基于基因共表达网络分析的癌症转录组数据处理方法与流程
1.本发明涉及一种基因数据处理方法,具体涉及一种基于基因共表达网络分析的癌症转录组数据处理方法。
背景技术:
2.近年来,癌症疾病的患病率越来越高,但是由于这类疾病治疗困难又极易复发,故对于癌症疾病的研究变得越来越重要。如果能利用生物信息学方法对癌症疾病进行功能基因模块的挖掘,并识别出其中的关键基因,必将能够进一步了解癌症疾病的患病机制,并对其临床治疗具有一定的帮助。
3.随着二代测序技术的快速发展,基因表达数据出现了爆炸式的增长,如何从大量数据中挖掘出隐藏的知识成为了后基因组时代的重要任务之一。与此同时,随着研究的深入,人们逐渐发现在细胞环境中,各种生物因子不是单独地行使作用,而是相互合作完成各种复杂的生物功能。故将各类生物数据采用适当的方法转化为生物网络,从而利用图论以及复杂网络理论的相关知识对其进行分析与挖掘,已成为处理海量生物数据的有效方法。生物网络是在生物学领域的科学问题中,利用生物元素构建的网络,网络中的结点代表生物元素,如蛋白质、基因等,而网络中的边则代表生物元素在生化、物理或功能上的相互作用关系。基因共表达网络是一种常用的生物网络,它的出现为基因组学的发展开辟了一个新的方向。
技术实现要素:
4.为了对癌症转录组数据进行有效处理,本发明提供一种基于基因共表达网络分析的癌症转录组数据处理方法。
5.本发明为解决技术问题所采用的技术方案如下:
6.本发明的基于基因共表达网络分析的癌症转录组数据处理方法,主要包括以下步骤:
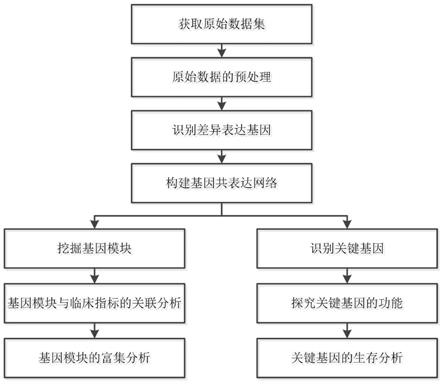
7.步骤一、获取原始数据集;
8.步骤二、原始数据的预处理;
9.步骤三、识别差异表达基因;
10.步骤四、构建基因共表达网络;
11.步骤五、挖掘基因模块;
12.步骤六、基因模块与临床指标的关联分析;
13.步骤七、基因模块的富集分析;
14.步骤八、识别关键基因;
15.步骤九、探究关键基因的功能;
16.步骤十、关键基因的生存分析。
17.进一步的,步骤一中,所述原始数据集来源于tcga数据库或geo数据库;所述原始
数据集包括癌症组织样本中的基因表达数据、癌旁组织样本中的基因表达数据和每个样本对应的临床数据。
18.进一步的,步骤二中,首先过滤掉低表达基因,然后对样本进行层次聚类,删除离群样本。
19.进一步的,步骤三中,利用fc-t算法识别出满足限定条件的所有差异表达基因。
20.进一步的,步骤四中,以差异表达基因在样本中的基因表达数据为基础,进行两两基因的皮尔森相关分析;设置限定条件对得到的所有关系进行筛选,将满足限定条件的两个基因视为存在共表达关系;将所有存在共表达关系的基因及其关系用一张图进行表示,即得到基因共表达网络。
21.进一步的,步骤五中,利用4种社团检测算法对基因共表达网络中的结点进行网络聚类,得到由功能相似的基因组成的社团即基因模块;使用“模块度”作为评价标准,选择最优的模块挖掘结果。
22.进一步的,步骤六中,对一个基因模块中所有的基因表达数据进行主成分分析,并将第一主成分定义为该基因模块的模块特征基因;将各基因模块的模块特征基因与不同的临床指标进行皮尔森相关分析,得到该基因模块与临床指标的关联矩阵。
23.进一步的,步骤七中,将感兴趣的基因模块中的基因与go数据库提供的生物过程、细胞成分和分子功能进行富集分析,同时将该基因与reactome数据库提供的信号通路进行富集分析。
24.进一步的,步骤八中,利用pagerank算法对基因共表达网络中所有结点的重要性进行打分,打分标准基于拓扑学原理,进而识别出在基因共表达网络中较为重要的结点,这些结点对应的基因即为关键基因。
25.进一步的,步骤九中,利用disgenet数据库检索与关键基因有关的疾病,对关键基因的功能进行探究。
26.进一步的,步骤十中,利用在线软件onclnc对关键基因进行生存分析,并绘制生存曲线。
27.本发明的有益效果是:
28.复杂网络理论在许多学科中都发挥着巨大的作用,近年来,其在计算机科学、物理学、社会学等学科中的应用都被广泛研究。生物体是一个高度复杂的体系,它的每一个生物学过程都需要许多物质的共同参与,研究单一的某个基因或蛋白难以从整体上去了解其背后所蕴藏的分子机制。由于癌症疾病的复杂性,现有的生物信息学分析方法难以对其转录组数据进行有效的分析与挖掘,因此,本发明将复杂网络理论应用到了生物学研究中,并具体应用到对癌症转录组数据的处理与分析方法中。
29.本发明提出了一种基于基因共表达网络分析的癌症转录组数据处理方法,主要包括:获取原始数据集;原始数据的预处理;识别差异表达基因;构建基因共表达网络;挖掘基因模块;基因模块与临床指标的关联分析;基因模块的富集分析;识别关键基因;探究关键基因的功能;关键基因的生存分析。由go/reactome富集分析结果可知,使用该方法划分的基因模块具有显著的生物学意义;由disgenet数据库对于关键基因的验证结果可知,使用该方法识别出的关键基因大部分均与肿瘤疾病有关。由此可证明本发明提供的一种基于基因共表达网络分析的癌症转录组数据处理方法在基因模块的挖掘及关键基因的识别方面
具有良好的效果。本发明的一种基于基因共表达网络分析的癌症转录组数据处理方法可以作为癌症疾病转录组数据的一个重要工具,该方法的应用也为进一步了解癌症疾病的患病机制提供了新的方向。
附图说明
30.图1为本发明的一种基于基因共表达网络分析的癌症转录组数据处理方法的流程图。
31.图2为实施例1中数据获取和预处理流程图。
32.图3为实施例1中癌症组织样本层次聚类树。
33.图4为实施例1中差异表达基因识别流程图。
34.图5为实施例1中差异表达基因火山图。
35.图6为实施例1中基因共表达网络构建流程图。
36.图7为实施例1中基因共表达网络及若干小网。
37.图8为实施例1中基因模块的挖掘流程图。
38.图9为实施例1中eigenvector算法的模块挖掘结果。
39.图10为实施例1中基因模块与临床指标的关联分析流程图。
40.图11为实施例1中基因模块与临床指标的关联矩阵。
41.图12为实施例1中基因模块的go/reactome富集分析流程图。
42.图13为实施例1中基因模块m1的bp富集结果。
43.图14为实施例1中基因模块m1的cc富集结果。
44.图15为实施例1中基因模块m1的mf富集结果。
45.图16为实施例1中关键基因的识别流程图。
46.图17为实施例1中基因naa40的生存曲线。
具体实施方式
47.以下结合附图对本发明作进一步详细说明。
48.本发明的一种基于基因共表达网络分析的癌症转录组数据处理方法,如图1所示,具体包括以下步骤:
49.步骤一、获取原始数据集
50.从tcga数据库(https://cancergenome.nih.gov/)或者geo数据库(https://www.ncbi.nlm.nih.gov/geoprofiles)获取原始数据集,原始数据集主要包括癌症组织样本中的基因表达数据、癌旁组织样本中的基因表达数据和每个样本对应的临床数据。
51.步骤二、原始数据的预处理
52.首先过滤掉其中的低表达基因,即将在癌症组织或癌旁组织中基因表达量(fpkm)的最大值小于1的低表达基因删除,然后将剩余的基因在癌症组织或癌旁组织中的表达量进行关于样本的层次聚类,并将离群样本进行删除,从而获得用于进一步挖掘的原始数据集。
53.步骤三、识别差异表达基因
54.利用fc-t算法识别出满足限定条件的所有差异表达基因。所说的限定条件为:fc》
=threshold||fc《=threshold&&p《=threshold,其中,fc表示threshold表示差异变化倍数,p表示t-test的统计学显著性。
55.步骤四、构建基因共表达网络
56.以差异表达基因在样本中的基因表达数据为基础,进行两两基因的皮尔森相关分析,得到其皮尔森相关系数(pcc)及p值;进一步地,设置限定条件|pcc|》=threshold&&p《threshold对得到的所有关系进行筛选,将满足限定条件的两个基因视为存在共表达关系;最后,将所有存在共表达关系的基因及其关系用一张图进行表示,即得到基因共表达网络。
57.步骤五、挖掘基因模块
58.利用4种社团检测算法(eigenvector、label-propagation、map-equation、edge-betweenness)对基因共表达网络中的结点(基因)进行网络聚类,得到由功能相似的基因组成的社团即基因模块;进一步地,使用“模块度”作为评价标准,选择最优的模块挖掘结果。
59.所说的4种社团检测算法(eigenvector、label-propagation、map-equation、edge-betweenness)分别使用r软件“igraph”程序包的函数leading.eigenvector.community()、label.propagation.community()、infomap.community()、edge.between ness.community()实现。
60.步骤六、基因模块与临床指标的关联分析
61.对一个基因模块中所有的基因表达数据进行主成分分析,并将第一主成分定义为该基因模块的模块特征基因(me);进一步地,将各基因模块的模块特征基因(me)与不同的临床指标进行皮尔森相关分析,取皮尔森相关系数(pcc)的绝对值,得到该基因模块与临床指标的关联矩阵。
62.步骤七、基因模块的go/reactome富集分析
63.将感兴趣的基因模块中的基因与go数据库(http://geneontology.org/)提供的生物过程(bp)、细胞成分(cc)和分子功能(mf)进行富集分析,同时将该基因与reactome数据库(https://reactome.org/)提供的信号通路进行富集分析。
64.步骤八、识别关键基因
65.利用pagerank算法对基因共表达网络中所有结点的重要性进行打分,打分标准基于拓扑学原理,进而识别出在基因共表达网络中较为重要的结点,这些结点对应的基因即为关键基因。可根据情况选择打分最高的前n个基因作为该疾病的关键基因。
66.步骤九、探究关键基因的功能
67.利用disgenet数据库(http://www.disgenet.org/)检索与关键基因有关的疾病,对关键基因的功能进行探究。
68.步骤十、关键基因的生存分析
69.利用在线软件onclnc(http://www.oncolnc.org/)对关键基因进行生存分析,并绘制生存曲线。
70.下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
71.实施例1乳腺浸润癌转录组数据分析
72.利用本发明的一种基于基因共表达网络分析的癌症转录组数据处理方法,对“肝细胞癌”的转录组进行数据分析,具体包括以下步骤:
73.(1)原始数据集的获取和预处理
74.如图2所示,具体包括以下步骤:
75.①
从tcga数据库(https://cancergenome.nih.gov/)中下载乳腺浸润癌(br ca)组织及其癌旁组织的表达谱数据,其中乳腺浸润癌组织样本共有1235个,癌旁组织样本共有149个,每个样本均包含60,483个基因。
76.②
将在乳腺浸润癌组织或癌旁组织中基因表达量(fpkm)的最大值小于1的低表达基因删除,共剩余14,129个基因。
77.③
将过滤后剩余的所有基因在乳腺浸润癌组织或癌旁组织中的表达量进行关于样本的层次聚类,层次聚类树如图3所示。由图3可知,共存在3个离群样本:tcga-dd-aaeb、tcga-cc-5259、tcga-fv-a4zp,将其删除后得到用于进一步分析的原始数据集。部分预处理后的原始数据(基因表达量)如表1所示。
78.表1预处理后的乳腺浸润癌组织基因表达数据
79.基因编号样本1样本2样本3样本4样本5ensg000001675782.9829626312.4269241782.1805546261.7044878431.574196206ensg000000782371.5114164092.9625679283.4967697942.5905459010.977175405ensg0000014608315.4236146734.185837527.123274776.7271153624.062698315ensg00000198242105.7124415207.8535728193.8028654113.9189313112.3564048ensg0000013410817.9167788819.2378533334.4252203826.488183514.67476604ensg0000016770025.3013904315.7393983912.300617185.99361125158.71591007ensg000000606425.2087184566.7045609236.2939901736.50486653411.32913478ensg0000016639100.2687484680.1949186840.15793994820.1794968ensg000000700872.8129582366.29303503217.2105787911.382587590.345172284ensg000001535618.7578266936.30464078915.1377772511.035223215.45512232
80.(2)差异表达基因的识别
81.如图4所示,具体包括以下步骤:
82.①
利用fc-t算法计算所有基因的fc值及p值,部分计算结果如表2所示。
83.②
设置限定条件fc》=2||fc《=0.5&&p《=0.05识别差异表达基因,共识别出4130个上调基因和471个下调基因。
84.③
利用r软件程序包“ggplot2”绘制差异表达基因火山图,以直观地显示差异表达基因的筛选结果,差异表达基因火山图如图5所示。
85.由表2及图4可知,与正常组织相比,乳腺浸润癌组织中有大量的基因发生显著的差异表达。
86.表2 fc-t算法计算结果
87.基因编号fcpensg000001460832.7009965211.13e-40ensg000001982422.3607430083.45e-33ensg000001677002.4812087844.38e-28
ensg000001663910.3024733581.03e-17ensg000001275112.2830661861.44e-46ensg000000646012.935507673.52e-58ensg000002277662.4440284433.37e-09ensg000000085172.3970338742.05e-13ensg000000700812.1845249792.20e-27ensg000002754793.4511668281.91e-23
88.(3)基因共表达网络的构建
89.如图6所示,具体包括以下步骤:
90.①
对于每个差异表达基因,计算其与其他差异表达基因的皮尔森相关系数(pcc)及p值,部分计算结果如表3所示。
91.②
设置限定条件|pcc|》=0.65&&p《0.05对得到的关系进行筛选,将满足限定条件的两个基因视为存在共表达关系。
92.③
将将所有存在共表达关系的基因及其关系导入cytoscape软件进行可视化,如图7所示。
93.④
根据可视化结果,将小网的基因删去,剩余的大网即为基因共表达网络。
94.表3皮尔森相关分析结果
[0095][0096][0097]
(4)基因模块的挖掘
[0098]
如图8所示,具体包括以下步骤:
[0099]
①
利用r软件“igraph”程序包的函数leading.eigenvector.community()、label.propagation.community()、infomap.community()、edge.betweenness.community()对基因共表达网络中的结点(基因)进行网络聚类,得到由功能相似的基因组成的社团即基因模块。
[0100]
②
计算4种社团检测算法(eigenvector、label-propagation、map-equation、
edge-betweenness)聚类结果的模块度,并选择模块度最大的结果进行进一步研究。本实施例中,eigenvector算法的模块度最高,故在此选择eigenvector算法得到的模块挖掘结果进行进一步研究。
[0101]
③
删除基因数量过小的社团(本实施例中删除基因数量小于50的社团),共剩余9个社团,对应于9个基因模块。eigenvector算法的模块挖掘结果如图9所示。
[0102]
(5)基因模块与临床指标的关联分析
[0103]
如图10所示,具体包括以下步骤:
[0104]
①
对各基因模块中所有的基因表达数据进行主成分分析以得到各基因模块的模块特征基因(me)。
[0105]
②
将各基因模块的模块特征基因(me)与4个临床指标event、t、n、m(其中,event表示患者的生存状态,t、n、m表示肿瘤分期)进行皮尔森相关分析,取皮尔森相关系数(pcc)的绝对值,得到基因模块与临床指标的关联矩阵,如图11所示。由图11可知,基因模块m1、m2、m3、m7与临床指标具有较高的相关性。
[0106]
(6)基因模块的go/reactome富集分析
[0107]
如图12所示,具体包括以下步骤:
[0108]
①
将基因模块m1、m2、m3、m6所包含的基因分别与go数据库所提供的生物过程(bp)、细胞成分(cc)、分子功能(mf)进行富集分析,并选择p值最小的10个term进行研究。基因模块m1的富集分析结果如图13~15所示。
[0109]
②
将基因模块m1、m2、m3、m6所包含的基因分别与reactome数据库所提供的信号通路进行富集分析,并选择p值最小的10个信号通路进行研究。基因模块m1的富集分析结果如表4所示。
[0110]
由各基因模块的富集结果可知,各基因模块的富集结果均具有较大的特异性,并且大多与肿瘤疾病有关,以此可证明模块挖掘结果的可靠性。
[0111]
表4基因模块m1的reactome富集结果
[0112]
通路编号通路名称富集数量pr-hsa-69278cell cycle,mitotic641.11e-16r-hsa-1640170cell cycle711.11e-16r-hsa-453279mitotic g1 phase and g1/s transition242.57e-12r-hsa-73886chromosome maintenance196.22e-12r-hsa-69205g1/s-specific transcription133.12e-11r-hsa-69206g1/s transition203.50e-10r-hsa-68886m phase324.48e-10r-hsa-69190dna strand elongation111.62e-09r-hsa-73894dna repair282.67e-08r-hsa-69242s phase193.58e-08
[0113]
(7)关键基因的识别
[0114]
如图16所示,具体包括以下步骤:
[0115]
①
利用pagerank算法对基因共表达网络中的所有基因的重要性进行基于拓扑结构的打分。
[0116]
②
将所有基因按照打分结果降序排列。
[0117]
③
选择前20个基因为肝细胞癌的关键基因。本实施例中的20个关键基因为:fabp7、cxcl3、loc284578、capn6、nrg2、hcfc1、ilf3、kansl1、naa40、ncoa6、pcdhb2、grik2、frmd7、ccser1、pcdhga1、pcdha1、lrrc37a6p、pcdhga12、znf486和pcdhgb5。
[0118]
(8)关键基因的功能探究
[0119]
将所有关键基因依次输入disgenet数据库(http://www.disgenet.org/)进行相关疾病的检索。其中,基因naa40的检索结果如表5所示。
[0120]
由disgenet数据库的检索结果可知,20个hub基因中大部分基因均与肿瘤疾病有关,以此可以证明本发明提出的基于基因共表达网络分析的癌症转录组数据处理方法的可靠性。
[0121]
表5基因naa40的检索结果
[0122][0123]
(9)关键基因的生存分析
[0124]
将所有关键基因利用在线软件onclnc(http://www.oncolnc.org/)进行生存分析并绘制生存曲线(cancer选择“brca”)。其中,基因naa40的生存曲线如图17所示。
[0125]
由关键基因的生存分析可知,20个关键基因均与患者的生存存在显著的相关性,这进一步证明了本发明提出的基于基因共表达网络分析的癌症转录组数据处理方法在关键基因识别方面具有显著的作用。
[0126]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1