一种优化的kraken2算法及其在二代测序中的应用的制作方法
1.本发明涉及生物信息学领域,特别是涉及一种优化的kraken2算法及其在二代测序中的应用。
背景技术:
2.宏基因组群落复杂庞大,需要对大量的dna进行测序,illumina二代测序技术是一种大规模平行测序技术,具有通量高,测序准确度高,时效短等特点,正好完美的匹配了宏基因组学的需求,成就了宏基因组学在感染检测的广泛应用。
3.测序之后微生物群落的物种检测,是宏基因组学研究中的最为重要工作,只有准确可靠地对微生物群落进行精确定位,才能关联宏基因组与研究的关联,比如研究患者的发病是否是某种微生物感染(如某个人怀疑是疟疾,那么需要准确地检测出其血液中存在疟原虫才能最终给出明确诊断),宏基因组分析是一种快速,准确,先进的检测技术,目前在感染类疾病辅助诊断中发挥了重要作用。
4.kraken2应用于illumina二代宏基因组测序,具有分析速度快,灵敏度高的特点,但是特异度较低,往往会检测出很多假阳性结果,这是因为kraken2算法的特点。根据taxid与seqid关系,对选取的参考基因组序列快速构建固定长度的kmers(默认为35bp的读长),优先构建某个层级的特异kmers,比如肺炎链球菌streptococcus pneumoniae,kraken2会优先构建该物种的特异kmers,而链球菌属streptococcus多个物种也存在某个kmer,则将该kmer定位到链球菌属下,同样的原理,某个kmer存在于链球菌科streptococcaceae下多个属,则将该kmer定位在链球菌科下。鉴于kraken2的算法,对于某种dna序列较高的微生物,虽然会有一定的概率会发生错误比对,但是基本上不会干扰该物种的检出。由于二代测序具有读长短的特点,因此很容易出现序列发生错误比对,或者无法精确比对(比如某条来自肺炎链球菌的序列,错误比对到streptococcus mitis,或者只能比对到streptococcus属层级),这是影响物种检测准确度的最重要因素。
5.除此之外,由于数据库包含的序列很多,比如质粒/载体等也在内,因此比对这部分比对的结果也会给出输出,这部分结果基本上是无意义的(也可以算作假阳性检出)。
6.鉴于此,提出本发明。
技术实现要素:
7.本发明的目的是寻求一种能够降低测序分析假阳性,能够提高物种检测的准确度,适用于illumina二代宏基因组测序的生信分析方法。
8.为实现上述目的,本发明提出如下技术方案:
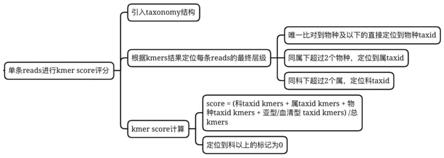
9.本发明首先提供了一种基于kraken2单条序列kmer评分和整体基于taxonomy结构统计的生信分析方法,所述方法包括如下步骤:
10.1)ngs测序数据使用kraken2进行序列比对,得到每条序列的taxid
‑
kmer结果;
11.2)基于taxonomy数据库建立taxid的层级关系,根据步骤1)taxid
‑
kmer结果获得
taxid,并关联taxonomy层级,再根据定位规则重定位taxid;
12.3)根据每条序列经过步骤2)定位的taxid和步骤1)的taxid
‑
kmer比对结果,计算每条序列的kmer score;
13.4)根据kmer score和taxonomy层级,对比对结果进行整体计算;
14.进一步的,还包括
15.5)根据4)的整体计算结果进行物种层级检测。
16.进一步的,所述步骤2)中层级关系包括血清型/亚型、种、属和/或科的一种或多种层级关系。
17.进一步的,所述步骤2)中定位规则包括如下:
18.通常情况下接受kraken2给出的taxid定位,以下情况除外:
19.某条序列根据taxid
‑
kmer结果获得唯一taxid且taxid低于种层级,则定位为该taxid所属的种层级taxid;
20.某条序列根据taxid
‑
kmer结果获得超过2个taxid时,分3种情况:
21.所有taxid,关联到种层级上只出现1个,其他taxid属于该种的血清型/亚型、属、科层级,则定位到该种层级taxid;
22.所有taxid,关联到种层级超过2个且属于同一属,则最终定位到属层级taxid;
23.所有taxid,关联到属层级超过2个(属层级没有分类也在内)且属于同一科,则最终定位到科层级taxid;
24.进一步的,所述步骤3)中所述计算的规则包括:
25.最终定位到科层级taxid以下的序列,其kmer score=(科taxid kmers+属taxid kmers+种taxid kmers+亚型/血清型taxid kmers)/总kmers;
26.最终定位到科层级taxid以上的序列,kmer score设定为0。
27.进一步的,所述步骤4)中整体计算包括:
28.a、设定一个过滤cutoff阈值,对每条序列根据kmer score进行过滤;
29.b、对a中经过过滤的序列,统计taxid的reads;
30.所述taxid的reads是一个样本出现的taxid的序列总数;
31.c、设定一个过滤阈值threshold,对b中定位到种层级的taxid进行过滤,计算其属相对比值,排除低于阈值的种层级taxid;
32.所述属相对比值为某个种层级taxid reads相对于同属reads最高的种层级taxid reads的比值;
33.进一步的,还包括:
34.d、经c过滤的种层级taxid,若缺乏属分类,则计算科相对比值,排除低于过滤阈值threshold种层级taxid;
35.所述科相对比值为某个种层级taxid reads相对于同科reads最高的种层级taxid reads的比值;
36.更进一步的,还包括:
37.e、经c,d过滤保留的种层级taxid reads校正1,计算属相对比值,将属层级taxid reads按照属相对比值计算种层级taxid属校正reads;
38.所述属相对比值为经c,d过滤后同属的种层级taxid reads总和,之后计算各种层
级taxid reads相对于总和的比例;
39.所述属层级taxid reads包括b中属层级taxid reads和c中未通过过滤阈值threshold的该属关联种层级taxid并入的reads;
40.f、经c,d过滤的种层级taxid reads校正2,计算科相对比值,将科层级taxid reads按照科相对比值计算种层级taxid科校正reads;
41.所述科相对比值为经c,d过滤后同科的种层级taxid reads总和,之后计算各种层级taxid reads相对于总和的比例;
42.所述科层级taxid的reads包含b中科层级taxid reads和d中未通过过滤阈值threshold的该科关联种层级taxid并入的reads。
43.进一步的,所述步骤5)物种层级检测,即等同于种层级taxid检测,根据c,d得到种层级taxid即为最终的物种taxid,取b,e,f得到的种层级taxid reads之和得到最终的物种taxid reads,并根据reads之和计算相对丰度。
44.进一步的,所述步骤1)中比对采用的数据库为nt、refseq或genbank数据库;优选的,所述数据库为nt数据库。
45.本发明首先还提供了基于kraken2单条序列kmer评分和整体基于taxonomy结构统计的方法在二代测序生信分析中的应用。
46.本发明还提供一种计算机可读介质,其存储有计算机程序,所述计算机程序被处理器执行时,实现权利要求上述任一项所述方法。
47.本发明还提供一种电子设备,其特征在于,包括处理器以及存储器,所述存储器上存储一条或多条可读指令,所述一条或多条可读指令被所述处理器执行时,实现权利要求上述任一项所述方法。
48.本发明有益的技术效果:
49.1)本发明所述生信方法能够降低生信分析的检出假阳性,能够提高物种检测的准确度,适用于二代宏基因组测序,包括单端和双端测序等。
50.2)本发明通过对单条序列精确定位和整体的系统性优化,保证了整体的灵敏度。
51.3)本发明通过引入taxonomy,排除了质粒/载体部分比对结果等,有效降低了无意义检出的情况。
附图说明
52.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
53.图1本发明体系简图;
54.图2单条序列进行taxid定位和kmer score评分示意图;
55.图3 9例spike
‑
in样本dna文库检出的假阳性物种比较图,opt代表本发明流程,confidence代表kraken2 confidence 0.5+bracken流程,kraken代表kraken2+bracken流程,s1
‑
s9代表9个样本;
56.图4 9例spike
‑
in样本rna文库检出的假阳性物种比较图,opt代表本发明流程,
confidence代表kraken2 confidence 0.5+bracken流程,kraken代表kraken2+bracken流程,s1
‑
s9代表9个样本;
57.图5 12例模拟样本和9例spike
‑
in样本灵敏度统计图,opt代表本发明流程,confidence代表kraken2 confidence 0.5+bracken流程,kraken代表kraken2+bracken流程,simulated代表12例模拟样本,spike
‑
in即9例spike
‑
in样本。
具体实施方式
58.下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限制本发明的范围,并且所述实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
59.部分术语定义
60.除非在下文中另有定义,本发明具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本发明。
61.如本发明中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由
…
组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。
62.本发明中的术语“大约”、“大体”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的
±
10%,优选
±
5%。
63.在提及单数形式名词时使用的不定冠词或定冠词例如“一个”或“一种”,“所述”,包括该名词的复数形式。
64.此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本发明描述的实施方案能以不同于本发明描述或举例说明的其它顺序实施。
65.本发明中的“read”或“每条reads”或“单条reads”,指高通量测序平台产生的一条核酸序列。
66.本发明中的术语“比对结果”:英文为“alignment”,指一条测序读出序列与一条参考序列之间的对应结果,一条测序读出序列可以同时具有多个比对结果。
67.本发明所述的“kmer”是指将一条序列连续切割,逐个碱基划动得到k个碱基的子字符串,例如reads长度为l,k
‑
mer长度设为k,则产生的k
‑
mers数目为:l
‑
k+1;再例如序列aactgact,设置k为3,则可以将其分割为aac、act、ctg、tga、gac、act共6个k
‑
mers。
68.本发明所述的“kraken2”是指本领域的一个基于kmer算法的高精度宏基因组序列分类软件,能够快速的将测序reads进行物种分类。
69.本发明所述的“kraken2优化算法”是指本发明开发的一种基于kraken2比对结果进行微生物物种层级检测的优化体系,旨在提高准确度和降低假阳性检出。
70.本发明所述的“nt比对数据库”是基于ncbi nt数据库建立的kraken2比对索引数
reads的比值;
97.8.经7过滤的种层级taxid,若缺乏属分类,则计算科相对比值,排除低于过滤阈值threshold种层级taxid;
98.所述科相对比值为某个种层级taxid reads相对于同科reads最高的种层级taxid reads的比值;
99.9.经7,8过滤保留的种层级taxid reads校正1,计算属相对比值,将属层级taxid reads按照属相对比值计算种层级taxid属校正reads;
100.所述属相对比值为经7,8过滤后同属的种层级taxid reads总和,之后计算各种层级taxid reads相对于总和的比例;
101.所述属层级taxid reads包括6中属层级taxid reads和7中未通过过滤阈值threshold的该属关联种层级taxid并入的reads;
102.10.经7,8过滤的种层级taxid reads校正2,计算科相对比值,将科层级taxid reads按照科相对比值计算种层级taxid科校正reads;
103.所述科相对比值为经7,8过滤后同科的种层级taxid reads总和,之后计算各种层级taxid reads相对于总和的比例;
104.所述科层级taxid的reads包含6中科层级taxid reads和8中未通过过滤阈值threshold的该科关联种层级taxid并入的reads。
105.11.微生物检测,即等同于种层级taxid检测,根据7,8得到种层级taxid即为最终的物种taxid,取6,9,10得到的种层级taxid reads之和得到最终的物种taxid reads,并根据reads之和计算相对丰度。
106.以下描述仅是为了帮助理解本发明而提供。这些描述不应被理解为具有小于本领域技术人员所理解的范围。
107.实施例1方法体系的设计优化
108.本实施例要解决的问题是如何通过数据分析方法,尽量保证kraken2比对结果的准确度。
109.1.首先,对于该问题,可以拆分为2个问题,如何降低假阳性物种检出以提高特异度,以及保证真实物种检出以获得较高的灵敏度,两者达到最佳平衡,即得到最好的准确度。通过分析kraken2是如何产生假阳性物种结果,以及灵敏度会降低的情况。
110.a)数据库的冗余,无论是refseq,genbank还是nt,都存在着大量的参考基因组冗余,这是造成假阳性检出的重要原因,错误的比对也同样会降低灵敏度;
111.b)序列相似性,典型的如大肠杆菌与志贺杆菌的序列度超过99%,这种情况在同属下的物种间尤为常见,这是造成假阳性检出的重要原因,也会干扰真实物种检出,降低了灵敏度;
112.c)宿主序列(通常指人源宿主)干扰,由于目前的宏基因组基本上来自于宿主样本采集,不可避免的会存在大量的宿主序列,这部分序列会一定程度上影响微生物的检测;
113.d)mngs序列较短,序列较短造成了每条序列得到的kmer较少,这在75bp单端测序上更为明显,容易发生错误比对。
114.2.其次,界定以上哪些情况是可以单独通过对样本测序结果的数据分析来解决的。
115.所谓单独通过对样本测序结果的数据分析,是指:所获得的信息只有一个样本序列文件(fastq),而不知道其他信息;通过一些算法,输出物种检测结果。
116.对于“a)数据库的冗余”,一方面可以通过在数据库整理,去冗余中来实现,但是数据库过度精简会造成假阴性的发生,所以除了数据库优化之外,还可以通过算法进行一定程度的降低,算法部分是本专利的考虑点;
117.对于“b)序列相似性”,一方面可以通过标准基因组更新和物种分类学的进步来解决,另外可以通过算法来进行一定程度的优化,这部分是本专利的考虑点;
118.对于“c)宿主序列(通常指人源序列)干扰”,这个一方面可以通过精准构建宿主的基因组序列和使用比对算法去除宿主基因组,但是并不能保证宿主基因组去除的是否准确彻底(去除不彻底仍会有一定的残留,另外去除过度会降低微生物检出的灵敏度),除此之外可以通过在算法上对单条序列比对结果进行精确分析,和整体上设定物种检出的阈值,得到一定程度的优化,这部分是本专利的考虑点;
119.对于“d)mngs序列较短”,这部分是技术硬性问题,需要通过技术的进步来提升。
120.3.kraken2算法的具体优化方案
121.针对2中的3个优化点,结合kraken2的比对原则,建立了如下的优化体系:
122.3.1单条序列的处理
123.a)引入taxonomy层级,根据单条序列taxid
‑
kmer比对结果依据定位规则重新定位其层级和对应的taxid;
124.定位规则包括:原则上接受kraken2给出的taxid定位,以下情况除外:
125.某条序列比对到唯一taxid且taxid低种层级,则定位为该taxid所属的种层级taxid(举例,某条序列长度76bp,35bp kmer长度则得到42个kmer,taxid
‑
kmer比对结果为:0:10,1313:32,除了不能比对的10个kmer之外,其余都比对到1313这个taxid上,通过taxonomy层级结构定位到taxid 1313streptococcus pneumoniae肺炎链球菌);
126.某条序列比对超过2个taxid,分为3种:
127.比对结果中的所有taxid,关联到种层级上只有1个物种,其他taxid属于该种的血清型/亚型,属、科层级,则定位到该种taxid(举例,某条序列长度76bp,35bp kmer长度则得到42个kmer,taxid
‑
kmer比对结果为:0:10,1313:20,1301:12,除了不能比对的10个kmer之外,20个kmer比对到1313这个taxid上,通过taxonomy层级结构对应streptococcus pneumoniae肺炎链球菌,另外12个kmer比对到1301streptococcus链球菌属,由于肺炎链球菌在链球菌属下,则该序列定位到种taxid 1313streptococcus pneumoniae肺炎链球菌);
128.比对结果中的所有taxid,关联出同属不同种的结果,则最终定位到属taxid(举例,某条序列长度76bp,35bp kmer长度则得到42个kmer,taxid
‑
kmer比对结果为:0:10,1313:20,28037:5,1301:7,除了不能比对的10个kmer之外,20个kmer比对到1313这个taxid上,通过taxonomy层级结构对应streptococcus pneumoniae肺炎链球菌,另外5个kmer比对到28037streptococcus mitis轻型链球菌,7个kmer比对到1301streptococcus链球菌属,由于链球菌属下出现了2个种,则该序列定位到taxid 1301streptococcus链球菌属);
129.比对结果中的所有taxid,关联出同科不同属的结果,则最终定位到科taxid;
130.b)根据每条序列经过a)定位的taxid和taxid
‑
kmer比对结果,根据计算规则计算每条序列的kmer score;
131.计算规则为:
132.最终定位到科以下的序列,其kmer score=(科taxid kmers+属taxid kmers+种taxid kmers+亚型/血清型taxid kmers)/总kmers;
133.最终定位到科以上的序列,kmer score设定为0。
134.3.2整体的处理
135.c)设定一个score_cutoff,根据每条序列的kmer score判定其结果是否可信,未过score_cutoff视为不可信结果予以排除;
136.d)对于最终定位到噬菌体/质粒/载体的序列,全部标记为未比对结果;
137.e)对c)d)中经过过滤的序列,统计taxid的reads;
138.f)对e)中定位到种层级的taxid关联属和科,计算每个属下所有种的属相对比值(属内各种reads数相对于属内reads最高种reads数的比值),设定一个threshold,将属相对比值低于过滤阈值的这部分种reads重新定位到属层级;
139.g)类同于f),对缺乏属信息但是有明确科信息的种,计算科相对比值(同科下每个种相对于该科内reads最高种的比值),将科相对比值低于f)中过滤阈值threshold这部分种reads重新定位到科层级;
140.h)对于最终比对到属层级的reads(包括原始比对到属层级的reads和步骤f)未通过过滤阈值threshold的该属下种reads之和),计算该属经f)过滤剩余种的reads总和,并计算该属剩余种相对总和的比值,之后根据各剩余种的比值,将最终比对到属层级的reads分配到各剩余种上;
141.i)对于最终比对到科层级的reads(包含原始比对到科层级的reads和g)步骤未通过过滤阈值threshold的该科下种reads之和),计算该科经过g)过滤剩余种的reads总和,并计算剩余种相对总和的比值,之后根据各剩余种的比值,将最终比对到科层级的reads分配到各剩余种上;
142.j)报出最终物种reads(即经过h),i)处理的种reads)和相对丰度时,设定一个reads过滤阈值reads_cutoff,低于某个阈值的物种,不予统计和报出。
143.4、kraken2的模拟数据测试和建立相应的各个阈值(score_cutoff,threshold,reads_cutoff)
144.建立测试数据集,测试优化方法,和初步建立方法中涉及的阈值
145.以选取的4种细菌(流感嗜血杆菌,肺炎链球菌,金黄色葡萄球菌,肺炎克雷伯菌),2种真菌(白色念球菌,烟曲霉),4种病毒(人类疱疹病毒,人乳头瘤病毒,甲型流感病毒,hiv病毒),按照一定的reads比例,加入到人源宿主reads(grc38.p13)中,序列长度设定为75bp,总数据量设定为10m,一共4组,每组由3个完全一致的样本组成。模拟样本使用的参考基因组如下表所示:
[0146][0147]
具体的模拟样本如下表所示(其中按照顺序3个为重复的样本,比如样本1,2,3为完全一致的样本):
[0148][0149][0150]
score_cutoff在不同阈值下统计样本的统计结果汇总,样本7
‑
12由于微生物总量
较低,重点关注,其错误率要高于整体层级,从统计结果来看设定高于此错误率的threshold就可以解决错误比对的发生,具体如下表所示:
[0151][0152]
鉴于score_cutoff在0.5和0.4两个数值达到错误率相同层级,对每个样本分为list物种假阳性物种(即错误比对检出的物种在10个模拟物种的同科),非list物种假阳性物种(错误比对检出的物种不在10个模拟物种的同科),统计其各自reads最高的物种,由于重复性样本检出完全一致,只列出代表样本结果,具体如下表所示:
[0153][0154]
由于优化方法中包含了对同属/科的校正,最终的reads_cutoff则是设定解决非同科检出的物种进行校正的,从score_cutoff不同值的比较来看,在允许稍低一点的比对率的前提下,score_cutoff设定为0.5,reads_cutoff设定到大于3即可消除非list假阳性物种检出(该值越低越有利于保证reads偏少检出的真实阳性物种的灵敏度)。
[0155]
综上,最终确定本发明的技术方案如下:
[0156]
1)基于kraken2比对结果进行;
[0157]
2)引入taxonomy层级,单条序列根据taxid
‑
kmer比对结果,依据定位规则重新定位其层级和对应的taxid;
[0158]
定位规则包括:
[0159]
原则上接受kraken2给出的taxid定位,以下情况除外:
[0160]
某条序列比对到唯一taxid且taxid低于物种层级,则定位为该taxid所属的物种taxid;
[0161]
某条序列比对超过2个taxid,分为3种:
[0162]
比对结果中的所有taxid,关联到物种层级上只有1个物种,其他taxid属于该物种的血清型/亚型,属、科层级,则定位到该物种taxid;
[0163]
比对结果中的所有taxid,关联出同属不同物种的结果,则最终定位到属taxid;
[0164]
比对结果中的所有taxid,关联出同科不同属的结果,则最终定位到科taxid;
[0165]
3)根据每条序列经过2)定位的taxid和taxid
‑
kmer比对结果,根据计算规则计算每条序列的kmer score评分;
[0166]
计算规则为:
[0167]
最终定位到科以下的序列,其kmer score=(科taxid kmers+属taxid kmers+物种taxid kmers+亚型/血清型taxid kmers)/总kmers;
[0168]
最终定位到科以上的序列,kmer score设定为0。
[0169]
4)设定score_cutoff为0.5,根据每条序列的kmer score判定其结果是否可信,kmer score未过score_cutoff视为不可信结果予以排除;
[0170]
5)对于最终定位到噬菌体/质粒/载体的序列,全部标记为未比对结果;
[0171]
6)对4)5)中经过过滤的序列,统计taxid的reads;
[0172]
7)对6)中定位到物种层级的taxid关联属和科,计算每个属下所有物种的属相对比值(属内各物种reads数相对于属内reads最高物种reads数的比值),设定一个threshold,其值设定为0.1,将属相对比值低于过滤阈值的这部分物种reads重新定位到属层级;
[0173]
8)类同于7),对缺乏属信息但是有明确科信息的物种,计算科相对比值(同科下每个物种相对于该科内reads最高物种的比值),将科相对比值低于7)中过滤阈值threshold这部分物种reads重新定位到科层级;
[0174]
9)对于最终比对到属层级的reads(包括6)属层级taxid的reads和步骤7)未通过过滤阈值threshold的该属物种reads之和),计算该属经7)过滤剩余物种的reads总和,并计算该属剩余物种相对总和的比值,之后根据各剩余物种的比值,将最终比对到属层级的reads分配到各剩余物种上;
[0175]
10)对于最终比对到科层级的reads(包含6)科层级taxid的reads和8)步骤未通过过滤阈值threshold的该科物种reads之和),计算该科经过8)过滤剩余物种的reads总和,并计算剩余物种相对总和的比值,之后根据各剩余物种的比值,将最终比对到科层级的reads分配到各剩余物种上;
[0176]
11)报出最终物种reads和相对丰度时,设定一个reads过滤阈值reads_cutoff,该值设定为3,reads低于reads_cutoff的物种,不予统计和报出。最后报出物种层级的reads和相对丰度。
[0177]
实施例2与传统kraken2方法的效果比较
[0178]
1、根据优化方法最终检测的模拟样本结果整理假阳性与假阴性检出如下:
[0179][0180]
统计指标:
[0181]
灵敏度为117/120(非人源物种总数)=97.5%;
[0182]
假阳性物种检出3例。
[0183]
2.2根据kraken2 confidence 0.5+braken流程检测的结果整理假阳性检出和假阴性检出如下表所示:
[0184]
样本taxid物种reads相对丰度结果样本1340412aspergillus novofumigatus10.00011假阳性样本1984962heterobasidion irregulare10.00011假阳性样本1145522nannochloropsis oceanica40.00044假阳性样本128037streptococcus mitis20.00022假阳性样本12656787venustampulla echinocandica10.00011假阳性样本1086049cladophialophora carrionii10.00011假阳性样本1010376human gammaherpesvirus 410.00011假阳性样本101873960pseudocercospora fijiensis20.00022假阳性样本102656787venustampulla echinocandica10.00011假阳性样本10727haemophilus influenzae00假阴性样本10573klebsiella pneumoniae00假阴性样本11727haemophilus influenzae00假阴性样本11573klebsiella pneumoniae00假阴性样本12727haemophilus influenzae00假阴性样本12573klebsiella pneumoniae00假阴性样本4145522nannochloropsis oceanica20.00022假阳性样本499802spirometra erinaceieuropaei20.00022假阳性样本71220188aspergillus tanneri10.00011假阳性样本745219guanarito mammarenavirus10.00011假阳性样本7145522nannochloropsis oceanica30.00033假阳性
[0185]
统计指标:
[0186]
灵敏度为114/120=95%;
[0187]
假阳性物种检出43例(其中reads大于3的为3例)。
[0188]
3、kraken2+bracken即不引入confidence阈值的检测结果中,统计其中的假阳性物种统计为5517例(其中reads大于3的为1188例),假阴性物种为出现在样本10
‑
12中的
klebsiella pneumoniae共3例。
[0189]
结论:kraken2不设置confidence阈值灵敏度与本专利方法一致,但是假阳性物种检出过多,而设置confidence阈值之后虽然假阳性物种检出会减少很多,但是灵敏度会降低。综合比较,本专利方法在保证了灵敏度基础上,降低了假阳性物种检出,效果更优。
[0190]
实施例3实际样本检测实验
[0191]
使用了9例spike
‑
in样本,各自建立dna文库和rna文库,进行上机测序,具体的样本及阳性物种见下表:
[0192]
[0193]
[0194]
[0195][0196]
本发明的流程未检出的阳性物种,kraken2 confidence 0.5+bracken流程未检出的阳性物种,kraken2+bracken流程未检出的阳性物种统计如下表所示(其中reads_opt,abundance_opt代表本发明流程的阳性物种检出,reads_confidence,abundance_confidence代表kraken2 confidence 0.5+bracken流程的阳性物种检出,reads_kraken,abundance_kraken代表kraken2+bracken流程的阳性检出):
[0197][0198]
阳性物种合计148,kraken2 confidence 0.5+bracken有2个物种未检出(灵敏度为98.6%),本发明流程和kraken2+bracken有一个物种未检出(灵敏度为99.3%),表现与模拟数据类似,灵敏度上本发明流程,和kraken2+bracken相同,稍高于kraken2 confidence 0.5+bracken的流程。
[0199]
dna文库各个流程检出的假阳性物种汇总统计如下表(对应图3结果,其中图片中
的opt代表本发明流程,confidence代表kraken2 confidence 0.5+bracken流程且对应表格中的第4列,kraken代表kraken2+bracken流程):
[0200][0201]
rna文库各个流程检出的假阳性物种汇总统计如下表(对应图4结果,其中图片中的opt代表本发明流程,confidence代表kraken2 confidence 0.5+bracken流程且对应表格中第4列,kraken代表kraken2+bracken流程):
[0202][0203][0204]
汇总模拟样本和spike
‑
in样本统计的灵敏度结果如下表(对应图5结果,其中图片中的opt代表本发明流程,confidence代表kraken2 confidence 0.5+bracken流程,kraken代表kraken2+bracken流程):
[0205][0206]
从以上的统计结果来看,本发明流程假阳性物种检出要远低于kraken2+bracken流程,且在保证了灵敏度高于kraken2 confidence 0.5+bracken基础上,假阳性检出要低于后者(即便是在reads>3才报出的情况下,仍然能降低大约1/3的假阳性物种检出)。
[0207]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,但本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1