一种药物共担载纳米颗粒及其制备方法和应用
1.本发明属于医疗领域,尤其涉及一种药物共担载纳米颗粒及其制备方法和应用。
背景技术:
2.肿瘤是指机体在各种致瘤因子作用下,局部组织细胞增生所形成的新生物,因为这种新生物多呈占位性块状突起,也称赘生物。根据新生物的细胞特性及对机体的危害性程度,又将肿瘤分为良性肿瘤和恶性肿瘤两大类,恶性肿瘤可分为癌和肉瘤,癌是指来源于上皮组织的恶性肿瘤,肉瘤是指间叶组织,包括纤维结缔组织、脂肪、肌肉、脉管、骨和软骨组织等。
3.目前,肿瘤依然是危害人类生命健康的最大杀手,现在用于肿瘤治疗的传统手段有手术,化疗,放疗。其中,由于手术和放疗只能用于肿瘤局部治疗,而对转移瘤束手无策,化疗可以使药物随着血液循环遍布全身绝大多数器官和组织,成为治疗肿瘤特别是中晚期肿瘤的有效手段。然而,如何将治疗药物有效递送到肿瘤组织,是目前肿瘤化疗面临的最大挑战。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种药物共担载纳米颗粒及其制备方法和应用,采用本发明方法制备的药物共担载纳米颗粒的载药率高,能够在肿瘤部位有效富集,治疗效果好,副作用小。
5.本发明提供了一种药物共担载纳米颗粒的制备方法,包括以下步骤:
6.a)将高分子载体、至少两种小分子药物和有机溶剂混合,得到混合溶液;
7.所述高分子载体为甲氧基聚乙二醇改性聚谷氨酸;所述小分子药物为小分子疏水药物和/或小分子亲水药物;
8.b)将所述混合溶液滴加到水中,搅拌,透析,得到药物共担载纳米颗粒。
9.优选的,所述甲氧基聚乙二醇改性聚谷氨酸中的甲氧基聚乙二醇链段的数均分子量为500~10000da,聚谷氨酸链段的数均分子量为10000~70000da;
10.所述甲氧基聚乙二醇改性聚谷氨酸中甲氧基聚乙二醇所对应的重复单元与聚谷氨酸所对应的重复单元的摩尔比为(1~20):1。
11.优选的,所述小分子药物选择米托蒽醌、阿霉素、jq1、喜树碱、藤黄酸、吲哚菁绿、ir783、abt263和nlg919中的至少两种。
12.优选的,所述高分子载体与小分子药物的质量比为(0.01~100):1。
13.优选的,所述高分子载体和小分子药物在混合溶液中的合计浓度≥0.01mg/ml。
14.优选的,所述水的体积为混合溶液体积的5~20倍。
15.优选的,所述滴加的速度为0.1~5ml/min;所述搅拌的时间≥0.5min;所述透析的时间为0.5~3天。
16.本发明提供了一种药物共担载纳米颗粒,包括高分子载体和担载在所述高分子载
体上的至少两种小分子药物;
17.所述高分子载体为甲氧基聚乙二醇改性聚谷氨酸;
18.所述小分子药物为小分子疏水药物和/或小分子亲水药物。
19.优选的,所述药物共担载纳米颗粒的粒径为50~200nm。
20.本发明还提供了一种上述技术方案所述制备方法制得的药物共担载纳米颗粒或上述技术方案所述的药物共担载纳米颗粒在制备肿瘤治疗药物中的应用。
21.与现有技术相比,本发明提供了一种药物共担载纳米颗粒及其制备方法和应用。本发明提供的药物共担载纳米颗粒制备方法包括以下步骤:a)将高分子载体、至少两种小分子药物和有机溶剂混合,得到混合溶液;所述高分子载体为甲氧基聚乙二醇改性聚谷氨酸;所述小分子药物为小分子疏水药物和/或小分子亲水药物;b)将所述混合溶液滴加到水中,搅拌,透析,得到药物共担载纳米颗粒。本发明提供的方法利用甲氧基聚乙二醇改性聚谷氨酸(plg
‑
g
‑
mpeg)作为药物载体,通过静电作用和疏水作用,对多种亲/疏水小分子药物进行高效担载。相对比单独担载,这种多种药物的共担载方式能够明显提高药物,特别是疏水药物的担载效率;同时,本发明方法所制备的药物共担载纳米颗粒的粒径稳定可控,其粒径范围在50~200nm,在体内具有较好的高渗透长滞留效应(enhancedpermeability and retention effect,epr效应),能够实现药物在肿瘤部位的有效运输和富集,治疗效果好,副作用低;而且,本发明方法所制备的药物共担载纳米颗粒还具有一定的ph响应释放性能,可实现肿瘤部位特异性药物释放,从而进一步减少药物的副作用;此外,本发明方法所制备的药物共担载纳米颗粒可根据选择的药物进行不同的联合治疗,具有一定的普适性。本发明提供的制备方法工艺简单,条件温和,易于操作,适合大规模生产应用;采用该方法制备的药物共担载纳米颗粒具有十分优异的治疗效果,在有效抑制肿瘤生长的同时能极大地降低药物对机体的副作用,具有十分广阔的市场前景。
附图说明
22.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
23.图1是本发明实施例4提供的共担载纳米颗粒在不同ph值溶液中的mto释放速率曲线图;
24.图2是本发明实施例5提供的4t1细胞用不同药物处理后的细胞生存率柱状图;
25.图3是本发明实施例7提供的4t1细胞用不同药物处理后的细胞生存率柱状图。
具体实施方式
26.下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
27.本发明提供了一种药物共担载纳米颗粒的制备方法,包括以下步骤:
28.a)将高分子载体、至少两种小分子药物和有机溶剂混合,得到混合溶液;
29.b)将所述混合溶液滴加到水中,搅拌,透析,得到药物共担载纳米颗粒。
30.在本发明提供的制备方法中,步骤a)中,所述高分子载为甲氧基聚乙二醇改性聚谷氨酸(plg
‑
g
‑
mpeg),其由聚谷氨酸(plg)主链和接枝在主链上的甲氧基聚乙二醇(mpeg)支链组成,其具体结构可如式(i)所示:
[0031][0032]
在本发明提供的制备方法中,步骤a)中,所述甲氧基聚乙二醇改性聚谷氨酸中的甲氧基聚乙二醇链段的数均分子量优选为500~10000da,具体可为500da、1000da、2000da、3000da、4000da、5000da、6000da、7000da、8000da、9000da或10000da;所述甲氧基聚乙二醇改性聚谷氨酸中的聚谷氨酸链段的数均分子量优选为10000~70000da,具体可为10000da、15000da、20000da、21600da、25000da、30000da、35000da、40000da、45000da、50000da、55000da、60000da、65000da或70000da;所述甲氧基聚乙二醇改性聚谷氨酸中甲氧基聚乙二醇所对应的重复单元与聚谷氨酸所对应的重复单元的摩尔比优选为(1~20):1,更优选为(1~15):1,最优选为(1~10):1,具体可为1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1。
[0033]
在本发明提供的制备方法中,步骤a)中,所述甲氧基聚乙二醇改性聚谷氨酸可以为市售,液可以按照以下步骤制备得到:
[0034]
i)将甲氧基聚乙二醇(mpeg)和聚谷氨酸(plg)在有机溶剂中混合反应,得到反应产物;
[0035]
ii)将所述反应产物与缩合剂混合反应,后处理,得到甲氧基聚乙二醇改性聚谷氨酸。
[0036]
在本发明提供的上述甲氧基聚乙二醇改性聚谷氨酸制备步骤中,步骤i)中,所述甲氧基聚乙二醇的数均分子量优选为500~10000da,具体可为500da、1000da、2000da、3000da、4000da、5000da、6000da、7000da、8000da、9000da或10000da;所述聚谷氨酸的数均分子量优选为10000~70000da,具体可为10000da、15000da、20000da、21600da、25000da、30000da、35000da、40000da、45000da、50000da、55000da、60000da、65000da或70000da;所述甲氧基聚乙二醇和聚谷氨酸的重复单元摩尔比优选为(1~20):1,更优选为(1~15):1,最优选为(1~10):1,具体可为1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1。
[0037]
在本发明提供的上述甲氧基聚乙二醇改性聚谷氨酸制备步骤中,步骤i)中,所述混合反应的温度优选为30~50℃,更优选为40℃;所述混合反应的时间优选为1~5h,更优选为2h。
[0038]
在本发明提供的上述甲氧基聚乙二醇改性聚谷氨酸制备步骤中,步骤ii)中,所述缩合剂包括4
‑
二甲氨基吡啶(dmap)和/或二异丙基碳二亚胺;所述4
‑
二甲氨基吡啶与制备所述反应产物的原料物聚谷氨酸的用量比优选为0.48mmol:(1~5)g,更优选为0.48mmol:2g;所述二异丙基碳二亚胺与制备所述反应产物的原料物聚谷氨酸的用量比优选为
4.8mmol:(1~5)g,更优选为4.8mmol:2g。
[0039]
在本发明提供的上述甲氧基聚乙二醇改性聚谷氨酸制备步骤中,步骤ii)中,所述混合反应在搅拌条件下进行;所述混合反应的温度优选为15~35℃,更优选为25℃(室温);所述混合反应的时间优选为1~5天,更优选为3天。
[0040]
在本发明提供的上述甲氧基聚乙二醇改性聚谷氨酸制备步骤中,步骤ii)中,所述后处理的方式优选为:依次进行乙醚沉淀、洗涤、真空干燥、蒸馏水透析和冻干。
[0041]
在本发明提供的制备方法中,步骤a)中,所述小分子药物为小分子疏水药物和/或小分子亲水药物,其相对分子质量通常小于1000;所述小分子疏水药物包括但不限于抗肿瘤化疗药米托蒽醌、阿霉素在内的蒽环类药物,及具有肿瘤免疫微环境调节作用的小分子疏水药,所述小分子亲水药物包括但不限于含有磺酸基团的小分子成像试剂。在本发明中,所述小分子药物优选包括米托蒽醌(mto)、阿霉素(dox)、jq1、喜树碱、藤黄酸(ga)、吲哚菁绿、ir783、abt263和nlg919中的至少两种,其中,jq1、ir783、abt263和nlg919的化学结构如下所示:
[0042][0043]
在本发明提供的一个实施例中,步骤a)中,所述小分子药物选择米托蒽醌和jq1,所述米托蒽醌和jq1的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:9。
[0044]
在本发明提供的另一个实施例中,步骤a)中,所述小分子药物选择米托蒽醌和藤黄酸,所述米托蒽醌和藤黄酸的质量比优选为1:(0.5~5),具体可为1:0.5、1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5或1:5,最优选为1:2。
[0045]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择米托蒽醌和abt263,所述米托蒽醌和abt263的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:
9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:10。
[0046]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择米托蒽醌和阿霉素,所述米托蒽醌和阿霉素的质量比优选为1:(0.2~3),具体可为1:0.2、1:0.5、1:0.7、1:1、1:1.2、1:1.5、1:1.7、1:2、1:2.3、1:2.5、1:2.7或1:3,最优选为1:1。
[0047]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择米托蒽醌和nlg919,所述米托蒽醌和nlg919的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:10。
[0048]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择阿霉素和jq1,所述阿霉素和jq1的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:10。
[0049]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择阿霉素和藤黄酸,所述阿霉素和藤黄酸的质量比优选为1:(0.5~5),具体可为1:0.5、1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5或1:5,最优选为1:2。
[0050]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择阿霉素和abt263,所述阿霉素和abt263的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:10。
[0051]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择阿霉素和nlg919,所述阿霉素和nlg919的质量比优选为1:(5~15),具体可为1:5、1:6、1:7、1:8、1:9、1:10、1:11、1:12、1:13、1:14或1:15,最优选为1:10。
[0052]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择jq1和abt263,所述jq1和abt263的质量比优选为5:(10~50),具体可为5:10、5:15、5:20、5:25、5:30、5:35、5:40、5:45或5:50,最优选为5:25。
[0053]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择喜树碱和jq1,所述喜树碱和jq1的质量比优选为1:(0.5~5),具体可为1:0.5、1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5或1:5,最优选为1:2。
[0054]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择喜树碱和abt263,所述喜树碱和abt263的质量比优选为1:(0.5~5),具体可为1:0.5、1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5或1:5,最优选为1:2。
[0055]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择藤黄酸和jq1,所述藤黄酸和jq1的质量比优选为2:(5~15),具体可为2:5、2:6、2:7、2:8、2:9、2:10、2:11、2:12、2:13、2:14或2:15,最优选为2:10。
[0056]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择吲哚菁绿和藤黄酸,所述吲哚菁绿和藤黄酸的质量比优选为1:(2~10),具体可为1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9或1:10,最优选为1:5。
[0057]
在本发明提供的其他实施例中,步骤a)中,所述小分子药物选择ir783和藤黄酸,所述ir783和藤黄酸的质量比优选为1:(2~10),具体可为1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9或1:10,最优选为1:5。
[0058]
在本发明提供的制备方法中,步骤a)中,所述高分子载体与小分子药物的质量比优选为(0.01~100):1,更优选为(0.05~10):1,最优选为(0.1~5):1,具体可为0.1:1、
0.12:1、0.15:1、0.16:1、0.18:1、0.2:1、0.23:1、0.25:1、0.27:1、0.3:1、0.32:1、0.34:1、0.37:1、0.4:1、0.42:1、0.45:1、0.47:1、0.5:1、0.52:1、0.55:1、0.57:1、0.6:1、0.62:1、0.65:1、0.67:1、0.7:1、0.75:1、0.8:1、0.85:1、0.9:1、0.95:1、1:1、1.2:1、1.5:1、1.7:1、2:1、2.5:1、3:1、3.5:1、4:1、4.5:1或5:1。
[0059]
在本发明提供的制备方法中,步骤a)中,所述有机溶剂可以与水互溶并能溶解载体和小分子药物,优选为甲醇和/或二甲基亚砜,更优选为二甲基亚砜。
[0060]
在本发明提供的制备方法中,步骤a)中,所述高分子载体和小分子药物在混合溶液中的合计浓度优选为≥0.01mg/ml,更优选为0.1~50mg/ml,最优选为1~20mg/ml,最最优选为10~20mg/ml。
[0061]
在本发明提供的制备方法中,步骤a)中,优选先将高分子载体和小分子药物分别与有机溶剂混合,然后再将得到的高分子载体溶液与小分子药物溶液混合。其中,所述高分子载体溶液的浓度优选为≥0.01mg/ml,更优选为0.1~50mg/ml,最优选为1~20mg/ml,最最优选为20mg/ml;所述小分子药物溶液的浓度优选为≥0.01mg/ml,更优选为0.1~50mg/ml,最优选为1~20mg/ml,最最优选为10mg/ml。
[0062]
在本发明提供的制备方法中,步骤a)中,所述混合的温度优选为15~35℃,更优选为25℃(室温);所述混合的时间没有特别限定,可使各组分充分溶解并混合均匀即可。
[0063]
在本发明提供的制备方法中,步骤b)中,所述水优选为超纯水;所述水的体积优选为混合溶液体积的5~20倍,更优选为5~15倍,最优选为10倍。
[0064]
在本发明提供的制备方法中,步骤b)中,所述滴加的温度优选为15~35℃,更优选为25℃(室温);所述滴加的速度优选为0.1~5ml/min,更优选为0.1~3ml/min,最优选为1~2ml/min。
[0065]
在本发明提供的制备方法中,步骤b)中,所述搅拌优选为快速搅拌;所述搅拌的温度优选为15~35℃,更优选为25℃(室温);所述搅拌的时间优选为≥0.5min,更优选为3~20min,最优选为5min。
[0066]
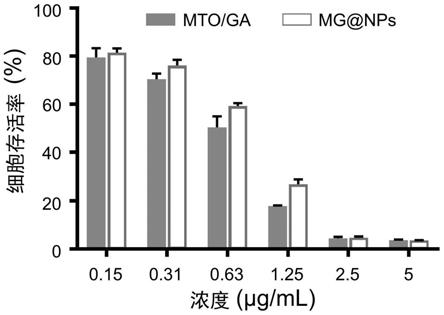
在本发明提供的制备方法中,步骤b)中,所述透析所采用的透析袋的截留分子量优选为2000~5000,更优选为3500;所述透析的温度优选为15~35℃,更优选为25℃(室温);所述透析的时间优选为0.5~3天,更优选为1~3天,最优选为2天。
[0067]
本发明还提供了一种采用上述技术方案所述方法制备的药物共担载纳米颗粒。本发明提供的药物共担载纳米颗粒包括高分子载体和担载在所述高分子载体上的至少两种小分子药物;其中,所述高分子载体和小分子药物在上文中已经介绍,在此不再赘述;所述药物共担载纳米颗粒的粒径优选为50~200nm,具体可为50nm、53.8nm、55nm、60.8nm、65nm、66.1nm、70nm、75nm、78.2nm、80nm、85nm、90nm、90.2nm、95nm、100nm、105nm、110nm、113nm、115nm、117.8nm、118.6nm、120nm、123.8nm、125nm、130nm、135nm、136.9nm、137.9nm、140nm、145nm、150nm、155nm、155.5nm、160nm、165nm、169nm、170nm、171.6nm、175nm、180nm、181.7nm、185nm、190nm、195nm或200nm,在上粒径范围内的纳米颗粒在体内具有较好的epr效应。
[0068]
本发明还提供了一种上述技术方案所述的药物共担载纳米颗粒或上述技术方案所述制备方法制得的药物共担载纳米颗粒在制备肿瘤治疗药物中的应用。此外,虽然本发明强调了其在制备肿瘤治疗药物中的应用,但所述药物共担载纳米颗粒的应用范围并不限
于肿瘤治疗领域,可根据选择担载的药物疗效应用于不同领域。
[0069]
本发明提供的技术方案利用甲氧基聚乙二醇改性聚谷氨酸作为药物载体,通过静电作用和疏水作用,对多种亲/疏水小分子药物进行高效担载。相对比单独担载,这种多种药物的共担载方式能够明显提高药物,特别是疏水药物的担载效率;同时,本发明技术方案所制备的药物共担载纳米颗粒的粒径稳定可控,其粒径范围在50~200nm,在体内具有较好的高渗透长滞留效应(enhanced permeability and retention effect,epr效应),能够实现药物在肿瘤部位的有效运输和富集,治疗效果好,副作用低;而且,本发明技术方案所制备的药物共担载纳米颗粒还具有一定的ph响应释放性能,可实现肿瘤部位特异性药物释放,从而进一步减少药物的副作用;此外,本发明技术方案所制备的药物共担载纳米颗粒可根据选择的药物进行不同的联合治疗,具有一定的普适性。本发明提供的技术方案工艺简单,条件温和,易于操作,适合大规模生产应用;采用该技术方案制备的药物共担载纳米颗粒具有十分优异的治疗效果,在有效抑制肿瘤生长的同时能极大地降低药物对机体的副作用,具有十分广阔的市场前景。
[0070]
为更清楚起见,下面通过以下实施例进行详细说明。
[0071]
实施例1
[0072]
1)高分子载体plg
‑
g
‑
mpeg的制备:
[0073]
按照甲氧基聚乙二醇和聚谷氨酸的重复单元摩尔比5:1,将2g的聚谷氨酸(数均分子量21600da)和所需用量的甲氧基聚乙二醇(数均分子量5000da)溶解在80毫升无水dmf中,然后在40℃下反应2小时;反应结束后,待混合物的温度降为室温,然后在混合物中加入0.48mmol dmap和4.8mmol的二异丙基碳二亚胺,接着在室温下搅拌3天;搅拌结束后,用乙醚沉淀混合物并洗涤两次,真空干燥后,用蒸馏水透析2天,最后冻干,得到plg
‑
g
‑
mpeg。
[0074]
2)药物共担载纳米颗粒的制备:
[0075]
将步骤1)制备的plg
‑
g
‑
mpeg高分子载体在二甲基亚砜中溶解,溶解浓度为20mg/ml;然后将小分子药物(米托蒽醌、jq1)分别溶于二甲基亚砜,溶解浓度为10mg/ml;将载体溶液和药物溶液按照相应的用量比例在室温下混合,之后将混合溶液缓慢滴加(2.0ml/min)至超纯水中,所用超纯水体积为二甲基亚砜体积的十倍;滴加结束后,在室温下搅拌5min,然后将搅拌后的混合液置于截留分子量为3500的透析袋中,在室温下透析2天,得到药物共担载纳米颗粒。
[0076]
3)药物共担载纳米颗粒载药率的测定:
[0077]
将步骤2)制备的药物共担载纳米颗粒分散在水溶液中,浓度1mg/ml,后用二甲基亚砜溶剂稀释200μl纳米颗粒分散水溶液,涡旋振荡15s后,使用紫外分光光度计检测药物米托蒽醌(mto)在621nm处的吸收峰,用于米托蒽醌的载药量测定;使用同样的方法溶解游离药,测定不同浓度游离药的吸收峰值,用于标准曲线绘制。对于药物jq1的载药率,将制备的药物共担载纳米颗粒分散在水溶液中,后将分散液冻干,取10mg用于元素分析检测,通过检测s元素定量纳米药物中jq1的含量。
[0078]
不同plg
‑
g
‑
mpeg与药物用量比例的药物共担载纳米颗粒的载药率测定结果详见表1:
[0079]
表1实施例1药物共担载纳米颗粒的载药率
[0080][0081]
表1中,dle表示载药率。
[0082]
通过表1数据可以看出,在载体plg
‑
g
‑
mpeg:mto:jq1比例为2:1:9时具有最优的药物担载率。此外,使用同样比例载体单独担载米托蒽醌和jq1,米托蒽醌载药量为31.1%,没有明显下降;但是jq1载药率下降为15.3%,载药过程中出现明显沉淀,这说明药物共担载体系能够明显提高疏水药的载药率。
[0083]
实施例2
[0084]
1)药物共担载纳米颗粒的制备:
[0085]
将实施例1步骤1)制备的plg
‑
g
‑
mpeg高分子载体在二甲基亚砜中溶解,溶解浓度为20mg/ml;然后将小分子药物(米托蒽醌、藤黄酸)分别溶于二甲基亚砜,溶解浓度为10mg/ml;将载体溶液和药物溶液按照相应的用量比例在室温下混合,之后将混合溶液缓慢滴加(2.0ml/min)至超纯水中,所用超纯水体积为二甲基亚砜体积的十倍;滴加结束后,在室温下搅拌5min,然后将搅拌后的混合液置于截留分子量为3500的透析袋中,在室温下透析2天,得到药物共担载纳米颗粒。
[0086]
3)药物共担载纳米颗粒载药率的测定:
[0087]
将步骤2)制备的药物共担载纳米颗粒分散在水溶液中,浓度1mg/ml,后用二甲基亚砜溶剂稀释200μl纳米颗粒分散水溶液,涡旋振荡15s后,使用紫外分光光度计检测药物米托蒽醌(mto)在621nm处的吸收峰,用于米托蒽醌的载药量测定;使用同样的方法溶解游离药,测定不同浓度游离药的吸收峰值,用于标准曲线绘制。对于药物藤黄酸(ga)的载药率,同样使用紫外分光光度法进行测定,药物ga的吸收峰检测在363nm处。
[0088]
不同plg
‑
g
‑
mpeg与药物用量比例的药物共担载纳米颗粒的载药率测定结果详见表2:
[0089]
表2实施例2药物共担载纳米颗粒的载药率
[0090]
[0091]
表2中,dle表示载药率。
[0092]
通过表2数据可以看出,在载体plg
‑
g
‑
mpeg:mto:ga比例为1.5:1:0.5时两种药物载药量最高。此外,使用同样比例载体单独担载ga和米托蒽醌,米托蒽醌载药量为36.4%,没有明显下降;但是藤黄酸载药率下降为5.3%,载药过程中出现明显沉淀,这说明药物共担载体系能够明显提高疏水药的载药率。
[0093]
实施例3
[0094]
药物共担载纳米颗粒的制备:
[0095]
将实施例1步骤1)制备的plg
‑
g
‑
mpeg高分子载体在二甲基亚砜中溶解,溶解浓度为20mg/ml;然后将不同的小分子药物分别溶于二甲基亚砜,溶解浓度为10mg/ml;将载体溶液和药物溶液按照相应的用量比例在室温下混合,之后将混合溶液缓慢滴加(2.0ml/min)至超纯水中,所用超纯水体积为二甲基亚砜体积的十倍;滴加结束后,在室温下搅拌5min,然后将搅拌后的混合液置于截留分子量为3500的透析袋中,在室温下透析2天,得到药物共担载纳米颗粒。不同担载药物以及plg
‑
g
‑
mpeg与药物的用量比例,详见表3:
[0096]
表3实施例3的原料信息表
[0097]
编号药物1药物2plg
‑
g
‑
mpeg、药物1、药物2的质量比1米托蒽醌jq12:1:92米托蒽醌藤黄酸2:1:23米托蒽醌abt2632:1:104米托蒽醌阿霉素2:1:15米托蒽醌nlg9192:1:106阿霉素jq12:1:107阿霉素藤黄酸2:1:28阿霉素abt2632:1:109阿霉素nlg9192:1:1010jq1abt2633:5:2511喜树碱jq12:1:212喜树碱abt2632:1:213藤黄酸jq12:2:1014吲哚菁绿藤黄酸2:1:515ir783藤黄酸2:1:5
[0098]
使用电位粒径仪对所制备的药物共载纳米颗粒进行粒径检测,结果如表4所示:
[0099]
表4实施例3制备的药物共载纳米颗粒的粒径
[0100]
编号共载纳米颗粒粒径(nm)11132169360.8478.25118.66137.9
7123.8853.89136.91066.111171.612281.713117.81490.215155.5
[0101]
通过表4可以看出,上述实施例制备的药物共载纳米颗粒的粒径稳定在50~200nm,因此其在体内能具有良好的epr效应。
[0102]
实施例4
[0103]
将实施例3编号1制备的共担载纳米颗粒分散在水溶液中,浓度为0.5mg/ml,体积为3ml,后将纳米颗粒水溶液置于截留分子量为3500的透析袋中,分别置于ph=7.2、6.5、5.0缓冲液中,于37℃恒温震荡箱中震荡,在0h、1h、2h、4h、6h、8h、12h、24h、48h和72h取上清缓冲液2ml用于药物检测,每次取点的同时补加2ml新的缓冲液。通过紫外分光光度计检测上清缓冲液621nm波长的吸收值,进而推算出不同时间米托蒽醌(mto)的药物释放量。
[0104]
实验结果如图1所示,图1是本发明实施例4提供的共担载纳米颗粒在不同ph值溶液中的mto释放速率曲线图。通过图1可以看出,在ph=7.2缓冲液中,米托蒽醌药物释放的最慢;ph=6.5条件下,其释放量与ph=7.2无明显差别;但是在更酸的条件下,ph=5.0缓冲液中,米托蒽醌快速释放,48h释放量是ph=7.2缓冲液的2倍。这个实验现象说明,对于蒽环类药物,本发明提供的方法制备的纳米药物具有一定的ph响应释放性能,这种性能有利于实现药物在肿瘤部位的特异性释放,减少药物在体内的副作用。
[0105]
实施例5
[0106]
将实施例3编号1所制备的药物共载纳米颗粒用于体外抗肿瘤治疗。选用4t1细胞用于体外抑瘤实验。将4t1细胞按照每孔1
×
104的密度种植于96孔板中,培养过夜。后将不同浓度的药物加入细胞中,培养4h后,将培养基上清移去,更换新鲜的培养基,继续培养44h,后在96孔板每孔加入20μl噻唑蓝溶液(5mg/ml),37℃继续培养4h,加入二甲基亚砜溶解,于490nm下测定每孔吸光度值。细胞存活率采用以下公式计算:
[0107]
细胞存活率(%)=a样品/a空白
×
100。
[0108]
实验结果如图2所示,图2是本发明实施例5提供的4t1细胞用不同药物处理后的细胞生存率柱状图;图2中,mto为游离态的化疗药米托蒽醌;jq1为游离态的小分子抑制剂jq1;mto+jq1为游离态的上述两种药物复配,质量比为mto:jq1=1:9;mj@nps为实施例3编号1所制备的药物共载纳米颗粒;每种药物的柱形图中从左到右依次对应的药物浓度为2μg/ml、1μg/ml、0.5μg/ml、0.25μg/ml、0.125μg/ml、0.0625μg/ml、0.032μg/ml。
[0109]
通过以上实验可以得到两个结论:1)实验结果表明,化疗药米托蒽醌和小分子抑制剂jq1具有非常好的药物协同效果,药物协同指数为0.48,两种药物共载对肿瘤细胞具有更强的杀伤能力;2)相对比游离药,形成的共载纳米药物具有相对较弱的肿瘤细胞杀伤能力,这可能与药物缓慢释放有关,这有利于降低纳米药物对正常组织的副作用。
[0110]
实施例6
[0111]
将实施例3编号1所制备的药物共载纳米颗粒用于体内抗肿瘤治疗。体内抗肿瘤实验选用4t1肿瘤模型,采用20g左右的balb/c小白鼠,肿瘤接种前,取对数生长期的4t1细胞,用胰蛋白酶消化,然后用细胞培养液混合胰蛋白酶,1
×
103rpm离心5min,pbs洗涤两次,用pbs悬浮细胞。按每只小鼠2
×
106细胞接种于小鼠腋下。7天后,瘤径平均长大至80mm3时进行体内抗肿瘤治疗。将实施例3编号1制备的纳米药物或等当量的游离药通过尾静脉注入老鼠体内,给药量为米托蒽醌5mg/kg,jq135mg/kg,仅进行一次给药。给药后跟踪肿瘤体积大小和老鼠体重的变化,整个实验过程共跟踪20天。治疗后的肿瘤生长情况见表5:
[0112]
表5进行不同治疗后的肿瘤重量
[0113]
给药治疗后肿瘤重量(g)pbs1.92游离mto0.5游离jq10.53游离mto+jq10.19实施例3编号1纳米颗粒0.07
[0114]
体内抑瘤实验结果表明,1)游离的米托蒽醌单药(mto)和jq1单药对肿瘤生长具有明显的抑制作用;2)游离的两种药结合治疗后,肿瘤重量是两种单药治疗的0.4倍,说明两种药物具有很好的药物协同治疗效果;3)实施例3编号1纳米颗粒治疗后,肿瘤生长被明显抑制,是所有治疗中抑瘤效果最好的,肿瘤重量是游离药组的0.37倍,说明相对比游离药,纳米颗粒由于epr效应能更好的在肿瘤部位蓄积,有效运载纳米药物至肿瘤部位,杀伤肿瘤。
[0115]
实施例7
[0116]
将实施例3编号2所制备的药物共载纳米颗粒用于体外抗肿瘤治疗。选用4t1细胞用于体外抑瘤实验。将4t1细胞按照每孔1
×
104的密度种植于96孔板中,培养过夜。后将不同浓度的药物加入细胞中,培养4h后,将培养基上清移去,更换新鲜的培养基,继续培养44h,后在96孔板每孔加入20μl噻唑蓝溶液(5mg/ml),37℃继续培养4h,加入二甲基亚砜溶解,于490nm下测定每孔吸光度值。细胞存活率采用以下公式计算:
[0117]
细胞存活率(%)=a样品/a空白
×
100。
[0118]
实验结果如图3所示,图3是本发明实施例7提供的4t1细胞用不同药物处理后的细胞生存率柱状图;图3中,mto/ga为游离态的米托蒽醌(mto)和藤黄酸(ga)复配,质量比为mto:ga=1:2;mg@nps为实施例3编号2所制备的药物共载纳米颗粒。
[0119]
通过图3可以看出,相对比游离药,形成的共载纳米药物具有相对较弱的肿瘤细胞杀伤能力,这可能与药物缓慢释放有关,这有利于降低纳米药物对正常组织的副作用。
[0120]
实施例8
[0121]
将实施例3编号6制备的药物共担载纳米颗粒分散在水溶液中,浓度1mg/ml,后用二甲基亚砜溶剂稀释200μl纳米颗粒分散水溶液,涡旋振荡15s后,使用紫外分光光度计检测阿霉素(dox)的载药率;使用同样的方法溶解游离药,测定不同浓度游离药的吸收峰值,用于标准曲线绘制。对于药物jq1的载药率,将制备的药物共担载纳米颗粒分散在水溶液中,后将分散液冻干,取10mg用于元素分析检测,通过检测s元素定量纳米药物中jq1的含
量。该药物共担载纳米颗粒的载药率详见下表:
[0122]
表6实施例3编号6药物共担载纳米颗粒的载药率
[0123]
药物载药率(%)dox30.4jq166.7
[0124]
此外,在同样药物比例下,单独担载两种药物,dox和jq1的载药量分别为29.7%和12.6%,载药过程中有明显沉淀析出,说明共载能够有效提高疏水药的载药率。
[0125]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1