一种同源重组修复缺陷的评估方法、装置和存储介质与流程
1.本技术涉及同源重组修复缺陷评估技术领域,特别是涉及一种同源重组修复缺陷的评估方法、装置和存储介质。
背景技术:
2.同源重组修复(homologous recombination repair,hrr)是dna双链断裂(double strand break,dsb)的首选修复方式。同源重组修复缺陷(homologous recombination deficiency,hrd)通常指细胞水平上的hrr功能障碍状态,可由hrr相关基因胚系突变或体细胞突变以及表观遗传失活等诸多因素导致,常存在于多种恶性肿瘤中,在卵巢癌、乳腺癌、胰腺导管癌、前列腺癌等肿瘤中尤其突出。hrd会产生特定的、可量化的、稳定的基因组改变,可通过建立基于基因组特征分析的评估体系来预测肿瘤hrd状态及其程度,已成为晚期卵巢癌患者临床应用聚腺苷二磷酸核糖聚合酶[poly(adp-ribose)polymerase,parp]抑制剂的新型生物标志物,也可能对乳腺癌、前列腺癌等肿瘤的parp抑制剂和铂类药物的临床用药具有指导价值。最近的上皮性卵巢癌parp抑制剂相关生物标志物检测的专家共识中指出,hrd检测被推荐用于指导一线新诊断卵巢癌的治疗方案选择,建议卵巢癌患者确诊就应进行hrd检测,包含brca基因检测,其结果对维持治疗的疗效预测及预后判断具有重要参考价值。目前,hrd作为反应肿瘤患者化疗敏感度、后期靶向治疗的最重要的生物标志物,已经成为卵巢癌治疗重要的一步。
[0003]
hrd临床检测所描述的肿瘤基因组特定改变,也被称为“基因组瘢痕”。自2012年以来,杂合性缺失(loss of heterozygosity,loh)、端粒等位基因不平衡(telomeric allelic imbalance,tai)、大片段迁移(large-scale state transition,lst)等被作为基因组瘢痕标志物,以量化基因组瘢痕的程度。loh定义为大于15mb且小于整个染色体长度的杂合性缺失;tai定义为延伸到其中一个亚端粒但不超过着丝粒且大于11mb的等位基因不平衡的染色体片段;lst定义为两个相邻区域之间的染色体断裂位点,肿瘤基因组截断点的总数可以用来描述基因组的不稳定性。其中,相邻区域是指两个区域长度均大于或等于10mb,且区域间距小于3mb。loh、tai和lst等3个指标都有独特的定义,在一定程度上能描述细胞hrd状态的程度。
[0004]
现有技术普遍采用基因芯片捕获测序或高深度全基因组测序进行同源重组修复缺陷的基因瘢痕评估。但是,基因芯片捕获测序存在数据误差较大的问题,而全基因组测序对测序深度要求较高。此外,国外文章shallowhrd:detection of homologous recombination deficiency from shallow whole genome sequencing中尝试采用低深度全基因组数据计算lst,但真实临床样本的性能差,并且,对发生全基因组复制的样本存在指标评分偏高、灵敏度低、准确率低等问题。因此,如何更简单、灵敏、准确的对同源重组修复缺陷的基因瘢痕进行评估,是目前亟待解决的问题。
技术实现要素:
[0005]
本技术的目的是提供一种新的同源重组修复缺陷的评估方法、装置和存储介质。
[0006]
为了实现上述目的,本技术采用了以下技术方案:
[0007]
本技术的第一方面公开了一种同源重组修复缺陷的评估方法,包括以下步骤:
[0008]
全基因组测序数据获取和比对步骤,包括获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件;
[0009]
全基因组测序数据质控步骤,包括对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据;
[0010]
污染数据过滤步骤,包括对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据;
[0011]
拷贝数变异分析步骤,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱;其中,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息;
[0012]
lst值计算步骤,包括根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cna cutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1;可以理解,lst的定义为相邻两个segments片段的间隔小于3mb,且长度均大于10mb;因此相邻两个segments要有断点,即高度差,cna cutoff是计算出来的高度差;当高度差越小,相应找到的lst会更多,则lst值越大;因此,本技术通过降低cna cutoff,使样本的lst增加;
[0013]
待测样本全基因组复制分析步骤,包括判断待测样本是否发生全基因组复制,如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值;
[0014]
同源重组修复缺陷状态评估步骤,包括根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。
[0015]
本技术中,局部最小值即对密度分布图取导数时,导数等于0对应的第一个局部最小值所在的点。
[0016]
需要说明的是,本技术的同源重组修复缺陷评估方法,可以直接采用低深度全基因组测序数据进行lst值计算,并采用全基因组复制情况对lst值进行校正,使得校正后的lst值可以直接单独与brca基因型别结合用于同源重组修复缺陷状态评估;并且,由于校正后的lst值计算更准确,提高了其作为parp抑制剂治疗及预后的中间参考数据的有效性和准确性。
[0017]
本技术的一种实现方式中,全基因组测序数据获取和比对步骤的低深度全基因组测序是指测序深度不超过5。
[0018]
需要说明的是,低深度全基因组测序是指测序深度不超过5的测序,进一步的,测序深度可以不超过3。
[0019]
本技术的一种实现方式中,采用bwa-mem2软件将去除接头的序列比对到参考基因组hg19上。
[0020]
本技术的一种实现方式中,全基因组测序数据质控步骤的比对率大于95%,深度大于0.8为合格;gc含量和重复率无阈值,仅用于对样本质量进行辅助判断。
[0021]
可以理解,以上具体数值仅仅是本技术的一种实现方式中具体采用的判断标准,在更严格或宽松的要求下,以上具体数值可以根据需求进行调整。
[0022]
本技术的一种实现方式中,污染数据过滤步骤的污染率阈值为使用群体性等位基因频率构建模型评估样本的污染率。
[0023]
本技术的一种实现方式中,污染率阈值为0.1。
[0024]
可以理解,污染率阈值为0.1只是本技术的一种实现方式中根据具体的群体性等位基因频率构建模型评估获得的阈值;在相同的发明构思下,采用不同的群体或模型所获得的污染率阈值可以不同,在此不作具体限定。
[0025]
本技术的一种实现方式中,待测样本全基因组复制分析步骤中,判断待测样本是否发生全基因组复制的方法包括,根据segments片段密度分布图,通过segments片段的极差情况判断样本是否发生全基因组复制,判断规则包括,
[0026]
a.当样本segments片段极差小于1时,样本不发生全基因组复制;
[0027]
b.当样本segments片段极差大于1,且峰值个数小于3时,样本不发生全基因组复制;
[0028]
c.当样本segments片段极差大于1,且峰值个数大于或等于3时,样本发生全基因组复制;
[0029]
d.当样本segments片段极差大于9,且峰值个数大于或等于2时,样本发生全基因组复制。
[0030]
需要说明的是,以上具体的判断待测样本是否发生全基因组复制的方法,能够准确有效的判断低深度全基因组测序样本的全基因组复制情况,从而更好的用于lst值校正。
[0031]
本技术的一种实现方式中,待测样本全基因组复制分析步骤还包括对segments片段密度分布图显示的峰进行判断,判断标准包括,(1)只有大于最大峰值15%的峰才算作峰;(2)初步判断峰值个数大于2时,则按以下规则判断:如果峰与左右两侧波谷的距离均大于最大峰值的4%,则该峰参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,则该峰不参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,下一个峰仍是相同情形,则记为一个峰。
[0032]
需要说明的是,以上segments片段密度分布图显示的峰的判断标准,通过对特殊情况峰的判断与处理,进一步的提高了全基因组复制判断的准确性和有效性;尤其适用于低深度全基因组测序样本的全基因组复制情况判断。
[0033]
本技术的一种实现方式中,同源重组修复缺陷状态评估步骤,具体判断同源重组修复缺陷状态的规则包括,当brca基因型别为突变型时,无论lst值如何,同源重组修复缺
陷状态均判断为阳性;当brca基因型别为野生型时,lst值大于或等于hrd生物学阈值,则判断同源重组修复缺陷状态为阳性,否则为阴性。
[0034]
本技术的一种实现方式中,hrd生物学阈值是以模型数据中brca突变样本中的95%均为hrd阳性确定的阈值;
[0035]
本技术的一种实现方式中,hrd生物学阈值为15。
[0036]
可以理解,hrd生物学阈值为15只是本技术的一种实现方式中根据具体的模型数据获得的阈值;在相同的发明构思下,采用不同的模型数据可能获得的具体的hrd生物学阈值有所不同,在此不作具体限定。
[0037]
本技术的第二方面公开了一种同源重组修复缺陷的评估装置,包括全基因组测序数据获取和比对模块、全基因组测序数据质控模块、污染数据过滤模块、拷贝数变异分析模块、lst值计算模块、待测样本全基因组复制分析模块和同源重组修复缺陷状态评估模块;
[0038]
全基因组测序数据获取和比对模块,包括用于获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件;
[0039]
全基因组测序数据质控模块,包括用于对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据;
[0040]
污染数据过滤模块,包括用于对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据;
[0041]
拷贝数变异分析模块,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息;
[0042]
lst值计算模块,包括用于根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cnacutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1;
[0043]
待测样本全基因组复制分析模块,包括用于判断待测样本是否发生全基因组复制,如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值;
[0044]
同源重组修复缺陷状态评估模块,包括用于根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。
[0045]
需要说明的是,本技术的同源重组修复缺陷评估装置,实际上就是通过各模块分别实现本技术同源重组修复缺陷评估方法中的各步骤;因此,各模块的具体限定可以参考
本技术同源重组修复缺陷的评估方法,在此不累述。例如,全基因组测序数据质控模块中,比对率大于95%,深度大于0.8为合格;污染数据过滤模块中,污染率阈值为使用群体性等位基因频率构建模型评估样本的污染率。又例如,通过segments片段的极差情况判断样本是否发生全基因组复制的具体判断规则,segments片段密度分布图显示的峰的判断标准,判断同源重组修复缺陷状态的规则等,都可以参考本技术的同源重组修复缺陷的评估方法。
[0046]
本技术的第三方面公开了一种评估同源重组修复缺陷的装置,该装置包括存储器和处理器;存储器包括用于存储程序;处理器包括用于通过执行存储器存储的程序以实现本技术的同源重组修复缺陷的评估方法。
[0047]
本技术的第四方面公开了一种计算机可读存储介质,该存储介质中存储有程序,该程序能够被处理器执行以实现本技术的同源重组修复缺陷的评估方法。
[0048]
由于采用以上技术方案,本技术的有益效果在于:
[0049]
本技术的同源重组修复缺陷评估方法和装置,利用低深度全基因组测序数据进行lst值计算,并采用全基因组复制情况对lst值进行校正;无需loh和tai等其他基因组瘢痕标志物,可以直接利用本技术校正后的lst值进行同源重组修复缺陷状态评估。并且,由于校正后的lst值计算更准确,提高了其作为parp抑制剂治疗及预后的中间参考数据的有效性和准确性。
附图说明
[0050]
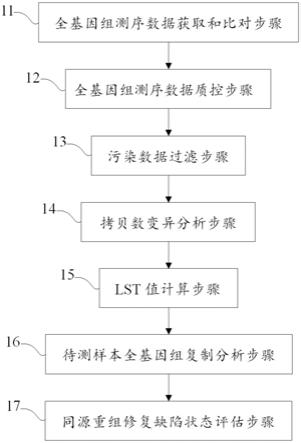
图1是本技术实施例中同源重组修复缺陷评估方法的流程框图;
[0051]
图2是本技术实施例中同源重组修复缺陷评估装置的结构框图;
[0052]
图3是本技术实施例中40例样本的生存曲线图。
具体实施方式
[0053]
下面通过具体实施方式结合附图对本技术作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本技术能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他装置、材料、方法所替代。在某些情况下,本技术相关的一些操作并没有在说明书中显示或者描述,是为了避免本技术的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
[0054]
目前尚未有准确有效的基于低深度全基因组测序进行同源重组修复缺陷评估的方法。虽然,国外有少数文章报道采用低深度全基因组数据计算lst,但真实临床样本的性能差、灵敏度低、准确率低。
[0055]
本技术创造性的提出,(1)采用ace替换了现有文献报道中的controlfreec进行基因拷贝数变异(cnv)分析。(2)用新算法计算cna cutoff值:将cna cutoff设置为密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加。cna cutoff是通过segment片段密度图来确定。原算法中,当cna cutoff为0.45时,lst score值会偏小,影响对hrd的判断。本技术针对这一情况进行了优化:当局部最小值大于0.45时,部分密度图在
0.45之前会出现导数值减小但未改变正负的小拐点;此时计算0.025到0.45之间密度图的导数,将导数的最小值对应的差值为新的cna cutoff。(3)判断wgd,对lst进行校正。
[0056]
通过以上三方面的优化和改进,提高对parpi用药有效性的预测。可以理解,基于以上改进,对患者的拷贝数变异拟合结果更准确,从而对lst score计算更准确,最后可以使对parpi用药有效性的预测更准确。
[0057]
基于以上研究和认识,本技术创造性的提出了一种同源重组修复缺陷的评估方法,如图1所示,包括全基因组测序数据获取和比对步骤11、全基因组测序数据质控步骤12、污染数据过滤步骤13、拷贝数变异分析步骤14、lst值计算步骤15、待测样本全基因组复制分析步骤16和同源重组修复缺陷状态评估步骤17。
[0058]
其中,全基因组测序数据获取和比对步骤11,包括获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件。
[0059]
本技术的一种实现方式中,具体的,使用bwa-mem2软件对截取掉umi的序列进行参考基因组比对,生成sam文件,参考基因组为hg19;使用samtools软件包将比对结果进行fixmate、sort、markdup,具体的,samtools fixmate-m${sample}.sam${sample}_fixmate.bam,该步骤将上述sam文件作为输入文件,修复表头文件信息得到fixmate.bam文件;samtools sort${sample}_fixmate.bam-o${sample}_sort.bam,将fixmate.bam文件排序用于下一步分析;samtools markdup-r${sample}_sort.bam${sample}_markdup.bam,将排好序的bam文件过滤掉pcr产生的重复序列,得到最终的bam文件用于下一步分析。
[0060]
全基因组测序数据质控步骤12,包括对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据。
[0061]
本技术的一种实现方式中,使用bamdst软件对上一步最终生成的bam文件进行质量分析,该软件提供所需bam文件以及bed文件(即位点信息文件)即可产生对应样本的质量信息,包括比对率、测序深度、gc含量、重复率。比对率大于95%,深度大于0.8为合格。gc含量与重复率无阈值,仅对样本质量进行辅助判断。
[0062]
污染数据过滤步骤13,包括对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据。
[0063]
本技术的一种实现方式中,污染率分析软件为verifybamid2.0.1,将前述最终生成的bam文件使用verifybamid2.0.1软件进行处理,该软件通过使用群体性等位基因频率构建模型评估样本的污染率,污染率小于0.1为合格。
[0064]
拷贝数变异分析步骤14,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱;其中,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息。
[0065]
lst值计算步骤15,包括根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cna cutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值
小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1。本技术的一种实现方式中,lst值具体使用shallowhrd软件进行计算。
[0066]
待测样本全基因组复制分析步骤16,包括判断待测样本是否发生全基因组复制,如果有发生全基因组复制,且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值。
[0067]
本技术的一种实现方式中,待测样本全基因组复制分析步骤还包括对segments片段密度分布图显示的峰进行判断,判断标准包括,(1)只有大于最大峰值15%的峰才算作峰;(2)初步判断峰值个数大于2时,则按以下规则判断:如果峰与左右两侧波谷的距离均大于最大峰值的4%,则该峰参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,则该峰不参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,下一个峰仍是相同情形,则记为一个峰。
[0068]
判断待测样本是否发生全基因组复制的方法包括,根据segments片段密度分布图,通过segments片段的极差情况判断样本是否发生全基因组复制,判断规则如下:
[0069]
a.当样本segments片段极差小于1时,样本不发生全基因组复制;
[0070]
b.当样本segments片段极差大于1,且峰值个数小于3时,样本不发生全基因组复制;
[0071]
c.当样本segments片段极差大于1,且峰值个数大于或等于3时,样本发生全基因组复制;
[0072]
d.当样本segments片段极差大于9,且峰值个数大于或等于2时,样本发生全基因组复制。
[0073]
同源重组修复缺陷状态评估步骤17,包括根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。
[0074]
本技术的一种实现方式中,判断同源重组修复缺陷状态的规则为:当brca基因型别为突变型时,无论lst值如何,同源重组修复缺陷状态均判断为阳性;当brca基因型别为野生型时,lst值大于或等于hrd生物学阈值,则判断同源重组修复缺陷状态为阳性,否则为阴性。并且,以模型数据中brca突变样本中的95%均为hrd阳性,确定hrd生物学阈值为15;即当brca基因型别为野生型时,lst值大于或等于15,则判断同源重组修复缺陷状态为阳性,否则为阴性。
[0075]
本领域技术人员可以理解,上述方法的全部或部分功能可以通过硬件的方式实现,也可以通过计算机程序的方式实现。当上述方法中全部或部分功能通过计算机程序的方式实现时,该程序可以存储于一计算机可读存储介质中,存储介质可以包括:只读存储器、随机存储器、磁盘、光盘、硬盘等,通过计算机执行该程序以实现上述功能。例如,将程序存储在设备的存储器中,当通过处理器执行存储器中程序,即可实现上述全部或部分功能。另外,当上述实施方式中全部或部分功能通过计算机程序的方式实现时,该程序也可以存储在服务器、另一计算机、磁盘、光盘、闪存盘或移动硬盘等存储介质中,通过下载或复制保存到本地设备的存储器中,或对本地设备的系统进行版本更新,当通过处理器执行存储器
中的程序时,即可实现上述方法中全部或部分功能。
[0076]
因此,基于本技术的同源重组修复缺陷的评估方法,本技术提出了一种同源重组修复缺陷的评估装置,如图2所示,包括全基因组测序数据获取和比对模块21、全基因组测序数据质控模块22、污染数据过滤模块23、拷贝数变异分析模块24、lst值计算模块25、待测样本全基因组复制分析模块26和同源重组修复缺陷状态评估模块27。
[0077]
其中,全基因组测序数据获取和比对模块21,包括用于获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件。例如,参考bwa-mem2软件将序列比对到参考基因组hg19上,生成sam文件;以sam文件作为输入,参考samtools软件包将比对结果依序进行fixmate、sort、markdup,获得过滤了pcr扩增重复序列的bam文件用于下一步分析。
[0078]
全基因组测序数据质控模块22,包括用于对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据。例如,参考bamdst软件进行质量分析,将比对率大于95%,深度大于0.8数据视为合格。
[0079]
污染数据过滤模块23,包括用于对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据。例如,参考污染率分析软件verifybamid2.0.1进行污染率分析,并定义污染率小于0.1为合格。
[0080]
拷贝数变异分析模块24,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息。
[0081]
lst值计算模块25,包括用于根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cna cutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1。例如,参考shallowhrd软件进行lst值计算。
[0082]
待测样本全基因组复制分析模块26,包括用于判断待测样本是否发生全基因组复制,如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度分布图中峰值个数大于4,则lst值减9,作为最终的lst值。
[0083]
本技术的一种实现方式中,待测样本全基因组复制分析模块26还包括用于对segments片段密度分布图显示的峰进行判断,判断标准包括,(1)只有大于最大峰值15%的峰才算作峰;(2)初步判断峰值个数大于2时,则按以下规则判断:如果峰与左右两侧波谷的距离均大于最大峰值的4%,则该峰参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,则该峰不参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,下一个峰仍是相同情形,则记为一个峰。通过segments片段的极差情况判
断样本是否发生全基因组复制的判断规则与本技术的同源重组修复缺陷的评估方法相同。
[0084]
同源重组修复缺陷状态评估模块27,包括用于根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。同样的,具体判断规则与本技术的同源重组修复缺陷的评估方法相同。
[0085]
本技术的另一实现方式中还提供了一种评估同源重组修复缺陷的装置,该装置包括存储器和处理器;存储器,包括用于存储程序;处理器,包括用于通过执行存储器存储的程序以实现以下方法:全基因组测序数据获取和比对步骤,包括获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件;全基因组测序数据质控步骤,包括对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据;污染数据过滤步骤,包括对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据;拷贝数变异分析步骤,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱;其中,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息;lst值计算步骤,包括根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cna cutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1;待测样本全基因组复制分析步骤,包括判断待测样本是否发生全基因组复制,如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值;同源重组修复缺陷状态评估步骤,包括根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。
[0086]
本技术另一种实现方式中还提供一种计算机可读存储介质,该存储介质中包括程序,该程序能够被处理器执行以实现如下方法:全基因组测序数据获取和比对步骤,包括获取待测样本的低深度全基因组测序下机数据,去除接头,将其比对到参考基因组上,根据比对排序过滤pcr产生的重复序列,获得比对文件;全基因组测序数据质控步骤,包括对比对文件进行质量分析,获得包括比对率、测序深度、gc含量和重复率在内的质量信息,根据质量信息过滤获取合格的测序数据;污染数据过滤步骤,包括对测序数据进行污染率分析,分析其受污染情况,获取污染率小于污染率阈值的测序数据;拷贝数变异分析步骤,包括采用ace软件确定低深度全基因组测序样本的肿瘤纯度,并且生成total cna谱;其中,total cna谱包含样本名、染色体、起始位置、终止位置、拷贝数和segments片段信息;lst值计算步骤,包括根据total cna谱计算样本的lst值,具体的,删除小于3m的segments片段,绘制segments片段密度分布图;取第一个局部最小值为cna cutoff;若特定区间0.025到0.45之
间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加;当相邻两个segments片段小于cna cutoff,则将两个segments片段拟合;计算cna cutoff的规则包括,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1;待测样本全基因组复制分析步骤,包括判断待测样本是否发生全基因组复制,如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值;同源重组修复缺陷状态评估步骤,包括根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。
[0087]
实施例
[0088]
本实施例中涉及的肿瘤样本由北京吉因加医学检验实验室有限公司提供。共40例有parp疗效的真实临床样本,其中有20例brca野生型,20例brca突变型。本例的同源重组修复缺陷的评估方法包括:
[0089]
(1)核酸提取
[0090]
本例采用组织样本分别提取40例样本的dna,ta克隆连接接头建库,再用gene+seq2000进行全基因组测序,数据量为5g。得到原始数据。
[0091]
提取采用硅胶膜法,使用generead dna ffpe kitminelute核酸提取柱从福尔马林固定-石蜡包埋(ffpe)的组织中提取dna。
[0092]
提取后用diagenode bioruptor pico打断仪进行dna打断,使dna均匀打断到200-250bp。
[0093]
打断后的dna进行末端修复、加“a”、加gene+seq2000 4bp uid接头、引入index进行10cycles pcr,逐步构建成为测序可识别的全基因组dna文库。
[0094]
全基因组dna文库pooling后经过热变性、环化、消化、终止反应使线性文库成环。
[0095]
环化后的文库在gene+seq2000平台进行测序上机,测序深度为5。
[0096]
(2)全基因组测序数据获取和比对
[0097]
包括获取待测样本的低深度全基因组测序下机数据,去除接头。使用bwa-mem2软件对截取掉umi的序列进行参考基因组比对,生成sam文件,参考基因组为hg19,使用samtools软件包将比对结果进行fixmate、sort、markdup,samtools fixmate-m${sample}.sam${sample}_fixmate.bam,该步骤将上述sam文件作为输入文件,修复表头文件信息得到fixmate.bam文件;samtools sort${sample}_fixmate.bam-o${sample}_sort.bam,将fixmate.bam文件排序用于下一步分析;samtools markdup-r${sample}_sort.bam${sample}_markdup.bam,将排好序的bam文件过滤掉pcr产生的重复序列,得到最终的bam文件用于下一步分析。
[0098]
(3)全基因组测序数据质控
[0099]
使用bamdst软件对上一步最终生成的bam文件进行质量分析,该软件提供所需bam文件以及bed文件(即位点信息文件)即可产生对应样本的质量信息,包括比对率、测序深
度、gc含量、重复率。本例比对率大于95%,深度大于0.8为合格。gc含量与重复率无阈值,仅对样本质量进行辅助判断。本例的40例样本均合格。
[0100]
(4)污染数据过滤
[0101]
对数据进行污染率分析,污染率分析软件为verifybamid2.0.1,分析样本受污染的情况。具体的,将“(2)全基因组测序数据获取和比对”中最终生成的bam文件,使用verifybamid2.0.1软件进行处理,该软件通过使用群体性等位基因频率构建模型评估样本的污染率。本例具体的,污染率小于0.1为合格,40例样本均合格。
[0102]
(5)拷贝数变异分析
[0103]
cnv分析模块采用ace软件。该部分主要用于确定低深度wgs样本的肿瘤纯度,并且生成每个样本的total cna谱。total cna谱每一行为一个50kb窗口所包含的信息,包括样本名,染色体,起始位置,终止位置,拷贝数与片段信息(即segments片段),用于下一步分析。
[0104]
分析结果显示,本例的40例样本的肿瘤纯度在0.27-0.87之间,如表2所示。部分total cna谱信息如表1所示。
[0105]
表1 total cna谱信息
[0106]
samplechrstartendcopynumberssegments179008702td18500019000000.5316250130.546180615179008702td19000019500000.540312140.546180615179008702td195000110000000.5484816460.546180615179008702td1100000110500000.6005030210.546180615179008702td1105000111000000.6497483170.546180615179008702td1110000111500000.6442053270.546180615
[0107]
(6)lst值计算
[0108]
通过“(5)拷贝数变异分析”生成的total cna谱计算样本的lst值,本例计算lst使用的是shallowhrd软件进行计算,删除小于3m的segments片段。当相邻segments小于cna cutoff,则将2段segments拟合。cna cutoff是通过segment片段密度图来确定。取密度分布图第一个局部最小值为cna cutoff;若特定区间0.025到0.45之间不存在局部最小值,则将cna cutoff设置为segments片段密度曲线第一个峰中出现的小拐点,通过降低cna cutoff,使样本的lst增加。计算cna cutoff的规则如下,若局部最小值大于0.025且小于0.45,取第一个局部最小值作为cna cutoff;若局部最小值小于0.025时,取0.025作为cna cutoff;若局部最小值大于0.45时,取0.45作为cna cutoff;当局部最小值大于0.45时,且密度图在0.45之前出现导数值减小但未改变正负的小拐点时,计算0.025到0.45之间密度图的导数,导数的最小值对应的差值作为cna cutoff;当相邻两个segments片段差值大于cna cutoff,同时间隔小于3mb,且长度均大于10mb,则lst值加1。本例40例样本的lst值如表2所示。
[0109]
表2肿瘤纯度及lst值
[0110]
样本肿瘤纯度brca情况lst值样本肿瘤纯度brca情况lst值10.38+40210.43-3020.50+29220.32+32
30.58-10230.81-1940.45-20240.29+2950.64-38250.85-2460.36-33260.82+1970.87+19270.78+1980.80-25280.81+2490.38-26290.45-19100.80+28300.48+22110.27-33310.30+17120.34-33320.69-24130.40-0330.50+30140.44-34340.45+28150.61+23350.54+20160.61-23360.87+29170.77+22370.71-21180.85-9380.44+26190.40-22390.70+28200.64+30400.64-28
[0111]
表2中“brca情况”栏,“+”表示突变型,
“‑”
表示野生型。
[0112]
(7)待测样本全基因组复制分析
[0113]
本例通过拷贝数片段信息的极差情况判断样本是否发生wgd。对密度分布图中峰的规则进行判断与制定,特殊情况峰进行判断与处理等;最后,综合峰值个数与片段信息极差情况判断样本是否发生wgd。具体如下:
[0114]
对segments片段密度分布图显示的峰进行判断,判断标准包括,(1)只有大于最大峰值15%的峰才算作峰;(2)初步判断峰值个数大于2时,则按以下规则判断:如果峰与左右两侧波谷的距离均大于最大峰值的4%,则该峰参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,则该峰不参与峰值个数的计数;如果峰与左右仅一侧波谷的距离小于最大峰值的4%,下一个峰仍是相同情形,则记为一个峰。
[0115]
判断待测样本是否发生全基因组复制的方法包括,根据segments片段密度分布图,通过segments片段的极差情况判断样本是否发生全基因组复制,判断规则如下:
[0116]
a.当样本segments片段极差小于1时,样本不发生全基因组复制;
[0117]
b.当样本segments片段极差大于1,且峰值个数小于3时,样本不发生全基因组复制;
[0118]
c.当样本segments片段极差大于1,且峰值个数大于或等于3时,样本发生全基因组复制;
[0119]
d.当样本segments片段极差大于9,且峰值个数大于或等于2时,样本发生全基因组复制。
[0120]
如果有发生全基因组复制,并且样本segments片段极差大于2,同时segments片段密度图中峰值个数大于4,则lst值减9,作为最终的lst值。本例40例样本的全基因组复制判
断结果以及最终的lst值如表3所示。
[0121]
表3全基因组复制及校正的lst值
[0122][0123][0124]
表3中,0代表无wgd,1代表有wgd。
[0125]
(8)同源重组修复缺陷状态评估
[0126]
根据brca基因型别和最终的lst值判断同源重组修复缺陷状态为阳性或阴性。以模型数据中brca突变样本中的95%均为hrd阳性,确定hrd生物学阈值为15。具体的,同源重组修复缺陷状态评估规则为:当brca为突变型时,无论lst值为任何值,hrd均判断为阳性;当brca为野生型时,lst score大于等于15,hrd判断为阳性,否则为阴性。
[0127]
同源重组修复缺陷状态评估结果显示,40例样本中,5例brca野生型患者被判定为hrd阴性,35例为hrd阳性,与实际情况相符。通过临床提供的患者pfs信息,用r中的survival、survminer、dplyr包绘制生存曲线,如图3所示,p-value=0.0011,hr=0.193,说明本例的同源重组修复缺陷的评估方法对parpi的疗效(pfs)预测,在同源重组修复缺陷状态为阳性和阴性的样本之间存在统计学显著差异,即能够通过本例的同源重组修复缺陷状态评估,为parpi的疗效(pfs)预测提供更准确有效的参考依据。
[0128]
以上内容是结合具体的实施方式对本技术所作的进一步详细说明,不能认定本技术的具体实施只局限于这些说明。对于本技术所属技术领域的普通技术人员来说,在不脱离本技术构思的前提下,还可以做出若干简单推演或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1