因此聚合物的原料是透明质酸,具有优异生物相容性;第一重复单元具体结构如下:
[0063][0064]
透明质酸通过氧化反应,使第一重复单元中部分第一聚合单体开环醛基化形成第三聚合单体,获得第三重复单元,形成的聚合体结构如下:
[0065][0066]
进一步与多巴胺进行席夫碱反应,使第三重复单元中部分第三聚合单体与多巴胺以亚胺键接枝,形成第四聚合单体,获得第四重复单元,形成的聚合体结构如下:
[0067][0068]
最后进行还原反应,使第四重复单元中部分第四聚合单体的亚胺键还原形成第二聚合单体,获得第二重复单元,形成的聚合体结构如下:
[0069][0070]
需要说明的是,上述结构式中第二重复单元和第四重复单元以2位和3位碳都与多巴胺接枝的结构表示,但是应当也存在2位和3位碳上的其中一位与多巴胺接枝的结构,第二重复单元数和第四重复单元数范围根据多巴胺取代情况换算为该结构的数量。
[0071]
在氧化剂选择性断裂透明质酸聚合单体中邻二羟基处,并全部醛基化的基础上,上述聚合物中聚合单元的重复数理论上有如下关系:n=x+y;x=a+b;a=c+d。
[0072]
在聚合物制备过程中,可以通过调整化学反应条件从而改变以上四种重复单元的
比例,继而在后续得到不同粘附性能和降解性能的粘合剂。具体可以通过对醛基化、接枝多巴胺和亚胺键还原这3个化学反应过程中所使用药品(如多巴胺,氧化剂,还原剂)的使用量以及使用种类进行调控,可以得到具有功能差异性的产品;另外反应的时间也可以改变。
[0073]
根据本发明的另一具体实施方式,聚合物中多巴胺的摩尔取代度为0.35~0.65,其中接枝的多巴胺中还原多巴胺摩尔比为40~65%。
[0074]
具体地,透明质酸氧化反应生成醛基化透明质酸;醛基化透明质酸与多巴胺发生席夫碱反应,使聚合物中多巴胺摩尔取代度达到0.35~0.65,即理论上第二重复单元和第四重复单元总数a占聚合物中所有重复单元数为35~65%,其中第二重复单元数占第二重复单元和第四重复单元总数的40~65%,即还原率为40~65%。
[0075]
可以采用核磁共振波谱法(nmr)检测多巴胺摩尔取代度;再利用还原剂如硼氢化钠或氰基硼氢化钠使40~65%的亚胺键还原,可以采用核磁共振波谱法(nmr)检测还原率。上述核磁共振波谱法具体可以采用德国bruker amx400m核磁仪进行1h
‑
nmr扫描以检测。
[0076]
当聚合物中多巴胺的摩尔取代度为达到上述比例范围时,用做组织粘合剂湿态粘附性能良好,且还原率达到上述比例范围时,降解时间较长;经试验证明得到的组织粘合剂粘附强度能够25kpa以上,降解时间达到8天及以上。
[0077]
根据本发明的另一具体实施方式,第一重复单元的重复单元数占聚合物的所有重复单元的重复单元数的比例为10%
‑
25%。
[0078]
具体地,透明质酸氧化反应生成醛基化透明质酸,使醛基化率达到75~90%,从而使第一重复单元(没有被醛基化的部分)的重复单元数占聚合物的所有重复单元的重复单元数的比例为10%
‑
25%;可以采用核磁共振波谱法(nmr)检测醛基化率,具体可以采用德国bruker amx400m核磁仪进行1h
‑
nmr扫描以检测。
[0079]
根据本发明的另一具体实施方式,组织粘合剂为聚合物的水溶液在氧化剂的参与下彼此交联生成的水凝胶。
[0080]
具体地,组织粘合剂为水凝胶状态,聚合物制备成水溶液加入交联剂,交联生成水凝胶,其中聚合物的质量百分比浓度为2~25%,聚合物中含有邻苯二酚结构,因此可以在交联剂高碘酸钠、三价铁离子等存在的条件下发生交联以形成水凝胶,交联剂也可以为光引发剂;但是不同种类氧化剂所形成的凝胶粘附性能有差异。
[0081]
在凝胶粘合剂制备过程中,通过选用不同的聚合物溶度、交联剂配比以及交联时间可以得到具有差异性的组织粘合剂产品。
[0082]
根据本发明的另一具体实施方式,交联剂包括质量百分比浓度为2.5%
‑
10%的高碘酸钠的水溶液。聚合物可以在高碘酸钠的催化下快速交联形成水凝胶,其机理可能是聚合物中多巴胺的邻苯二酚互相连接形成。并且,高碘酸钠作为交联剂形成的凝胶性能较好。
[0083]
根据本发明的另一具体实施方式,聚合物的水溶液的质量百分比浓度为5%
‑
10%。
[0084]
根据本发明的另一具体实施方式,聚合物的水溶液与高碘酸钠的水溶液发生交联反应时的体积比为1:1,反应时间为20~40秒。
[0085]
以下对透明质酸作为原料制备组织粘合剂优选的工艺进行具体实施例说明,但本发明并不受以下实施例的任何限制。
[0086]
实施例1
[0087]
1.醛基化透明质酸的制备
[0088]
将1.0g透明质酸溶解于ph值为5.0的磷酸盐缓冲溶液(pbs缓冲溶液)中,加入0.4g的高碘酸钠,在25℃下避光条件下反应5h,最后加入乙二醇,乙二醇与未参与反应的高碘酸钠发生反应,从而终止氧化反应。反应结束后,将反应液装入透析膜(截留分子量为8000
‑
14000da)中用超纯水透析两天,冷冻干燥得到醛基化透明质酸固体。
[0089]
取适量醛基化透明质酸固体样品溶解于重水,随后采用核磁仪进行1h
‑
nmr扫描以检测醛基取代度。经检测,醛基化率为80%。
[0090]
2.利用席夫碱反应制备多巴胺接枝透明质酸
[0091]
取1.0g步骤1中得到的醛基化透明质酸固体溶解于ph值为5.0的磷酸盐缓冲溶液中,加入0.5g多巴胺盐酸盐,在25℃下反应12h。反应结束后,将反应液装入透析膜(截留分子量为8000
‑
14000da)中用超纯水透析两天,冷冻干燥得到多巴胺接枝透明质酸固体。
[0092]
取适量多巴胺接枝透明质酸固体样品溶解于重水,随后采用核磁仪进行1h
‑
nmr扫描以检测多巴胺摩尔取代度。经检测,多巴胺摩尔取代度为0.35。
[0093]
3.多巴胺接枝的醛基化透明质酸的还原
[0094]
取1.0g步骤2中得到的多巴胺接枝透明质酸溶解于ph=5.0的磷酸盐缓冲溶液(pbs缓冲溶液)中,然后用0.5g氰基硼氢化钠选择性还原席夫碱,在25℃下反应4h,反应结束后,将反应液装入透析膜(截留分子量为8000
‑
14000da)中用超纯水透析两天,冷冻干燥得到部分还原的还原多巴胺接枝透明质酸固体。
[0095]
取适量部分还原的还原多巴胺接枝透明质酸固体样品溶解于重水,随后采用核磁仪进行1h
‑
nmr扫描以检测还原率。经检测,席夫碱还原率为40%。
[0096]
4.粘合剂的制备
[0097]
取1.0g步骤3的还原多巴胺接枝透明质酸固体,用去离子水配成5%质量百分比溶度的溶液,另外配2.5%质量百分比溶度的高碘酸钠溶液,等体积混合,静置30s,快速交联得到组织粘合剂。
[0098]
实施例2
[0099]
步骤参照实施例1,不同的是,步骤1醛基化透明质酸的制备中,加入0.5g的高碘酸钠,反应时间为4h,使醛基化率为80%;步骤2席夫碱反应制备多巴胺接枝透明质酸中,加入0.6g的多巴胺盐酸盐,反应时间为10h,使多巴胺摩尔取代度为0.35;步骤3多巴胺接枝的醛基化透明质酸的还原中,加入氰基硼氢化钠反应时间为5h,席夫碱还原率为43%;步骤4粘合剂的制备中,还原多巴胺接枝透明质酸固体配成8%质量百分比溶度的溶液,配制高碘酸钠溶液的质量百分比溶度为5%,静置时间为40s。
[0100]
实施例3
[0101]
步骤参照实施例1,不同的是,步骤1醛基化透明质酸的制备中,加入0.5g的高碘酸钠,反应时间为6h,使醛基化率为85%;步骤2席夫碱反应制备多巴胺接枝透明质酸中,加入0.7g的多巴胺盐酸盐,反应时间为13h,使多巴胺摩尔取代度为0.5;步骤3多巴胺接枝的醛基化透明质酸的还原中,加入0.75g氰基硼氢化钠,反应时间为5h,席夫碱还原率为50%;步骤4粘合剂的制备中,多巴胺接枝透明质酸固体配成5%质量溶度的溶液,静置时间为20s。
[0102]
实施例4
[0103]
步骤参照实施例1,不同的是,步骤1醛基化透明质酸的制备中,加入0.3g的高碘酸
钠,反应时间为5h,使醛基化率为75%;步骤2席夫碱反应制备多巴胺接枝透明质酸中,加入0.8g的多巴胺盐酸盐,反应时间为12h,使多巴胺摩尔取代度为0.5;步骤3多巴胺接枝的醛基化透明质酸的还原中,加入0.6g氰基硼氢化钠,反应时间为6h,席夫碱还原率为50%;步骤4粘合剂的制备中,多巴胺接枝透明质酸固体配成10%质量溶度的溶液,配置高碘酸钠溶液的质量百分比溶度为5%,静置时间为30s。
[0104]
实施例5
[0105]
步骤参照实施例1,不同的是,步骤1醛基化透明质酸的制备中,加入0.6g的高碘酸钠,反应时间为5h,使醛基化率为90%;步骤2席夫碱反应制备多巴胺接枝透明质酸中,加入0.9g的多巴胺盐酸盐,反应时间为14h,使多巴胺摩尔取代度为0.65;步骤3多巴胺接枝的醛基化透明质酸的还原中,加入0.8g氰基硼氢化钠,反应时间为6h,席夫碱还原率为62%。
[0106]
实施例6
[0107]
步骤参照实施例1,不同的是,步骤1醛基化透明质酸的制备中,加入0.5g的高碘酸钠,反应时间为6h,使醛基化率为90%;步骤2席夫碱反应制备多巴胺接枝透明质酸中,加入1.0g的多巴胺盐酸盐,反应时间为12h,使多巴胺摩尔取代度为0.65;步骤3多巴胺接枝的醛基化透明质酸的还原中,加入1.0g氰基硼氢化钠,反应时间为5h,席夫碱还原率为65%;步骤4粘合剂的制备中,多巴胺接枝透明质酸固体配成5%质量溶度的溶液,配置高碘酸钠溶液的质量百分比溶度为10%,静置时间为40s。
[0108]
以下对实施例1
‑
6获得的组织粘合剂进行性能测定:
[0109]
(1)粘附强度测试:
[0110]
在室温下,参考astm f2255
‑
05测试标准,通过新鲜猪皮模拟生物体组织,使用万能材料试验机(instron 5848)进行搭载剪切强度测试。具体操作如下:取新鲜猪皮,小心刮去表面脂肪层,将猪皮切成宽2cm,长6cm的条状,用ph=7.4的磷酸盐缓冲溶液(pbs)润湿猪皮。测试时,在一块猪皮前端涂抹0.2ml实施例溶度的多巴胺接枝的醛基化透明质酸溶液,同时在另一块猪皮前端涂抹0.2ml实施例溶度的高碘酸钠/氢氧化钠水溶液(ph=8.0
‑
9.0),猪皮表面涂抹区域长度为1cm。接触两块猪皮,形成1
×
2cm2的粘合区域,同时用200g的砝码按压5min。利用万能材料试验机以1mm/min的速率进行拉伸测试。每组测量5个平行样。
[0111]
(2)爆破压力测试:
[0112]
在室温下,参考astm f2392
‑
04标准进行爆破压力测试。具体操作如下:取新鲜猪皮并切成4
×
4cm的形状,小心刮去表面脂肪层。用手术刀在猪皮中心制作直径5mm的圆形切口,并用ph=7.4的磷酸盐缓冲溶液(pbs)润湿猪皮。测试时,同时向圆形切口中注入等体积实施例溶度的多巴胺接枝的醛基化透明质酸溶液和碱性高碘酸钠溶液,静置5分钟后进行爆破压力测试。使用注射器以5ml/min的速度注入空气来持续增加压力,利用数字压力计实时监测爆破压力并读取数据,当压力值下降时认为达到最大爆破压力。每组测量5个平行样。
[0113]
(3)体外降解性能测试:
[0114]
将实施例得到的凝胶粘合剂冷冻干燥,称重并记录为m0。37℃下,将干燥后凝胶浸泡在含有溶度为0.125mg/ml的透明质酸酶的ph=7.4的磷酸盐缓冲溶液(pbs)中,以进行体外降解测试。在设定的时间点取出凝胶,冷冻干燥并称重记录质量为m
x
。同时,用新鲜的透
明质酸酶缓冲溶液替换。体外降解率(degradation rate,dr)的计算公式如下:
[0115][0116]
测试结果如下表:
[0117]
实施例粘附强度(kpa)爆破压力(kpa)降解时间(day)125.1
±
2.920.2
±
2.18235.6
±
4.824.7
±
3.19342.1
±
4.438.8
±
2.411455.8
±
3.949.3
±
3.910567.7
±
5.355.2
±
4.912676.5
±
4.763.8
±
2.913
[0118]
从上表可以看出,本发明获得以透明质酸为原料的组织粘合剂具有较高的粘附强度和降解性能。并且,实施例从1
‑
6,多巴胺摩尔取代度逐渐增大,获得组织粘合剂粘附强度也逐渐增大;并且席夫碱还原度增加,组织粘合剂稳定性增强,降解时间变长。表明本发明可以通过调节该重复单元的含量可实现组织粘合剂降解时间可控。
[0119]
以上内容是结合具体的实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1